
Rapid evolution of piRNA clusters in the Drosophila melanogaster ovary
Abstract
The piRNA pathway is a highly conserved mechanism to repress transposable element (TE) activity in the animal germline via a specialized class of small RNAs called piwi-interacting RNAs (piRNAs). piRNAs are produced from discrete genomic regions called piRNA clusters (piCs). Although the molecular processes by which piCs function are relatively well understood in Drosophila melanogaster, much less is known about the origin and evolution of piCs in this or any other species. To investigate piC origin and evolution, we use a population genomic approach to compare piC activity and sequence composition across eight geographically distant strains of D. melanogaster with high-quality long-read genome assemblies. We perform annotations of ovary piCs and genome-wide TE content in each strain. Our analysis uncovers extensive variation in piC activity across strains and signatures of rapid birth and death of piCs. Most TEs inferred to be recently active show an enrichment of insertions into old and large piCs, consistent with the previously proposed “trap” model of piC evolution. In contrast, a small subset of active LTR families is enriched for the formation of new piCs, suggesting that these TEs have higher proclivity to form piCs. Thus, our findings uncover processes leading to the origin of piCs. We propose that piC evolution begins with the emergence of piRNAs from individual insertions of a few select TE families prone to seed new piCs that subsequently expand by accretion of insertions from most other TE families during evolution to form larger “trap” clusters. Our study shows that TEs themselves are the major force driving the rapid evolution of piCs.
Animal genomes are parasitized by a horde of transposable elements (TEs) whose mutagenic activity can lead to sterility (Bingham et al. 1982; Bucheton et al. 1984). In the animal germline, the Piwi-interacting RNA (piRNA) pathway is a conserved small RNA-based mechanism regulating TE activity (Lau et al. 2006; Brennecke et al. 2007; Houwing et al. 2007; Grimson et al. 2008). piRNAs are 23- to 32-nucleotide RNAs produced from discrete loci called piRNA clusters (piCs) that guide effector Piwi proteins to silence TEs (Ozata et al. 2019). The piRNA pathway presents features of an adaptive defense system against TE invasion (Aravin et al. 2007; Brennecke et al. 2008; Khurana et al. 2011; Yu et al. 2019), but little is known about the processes driving its evolution. Many piRNA pathway genes encoding the proteins involved in TE silencing display signatures of adaptive evolution (positive selection) in several species’ lineages (Simkin et al. 2013; Yi et al. 2014; Palmer et al. 2018), which may indicate adaptation to a rapidly changing TE sequence pool and new invasions (Cosby et al. 2019). However, little is known about the means by which piRNA-producing loci, piCs, originate and evolve in flies or any other species.
piRNAs are produced from long noncoding RNA precursors that are transcribed from dispersed loci called piRNA clusters (Brennecke et al. 2007; Mohn et al. 2014). piCs occupy 0.1%–3% of the genome in fruit flies, mosquitoes, and mice and are enriched for TEs and other repeats such as satellites, as well as sometimes host gene sequences (Brennecke et al. 2007; Houwing et al. 2007; Chirn et al. 2015; Chen et al. 2021; Ma et al. 2021). The best-characterized function of piRNAs is to repress TEs. Because TE activity and composition vary significantly between and within species, TEs themselves may be important drivers of piC evolution, but this has not been thoroughly tested. TEs show high diversity in their mechanisms of transposition and their genomic distribution (Sultana et al. 2017; Wells and Feschotte 2020), as well as spatial and temporal activity (Pasquesi et al. 2020; Lawlor et al. 2021; Chang et al. 2022). Hence, it is likely that piCs evolve through diverse mechanisms to repress newly introduced TEs.
The organization of piCs is best characterized in Drosophila melanogaster. The genome-wide piC landscape in the D. melanogaster ovary is composed of tens of large (>10 kb) loci and hundreds of smaller (<10 kb) loci. It is also known that most large clusters (>10 kb) reside in pericentromeric and subtelomeric regions. Larger pericentromeric clusters consist of tens to hundreds of diverse TE insertions, whereas the small clusters (<10 kb) often contain recent TE insertions (Shpiz et al. 2014; Baumgartner et al. 2022; Miller et al. 2023). The architecture and composition of some large clusters suggest a “trap” model for the evolution of piCs, wherein TE insertions within clusters are selectively favored because of their production of piRNAs to repress all copies of the same active family (Bergman et al. 2006; Brennecke et al. 2007; Zanni et al. 2013; Kelleher et al. 2018; Kofler 2019). Over time, this process is predicted to result in enlargement of piCs, which serves as the host's archive of past TE activity. This is consistent with the observation of large piCs producing a bank of diverse piRNAs related to previously encountered TEs. It is important to note that recent studies reported bias in the ovarian piRNA sequence pool and piC loci composition toward younger TE insertions (Saint-Leandre et al. 2020; Gebert et al. 2021; Said et al. 2022). This indicates that evolutionary processes rapidly shape piC sequence composition to target the most recently active TEs.
In contrast to the large canonical piCs, individual TE insertions can also function as piCs (Olovnikov et al. 2013; Le Thomas et al. 2014; Akkouche et al. 2017). Recent studies have shown that piRNAs from large “trap”-like piCs are not essential for TE silencing, nor do they contribute the majority of piRNAs out of the total piRNA pool (Gebert et al. 2021; Genzor et al. 2021). Hence, piCs from individual TEs may produce a substantial quantity of piRNAs, which abrogates the need for active TEs to land into existing “trap” clusters to come under the control of the piRNA pathway. Although the “trap” model of piC evolution has received empirical support, its relative contribution to piC evolution in D. melanogaster is unclear, and the mechanisms underlying the origin and evolution of piCs remain broadly uncharacterized.
Results
Extensive variation in the genomic landscape of piCs
To quantify piC variation in D. melanogaster, we generated a comprehensive annotation of active piCs in eight highly inbred strains. Seven of these strains are derived from natural populations of distinct worldwide origins and have publicly available long-read genome assemblies (Chakraborty et al. 2019). For each of these seven strains, we constructed and sequenced libraries of small RNAs isolated from ovaries of two biological replicates sampled 6 mo apart (Supplemental Fig. S1; Supplemental Table S1). In addition, we analyzed two ovarian small RNA libraries for the reference iso-1 strain, generated as part of two independent studies (Shpiz et al. 2014; Asif-Laidin et al. 2017). Each small RNA library is analyzed separately using the pipeline discussed briefly here and in more detail in the Methods section, which lists three methods to define piCs (Supplemental Fig. S2). The restrictive and proTRAC methods serve the purpose of discovering moderately to highly expressed piCs using uniquely and multimapping piRNAs, respectively. The permissive method is performed mainly to validate low to moderately expressed piCs detected by proTRAC using multimapping piRNAs. Both restrictive and proTRAC methods yield highly reproducible piC coordinates across replicates of each strain with >88% of total piC count shared between two replicates overlapping over >75% of their respective length (Fig. 1A; Supplemental Fig. S3; Supplemental Table S3). However, interstrain pairwise comparisons reveal that pairs of strains share only an average of ∼40% of their total piCs counts. The high-confidence set of piCs from proTRAC, which either have high expression (>25) or were supported by uniquely mapping piRNAs, along with all piCs from the restrictive method for each replicate are combined to create a replicate-specific “master list” of piCs.
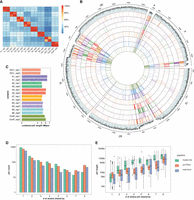
Interstrain variability of piCs in D. melanogaster strains. (A) Cross-strain and replicate overlap (percentage of total piCs count) of independently predicted piCs for each small RNA library using the restrictive method. Interstrain comparisons are performed after piCs were remapped to iso-1 coordinates, and overlap >1 bp between two piCs is deemed as shared. Intrastrain comparisons are performed similarly but in native genome assembly. (B) Genome-wide distribution of lifted-over piCs in seven DSPR strains and reference iso-1 strain. Bars along the circumference represent the presence of piCs in 10-kb bins for each chromosome. The outermost bar plot is piRNA mappability scores, followed by iso-1 piCs and then by piCs of seven DSPR strains. (C) Combined size of predicted piCs genome-wide from each replicate small RNA library independently in respective genome assemblies. (D) Population frequency of piCs quantified after liftover to the reference iso-1 genome. (E) piC length distribution by population frequency in kilobase pairs quantified after liftover to the iso-1 genome.
Genome-wide visualization of piC annotations in iso-1 coordinates across chromosomes reveals variability in the piC landscape across the eight strains (Fig. 1B; Supplemental Table S4). In aggregate, the total amount of genomic DNA covered by active piCs in each strain ranged from 4.8 Mb to 6.3 Mb (Fig. 1C), encompassing 3.4%–4.8% of their respective genome assemblies (Supplemental Fig. S4C). Although their piC landscape is broadly similar in terms of being denser within peri-centromeric and telomeric heterochromatic regions (characterized by low mappability scores) compared with euchromatic regions, it is readily apparent that many individual clusters are present in only one or a few strains, even within these heterochromatic regions. Smaller piCs, generally located in but not exclusive to euchromatic regions, are characterized by higher mappability scores and appear even more variable across strains (Fig. 1B). Thus, from this broad-scale view, it appears that the total span of the genome occupied by piCs within each strain is largely similar, but the positions of piCs are highly variable across strains.
Abundant strain-specific and strain-biased piCs
To quantify the frequency of piCs across the eight strains, we scored the overlap of piCs predicted independently for each of the eight strains using the master-list coordinates. To account for changes in size of piCs among strains, we required a minimum positional overlap of only 1 bp for piCs to be considered shared between strains. Even when using this relaxed criterion, we found that 568 (restrictive) to 906 (proTRAC) of piCs are active only in a single or a few strains, confirming that each strain has a unique piC landscape (Fig. 1D). The results are similar whether we used the piC predictions of the restrictive and proTRAC methods separately or the combined master list (Fig. 1D; Supplemental Table S2). All strains contained 35–60 piCs that are strictly unique to that strain and another approximately 30 piCs (<10%) that could not be lifted-over and therefore are likely to be strain specific also (Supplemental Fig. S4A). Thus, we can conservatively estimate that each strain possesses more than 50 piCs that are not shared by any of the other seven strains examined. In addition, 142 and 69 piCs (restrictive pipeline) are shared between two and three strains, respectively. All such piCs, shared by four strains or less, are together termed as “rare” piCs. Rare piCs not only are extremely abundant but also show significant piRNA expression ranging from 10–25 RPM, which is comparable to previously described canonical piCs like 80EF and the traffic jam 3′ UTR (Fig. 2; Supplemental Fig. S5). Additionally, despite their small size, in aggregate, strain-specific piCs contribute a substantial portion of the total genomic span of piCs (average of ∼1 Mb) and 15%–20% of the total piC genomic length of each strain (Supplemental Fig. S4B).

Natural variation in expression of uniquely mapping piRNAs from 42AB, 76C, and 76D. (A) Uniquely mapping piRNA abundance profiles of 42AB piCs for the four trains with two small RNA library replicates. The y-axis values are piRNA reads for 100-bp bins per million miRNA reads. Mappability scores (0–1) are shown for 100-bp bins of each respective 42AB genomic assembly in the heatmap. (B) Uniquely mapping piRNA expression profile of 76C, 76D, and 76E piCs for the six strains with two small RNA library replicates. The y-axis values are piRNA reads for 100-bp bins per million miRNA reads. Mappability scores (0–1) are shown for 100-bp bins of each respective 76C, 76D, and 76E genomic assembly in the heatmap. (C) Ribbon plot of multiple-sequence alignment for 42AB assembly in four strains. Only alignments of >80% identity and >1 kb in length are shown.
Next, we examined the relationship between the size of piCs and their level of sharing across strains. First, we note that the piC length predicted from each library is similar, with a median from 5.7 kb to 7.5 kb (Supplemental Fig. S4C). We find that piC size is positively correlated with the level of sharing across strains, and this correlation holds true for all prediction methods (Pearson's r = 0.56 for proTRAC, 0.58 for restrictive, and 0.76 for master-list, P-value < 2.2 × 10−16) (Fig. 1E). We also verified this correlation with differing piC overlap parameters to compute sharing across strains (Supplemental Fig. S11). In other words, piCs detected in a single or a minority of the strains (rare piCs) tend to be smaller (2–10 kb) than those shared by most of the strains (common piCs). If we posit that rare piCs compared with common piCs represent evolutionarily younger piCs, this relationship suggests that piCs are born relatively small and increase in size as they get older. Alternatively, larger piCs may be more evolutionarily stable than smaller ones. We note, however, that even large piCs can still be variable in activity across strains. For example, large well-known piCs like 42AB and 38C are still only active in six or seven of the eight strains (see below). Taken together, these results suggest that ovarian piCs are extremely labile and poorly conserved in activity across D. melanogaster strains.
Extensive variability in piRNA expression of piCs
To illustrate the differences in activity of piCs among the eight strains, we examined the piRNA coverage profiles for 42AB and 38C, two large canonical piCs, and three small piCs with varying activity across strains in 76C to 76E cytogenetic regions (Fig. 2). The 42AB piC has been extensively documented for its high piRNA expression (Brennecke et al. 2007; Klattenhoff et al. 2009). We present normalized coverage of uniquely mapping piRNAs to per million miRNAs (Fig. 2A) for the respective 42AB assemblies from both small RNA library replicates of four strains. Additionally, to examine differences in read coverage owing to mappability, theoretical mappability scores are visualized along the length of the cluster in 100-bp bins. Strains A1 and A7 have severely reduced (more than 20-fold) piRNA expression levels throughout 42AB compared with the other strains, whereas iso-1 and B6 show 42AB piRNA abundance similar to levels reported in other studies (Brennecke et al. 2007; Klattenhoff et al. 2009). Similarly, 38C, a highly active dual-stranded piC in iso-1, shows significant variability in uniquely mapping piRNAs across strains (Supplemental Fig. S5). Similar interstrain difference in normalized piRNA abundance was observed when piRNA coverage was normalized by sequencing depth, as exemplified by 38C (Supplemental Fig. S5) and additional small piCs for strain B6 (Supplemental Fig. S6). Because 42AB and 38C are active in six out of eight strains, it is most parsimonious to conclude that these piCs are relatively old but have lost activity in a subset of strains.
Two piCs in the 76C, 76D, and 76E cytogenetic regions are shown because each shows different levels of sharing across strains. 76C is a ∼15-kb piC, likely unistranded, exclusively active in strains A7 and B3, whereas 76D is a dual-stranded piC ranging from 6 kb to 20 kb in size but active in all the six strains shown (Fig. 2B). 76E, a unistranded piC with relatively low piRNA abundance, is considered active in all strains except A2. Comparison of such syntenic piCs between strains in their native genome assemblies provides validation of the variable activity of piCs across strains presented earlier from annotation pipelines (Fig. 1B). In addition, we also note that although genomic assemblies of 42AB from different strains are similar in size, there are structural rearrangements, which may contribute to differences in sequence composition of this piC across strains (Fig. 2C; Supplemental Figs. S8, S9).
Structural variation in piCs supports aspects of the “trap” model
To understand the mutational processes underlying the changes in structure and sequences of piCs among strains, we examined the contribution of interstrain structural variants (SVs), namely, insertions and deletions (indels). We detected indels for each of the strains relative to the iso-1 reference strain (Solares et al. 2018; Chakraborty et al. 2019). We mapped raw long sequencing reads for each strain to the iso-1 genome and called indels using three independent SV callers (see Methods). Indels from all strains were then collapsed to construct a list of unique SVs that consisted of 2273 putative insertions and 4409 deletions. Indels were then polarized into “true” insertions and deletions by comparison of each variant to the Drosophila simulans and Drosophila sechellia reference strains, which enabled inference of the ancestral state (see Methods) (Supplemental Fig. S10A,B). Polarization led to loss of ∼55% of indels as the ancestral or derived state of the loci could not be determined owing to conflicts in calls between the two outgroup species. After this filtering, 1183 insertions and 1873 deletions were retained for analysis (Supplemental Table S6).
We examined the size distribution of indels overlapping piC and non-piC regions of the genome to assess indels associated with piC variation. First, genome-wide, insertions range from 30 bp to 91 kb with a median of 612 bp, whereas deletions range from 30 bp to 7.6 kb with a lower median of 208 bp compared with insertions (Fig. 3A), which is consistent with previous SV profiling of D. melanogaster strains (Dopman and Hartl 2007; Zichner et al. 2013; Huang et al. 2014). However, insertions overlapping piCs have a significantly larger median length (2.2 kb vs. 0.512 kb) than insertions nonoverlapping piCs (Kruskal–Wallis test, χ2 = 10.812, df = 1, P-value = 0.001). Meanwhile, deletions overlapping piCs are not significantly different in length than those nonoverlapping piCs (Kruskal–Wallis test, χ2 = 0.72404, df = 1, P-value = 0.39) (Fig. 3B). We also compared the length distribution of indels in piCs grouped by the number of strains with which they are shared. We find that strain-specific or rare piCs (shared by less than four strains) are associated with relatively large insertions (median length of 5.2 kb), whereas common piCs (shared by more than half of the strains) have a median insertion length of <1 kb (Fig. 3C). In sum, rare piCs are associated with relatively large insertions, which is consistent with the idea that these piCs emerged from recent TE insertions.
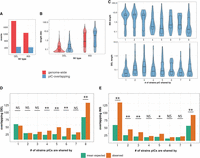
Common piCs show “trap” like sequence turnover. (A) Observed counts of indels genome-wide and overlapping with piCs. (B) Length distribution of indels genome-wide and overlapping with piCs. Significant differences are shown from Kruskal–Wallis test comparisons. (C) Length distribution of indels overlapping piCs grouped by the number of strains they are shared by (i.e., population frequency). (D,E) Enrichment analyses of deletion (DEL) and insertions (INS) variants in piCs performed. Variants overlapping with piCs of differing population frequencies are compared with the expected mean overlap counts genome-wide. Variant calls were shuffled 1000 times. (*) P < 0.05, (**) P < 0.005.
Next, we tested whether piCs are enriched for indels relative to the rest of the genome. To do this, we compared the indel counts overlapping piCs for each of the piC frequency categories with those expected based on 1000 sets of randomly shuffled indels. We find that deletions are significantly enriched in common piCs, but not in rare piCs (Fig. 3D). Insertions are strongly enriched both in rare and common piCs (Fig. 3E). These results may be confounded by the location of many piCs within constitutive heterochromatin, for which the rate of SVs is generally high (Montgomery et al. 1991; Chakraborty et al. 2021). However, we find that only ∼28% of all piCs lie within constitutive heterochromatin boundaries of the reference genome assembly, and indels are significantly enriched in piCs even when we compare them to heterochromatic regions (Supplemental Fig. S7; Riddle et al. 2011). Thus, the SV enrichment we observe within the common piCs is unlikely to be solely driven by their location within constitutive heterochromatin. Overall, we conclude that piCs are subject to a high rate of structural genomic change relative to the rest of the genome, which likely contributes to their rapid evolutionary turnover. Additionally, we find that common piCs are enriched for both insertions and deletions, which is consistent with these clusters evolving as “traps” that expand and contract. In contrast, rare piCs are only enriched for insertions, which supports the notion that these are generally young clusters born from presumably recent TE insertions.
TE reannotation of each strain uncovers ∼3 Mb of unannotated TE DNA
Although our analysis supports that SV events are important drivers of piC evolution, it does not directly address the role of TE activity. To assess the contribution of TEs to the composition and changes in the activity of piCs across strains, we performed de novo annotation of TEs in each of the strain genome assemblies. This was necessary because many TE consensus sequences present in the reference TE library for D. melanogaster (FlyBase release 2019_05) were discovered and curated more than two decades ago using primarily the iso-1 and Oregon-R strains (Bowen and McDonald 2001; Bartolomé et al. 2002; Kaminker et al. 2002). However, recent advances in long-read sequencing technology have provided a means to obtain a more unbiased view of the repetitive landscape of Drosophila genomes, revealing novel TE families (Ellison and Cao 2020; Han et al. 2022; Rech et al. 2022). We developed a TE annotation pipeline based on RepeatModeler2 (for discovery) (Smit 1999; Flynn et al. 2020), RepeatMasker (for annotation), and additional tools to distinguish novel TEs from known TEs and curate a comprehensive TE library for the eight strains used in this study (see Methods) (Fig. 4A).
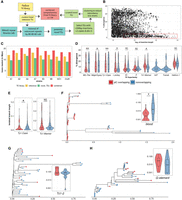
De novo TE annotation uncovers ∼3 Mb of hidden TEs and reveals strong associations of young LTR TEs with piCs more than any other TE subclasses. (A) TE annotation pipeline using RepeatModeler2 and RepeatMasker to create the comprehensive TE library. (B) Abundance of extremely similar and long TE insertions from RepeatMasker output of strain B6 using novel TE consensus library. (C) Differences in million base-pairs (Mbp) masked in RepeatMasker results using novel-only, reference-only, and combined TE library. (D) Divergence estimates for all defragmented iso-1 insertions (>200 bp) from RepeatMasker output. Insertions with >1-bp overlap with master-list iso-1 piCs are considered piC overlapping. Difference between groups is tested by Wilcoxon rank-sum test. (E) Terminal branch length for all iso-1 insertions from Ty1/Copia and Tc1/Mariner superfamilies from maximum likelihood trees. (F–H) Maximum likelihood trees constructed from all defragmented insertions for blood, Tc1-2, and G-element families, and the inset shows terminal branch length quantification. Difference between groups is tested by Wilcoxon rank-sum test: (*) P < 0.05, (**) P < 0.005, (***) P < 0.0005.
TE family sequence assemblies by RepeatModeler2 for each strain were aligned to the reference TEs using the 80-80-80 rule (Wicker et al. 2007). RepeatModeler2 sequences already present in the reference TE library were removed. Next, the remaining RepeatModeler2 sequences were used to remask the genomes to examine them for novel TE families. More than 5000 insertions (>500 bp in size, median of 676 bp), very similar (<5% divergence) to their respective RepeatModeler2 family consensus, were discovered for each strain (for an example of strain B6, see Fig. 4B). These novel insertions resulted in masking of an additional 2.5 Mb to 4 Mb in each genome assembly that would have been missed or misannotated as highly diverged insertions by masking with only the reference TE library (Fig. 4C). In summary, a refined and comprehensive TE library was created with a combination of 129 reference TE consensus sequences and 47 uncharacterized consensus (Supplemental Table S7) sequences that capture all TE insertions genome-wide and reflect their relative age.
Young LTR retrotransposons are enriched within rare piCs
To understand the coevolution of TE and piCs, we correlated TE insertion age and piC sharing across strains (as proxy for piC age). Using the new TE library described above, we sought to compare the age and composition of TEs within piCs to those in the rest of the genome. To infer the age of each family, we used the median sequence divergence of each insertion to their family consensus. To examine TE composition, we grouped TEs into the major subclasses and superfamilies represented in Drosophila: non-LTR retrotransposons (LINEs), LTR retrotransposons (Ty1/copia; Ty3/mdg4; BEL/Pao superfamilies), Rolling Circle (RC) transposons, and cut-and-paste DNA transposons. We found that TE copies from all three LTR superfamilies are significantly younger in piCs than non-piC regions (Fig. 4D). Conversely, TE copies from the LINE, RC, and DNA subclasses are not significantly different in age in piCs than in non-piC regions. To test these results using an independent method to date insertions, we built phylogenetic trees from all copies for one LTR superfamily (Ty1/copia) and one DNA transposon superfamily (Tc1/mariner) and used terminal branch lengths to estimate their relative age (Carr et al. 2012). We chose these superfamilies because they are of moderate abundance and therefore manageable for multiple sequence alignments and phylogenetic analyses. The results of these analyses yielded the same trend observed genome-wide using sequence divergence from consensus sequences: The Ty1/copia LTR retrotransposons (n = 135) overlapping piCs are significantly younger than nonoverlapping ones, whereas Tc1/mariner (n = 89) DNA transposons show no such bias (Fig. 4E).
To examine whether these trends hold at the level of individual TE families, we selected one family with moderate copy number from the LTR, LINE, and DNA subclasses and compared the age of piC overlapping and nonoverlapping copies within each family. We analyzed as a representative Ty3/mdg4 LTR superfamily, blood, a family with 63 copies in the iso-1 strain that is known to be transpositionally active (Bingham and Chapman 1986; Kofler et al. 2015). Consistent with the trend observed at the level of the LTR superfamily, we found that 43 out of 63 blood insertions are associated with piCs. Most of these are very recent insertions with a median terminal branch length of <0.002, which is significantly shorter than that of insertions not overlapping with piCs (Wilcoxon rank-sum test, P-value = 0.014) (Fig. 4F). In other words, piC overlapping blood insertions are significantly younger than the nonoverlapping ones. We analyzed, as a representative of the Tc1/mariner superfamily of DNA transposons, Tc1-2, a family with 35 copies in the iso-1 genome. Consistent with the trend observed at the level of the entire superfamily, the age of the Tc1-2 copies overlapping piC is not significantly different than that of non-piC overlapping copies (Wilcoxon rank-sum test, P-value = 0.903) (Fig. 4G). Analyzing the G-element LINE family, which counts 35 copies in iso-1 and is still active (Di Nocera et al. 1986), we found that the age of piC overlapping copies is not significantly different from nonoverlapping copies (Wilcoxon rank-sum test, P-value 0.855), and the youngest G-element insertions according to terminal branch length do not overlap piCs (Fig. 4H). Taken together, these results suggest that young LTR retrotransposon insertions tend to be enriched in piCs, but this trend is not observed for other TE subclasses and superfamilies.
A small subset of active LTR retrotransposon families give rise to young piCs
To test the central prediction of the “trap,” in which recent transposition events from active TEs must be enriched in piCs, we established a set of nonreference TE insertions in each DSPR strain using the raw long-read data available for each. Briefly, we applied TLDR (a long-read TE insertion detection tool) (Ewing et al. 2020) with a cutoff of at least two supporting reads per 10× genome coverage to remove false positives and enrich for germline insertions (see Methods). Using these parameters, we identified 285 to 857 nonreference TE insertions in each of the seven DSPR lines but only 75 insertions for iso-1, which is expected because the reference genome is also derived from the iso-1 strain. Presumably, the 75 nonreference insertions for iso-1 reflect the use of different isolates for the reference genome assembly and for the long-read data. Further clustering and parsing of all nonreference insertions across the eight strains resulted in a list of 3545 unique TE insertions of at least 200 bp in length (Supplemental Table S8). These insertions belong to 165 of the 184 different families in our TE library. Ninety-four of these 165 TE families were classified as “active” because each of these included at least 10 nonreference insertions shared by no more than two strains, whereas the other 98 families were classified as “inactive” (Supplemental Table S9).
We used this compendium of insertions to test whether active TEs are significantly enriched within piCs using a binomial test to compare the observed overlaps with piCs to the average overlaps expected from 1000 random reshufflings of TE insertions (see Supplemental Methods; Kapusta et al. 2013). This analysis revealed that seven active TE families are significantly enriched in piCs, whereas one active and 10 inactive families are significantly depleted in piCs (Fig. 5A). All the inactive TE families that are significantly depleted in piCs belong to either DNA or LINE subclasses, with the exception that one active family depleted in piCs was 17.6, a Ty1/copia superfamily member (Inouye et al. 1986) with 79 nonreference insertions. These results are consistent with the prediction of the “trap” model that piCs are enriched for active TE families but also composed of inactive families.
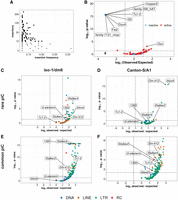
Insertions of only a few active LTR families associates with rare piCs. (A) Scatter plot of nonreference TE insertion counts and mean population frequencies of 176 TE families. Black dots highlight selected TEs classified as “active,” whereas gray dots are deemed “inactive.” (B) Enrichment analyses of nonreference insertions of TE families in master-list piCs using random shuffling of nonreference TE insertions. The y-axis P-values are from binomial tests conducted to compare observed counts to expected average overlaps of nonreference TE insertions to piCs for each family. (C–F) Enrichment analyses of TE families in master-list rare and common piCs of the A1/Can-S strain using random shuffling. All TE insertions (>200 bp) of strain iso-1 genome are randomly shuffled with constant piCs coordinates grouped into rare (shared by four strains or fewer) and common piCs (shared by five strains or more). P-values on y-axes are from binomial tests conducted to compare observed counts to expected average overlaps of TE insertions to piCs for each family. Statistically significant families are labeled.
Next, we sought to distinguish family-level enrichment of TE insertions within rare, recently arisen piCs (smaller piCs [<10 kb] shared by no more than three strains) and within common larger “trap”-like piCs (>10 kb, shared by at least five strains). To increase the statistical power for this analysis, we used all TE sequences annotated by RepeatMasker in each genome, instead of only nonreference insertions. For iso-1, we find that only four TE families are significantly enriched within young piCs. All four are LTR retrotransposon families of the Ty3/Mdg4 superfamily (blood, Dm-412, flea, Stalker-2), and all except flea belong to the Mdg1 lineage (Fig. 5C,D; Costas et al. 2001; Bertocchi et al. 2020). All four families are also classified as active in this study as well as previous studies that examined TE insertion frequency among D. melanogaster populations (Kelleher and Barbash 2013; Kofler et al. 2015). In contrast, we find that numerous active and inactive families from all the TE subclasses are significantly enriched in large and common “trap-like” piCs, largely representative of the overall TE landscape of D. melanogaster (Fig. 5C,D). In the A1 genome, 20 TE families are significantly enriched in rare piCs. Again, these are predominantly LTR retrotransposons (14 families), but four DNA transposon families and two LINE families are also significantly enriched. (Fig. 5E,F). 1360, a DNA transposon, is one of the four enriched DNA transposon families in A1 rare piCs but not in iso-1. It is possible these differences arise from changes in TE activity, the number of recent insertions, or strain background impacts TE's readily conversion into active piCs. blood insertions are neither enriched nor depleted in common piCs of both strains. Taken together, these analyses yield a contrasting portrait of TE composition in the two major types of piCs.
TE composition of piCs captures distinct steps in piC evolution
To further illuminate the evolution within piCs, we analyzed how the overall age and distribution of TEs of piCs change as they become more frequent in the population and presumably older. We plotted the mean percentage of divergence of individual TE insertions to their consensus sequences (a measure of TE age) across piCs and their flanking non-piC regions for each piC frequency class (strain-specific or shared by two to eight strains). We find that the divergence of TE insertions in rare piCs (shared by three or fewer strains) is markedly lower (3.5%–5%) than in their flanking regions (10%–15%) (Fig. 6A). In addition, the divergence of TE copies within piCs increases gradually with the frequency of the piCs to the extent that for the most common piCs (shared by seven and eight strains) the mean percentage of TE divergence is only slightly lower than in their flanking genomic regions (Fig. 6A). This apparent increase in average age of TE insertions as piCs become more common across strains provides weight to the inference that more common piCs represent evolutionarily older clusters relative to those that are strain specific or rare. This pattern also suggests that piCs are born from singleton TE insertions and grow by gradual accretion of TEs over time.
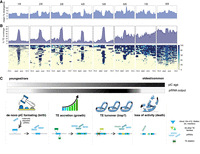
Age and distribution of TEs provide a portrait of intraspecific piC evolution. (A) TE age (by percentage of divergence) is plotted for piCs of increasing population frequency from left to right. Summary profile plot of mean percentage of TE insertion divergence in 500-bp windows in scaled piC regions and flanking non-piC regions of the reference genome. Each group represents piCs that are shared by 1/8 to 8/8 strains. (B) TE content of piCs of different stages are plotted. Summary profile plot and heatmap of mean TE coverage in 500-bp windows in scaled piC regions and flanking ±15-kb non-piC regions of the reference genome. Scaled piC regions are marked by start and end labels at the bottom of the heatmap. (C) New unified model of piC evolution: “Birth and death” is shown in four steps from left to right.
To further test this idea, we examined how TE coverage within piCs and surrounding regions changes as piCs become more common and presumably older (Fig. 6B). First, we observe that piCs generally show significantly higher TE coverage than their flanking genomic regions. Second, we find that strain-specific piCs, which presumably represent the youngest piCs, show high mean TE coverage in the middle at >60% (on average, 60 out every 100 bp is composed of TE sequence), which drops at the edges of piC coordinates to <20% (Fig. 6B). In contrast, more common piC groups show consistently higher TE coverage across their entire length. This pattern is consistent with a birth-and-growth process in which a piC emerges from individual TE insertion, but piRNA production spreads to flanking TE insertions as they insert near or within the piC.
Discussion
To study piC evolution at a fine-scale resolution in D. melanogaster, we used population genomic methods to characterize piC variation across eight inbred strains. A crucial asset was the availability of high-quality genome assemblies for these strains (Chakraborty et al. 2019). This enabled us to produce de novo annotation of piCs for each strain from mapping inferred piRNAs from ovarian small RNA libraries we constructed and sequenced for two biological replicates sampled six months apart. Our piC annotations for the two replicates showed high reproducibility with >88% of piCs annotated in one replicate found in the second replicate (Fig. 1A; Supplemental Fig. S3). Also, to understand variation in sequence composition and age of piCs, it was necessary to produce libraries of TE consensus sequences representative of the eight strains analyzed here. By performing de novo discovery and reannotation of TE families for each genome, we identified 47 novel TE families (Fig. 4). Although further investigation is required to examine their evolutionary origins and relationship to known TE families, it appears that many of the novel TE families we annotated were highly diverged from known families and often “hidden” in highly repetitive regions that would likely be poorly assembled in short-read genome assemblies. These results stress the benefits of high-quality genome assemblies and the necessity to perform de novo TE discovery when new strains or geographical isolates are considered. This is true even for model species like D. melanogaster, for which TEs have been extensively cataloged, because previous TE identifications were mostly based on a single reference genome. Robust annotation of piCs and TEs allowed us to compare the activity and TE composition of piCs across strains and derive general principles about piC origin and evolution.
Our findings recapitulate aspects of the “trap” model of piC evolution. First, we find enrichment of insertions of active TE families and depletion of inactive families in piC (Fig. 5B). In addition, we also find that “trap”-like large piCs are enriched for diverse TE families, which are relatively younger in piC than in flanking non-piC regions (Figs. 5D, 6A). This is consistent with the idea that “trap” piCs represent an archive of past TE activity, which biases piRNA production toward recently active TE families. Consistent with previously reported variation in piC activity (Rozhkov et al. 2010; Ellison and Cao 2020; Zhang et al. 2020), we find extensive intraspecific variation in piC activity, even for large “trap”-like piCs. For example, 42AB and 38C clusters show a significant loss in piRNA production in one or multiple strains (Fig. 2; Supplemental Fig. S6). What could cause the loss in piRNA production from large heterochromatic piCs? Our current lack of understanding of the cis-regulatory requirements for piC activity makes it difficult to determine whether changes in piRNA production are caused by genetic or epigenetic changes in the piCs. Consistent with previous studies (Ellison and Cao 2020; Wierzbicki et al. 2023), we observe considerable structural variation in large peri-centromeric piCs (Supplemental Fig. S7). It is possible that such structural changes result in changes in piRNA production, but further studies are needed to elucidate the mechanisms by which large and seemingly stable piCs lose their activity.
What can TE composition of piCs tell us about the coevolution of TEs and piCs? As previously reported (Kofler 2020; Wierzbicki et al. 2023), we find that diverse TE families (from all subclasses) are enriched in large, common piCs (Fig. 5). This enrichment may be explained by selection against new TE insertions in gene-rich euchromatic regions, which leads to accumulation of TEs in heterochromatic regions, where purifying selection is also weak (Charlesworth and Langley 1989; Bartolomé et al. 2002; Blumenstiel et al. 2002; Dolgin and Charlesworth 2006; Schrider et al. 2013). However, our genome-wide analysis revealed that SV enrichment in common piCs cannot be completely explained by their overlap with heterochromatin (Supplemental Fig. S8). Thus, the enrichment of diverse elements within large common piCs may be driven in part by the lesser deleterious effects of their accumulation in these regions and/or positive selection for their piRNA production.
Are particular TEs prone to give rise to piCs? To answer this question, we tested for enrichment of individual TE families within rare piCs, which likely represent young clusters. Only a small set of retrotransposon families are enriched within such clusters, and most belong to the mdg1 subclade of LTR retrotransposons such as blood and Dm-412 (Fig. 5). Why would these elements be prone to seed new piCs? There are two possibilities, which are not mutually exclusive. One possibility is that these TE families are readily licensed as piCs from maternally inherited piRNAs, thereby giving rise to the emergence of dispersed piC loci following recent transposition events (Le Thomas et al. 2014; Shpiz et al. 2014). Alternatively, it may be linked to their propensity to produce double-stranded RNA (dsRNA) and endogenous siRNAs, as endo-siRNAs production has been associated with the formation of piCs from transgenes (Olovnikov et al. 2013; Akulenko et al. 2018). Many LTR (including blood) and non-LTR retrotransposons are known to possess bidirectional promoters that can result in the formation of dsRNAs that stimulate the production of siRNAs (Watanabe et al. 2008; Russo et al. 2016; Hung and Slotkin 2021). It is possible that the inherent capacity of some retrotransposons to produce dsRNAs nucleate the formation of piCs. This idea has received support in a recent study showing that endogenous siRNA production precedes piC formation, and maternal inheritance of these siRNAs is required for licensing of piCs (Luo et al. 2023). However, we cannot rule out that these rare TE-specific piCs arose exclusively from maternally inherited piRNAs as piRNAs from such piCs could not be differentiated from the piRNAs produced from insertions in large common piCs (Supplemental Fig. S12).
In summary, a picture is emerging in which the rapid evolution of piCs among D. melanogaster strains is driven by the activity of TE families themselves. We propose a “birth-and-death” model of piC evolution, which combines components of the conventional “trap” model and features of piCs found in this study (Bergman et al. 2006; Khurana et al. 2011; Shpiz et al. 2014; Gebert et al. 2021). In this model (Fig. 6C), we posit that piCs form frequently throughout the genome, often from recent TE insertions, with certain LTR retrotransposon families making a stronger contribution to seeding new piCs. Newly emerged piCs may increase in frequency and size owing to natural selection or drift, depending on factors such as their propensity to trigger genomic autoimmunity (Blumenstiel et al. 2016; Huang et al. 2022), ectopic recombination (Petrov et al. 2003; Sentmanat and Elgin 2012), and the establishment of a chromatin environment conducive for piRNA production (Le Thomas et al. 2014). Over time, these stabilized clusters may grow by “trapping” additional TE insertions, which will eventually result in large heterochromatic clusters such as 42AB. Because of the host's limited capacity to maintain such piCs without incurring a fitness cost, those clusters that lack piRNAs targeting active TEs may gradually lose activity or become dispensable (Gebert et al. 2021). We can only speculate about the mechanisms that lead to the loss of activity of large piCs, but the rapid turnover of piC loci provide some possibilities. Because of re-establishment of female piC loci every generation from maternally deposited piRNAs, it is possible that a large piC may not get licensed if it only contains old insertions from inactive TE families, which are poorly represented in maternally deposited piRNAs. Because the maternally deposited piRNA pool is expected to diverge from generation to generation to incorporate new TEs, it may not contain enough homologous piRNAs for licensing of older piCs that lack recently active TEs. This seems consistent with high piRNA abundance of 42AB in five of the eight strains, low in two of the eight strains, and extremely low in one strain. The most parsimonious conclusion from such continuous variation can be drawn that 42AB activity is on the decline in D. melanogaster populations. Further studies are warranted to test this “birth-and-death” model. Our study provides a first in-depth view of piC evolution in D. melanogaster that is likely to stimulate other comparative studies of piRNA evolution.
Methods
Fly stocks
DSPR founder stocks of A1 (b1_paired), A2 (b3841_paired), A4 (b1_3852), A7 (t7_paired), B3 (b3864_paired), B6 (t1_paired), and Oregon-R were a gift from Anthony Long (UCI). All stocks were maintained on standard cornmeal medium at 22°C under a 12-h day–night cycle.
Small RNA library construction and sequencing
Small RNA libraries were constructed by size fractionation on urea-polyacrylamide gel electrophoresis as described by Ma et al. (2021), and additional details are provided in the Supplemental Methods. All libraries were quantified using Qubit 3.0, pooled into replicate-1 and replicate-2 groups, and analyzed on a Agilent Bioanalyzer. Single-end 75-bp Illumina sequencing was performed for all libraries on NextSeq 500 at the Cornell Biotechnology Resource Center.
piC annotation
Active piC annotation was conducted independently for each replicate of each strain using a custom pipeline adapted from previously described methods (Rosenkranz and Zischler 2012; Mohn et al. 2014). Detailed annotation steps of each pipeline are provided in the Supplemental Methods and outlined in Supplemental Figure S2.
Structural variation detection and filtering
Raw long reads for the seven DSPR strains, the iso-1 reference strain, D. simulans (wxd1), and D. sechellia (sech25) were mapped to the D. melanogaster iso-1 release 6 (GCA_000001215.4) without the Y Chromosome with minimap2.1 map-pb ‐‐N3, and resulting SAM file was converted to BAM and sorted (Li et al. 2009; Li 2018). Three SV callers—Sniffles-2.0 (Smolka et al. 2024), cuteSV-1.0.13 (Jiang et al. 2020), and svim-2.0 (Heller and Vingron 2019)—were used for SV detection. Filtering, genotyping, and collapsing of SV calls are detailed in the Supplemental Methods. Filtered and polarized SV calls are reported in Supplemental Table S5.
De novo TE annotation
To create a comprehensive and accurate representative TE library representing the TE insertions contained in the eight strains, de novo TE annotation was conducted using several computational tools. A summary of all major steps is presented in a flow chart in Figure 4A. Briefly, canonical FlyBase TE consensus sequences were filtered to include only TEs that best represent the TE insertion landscape of each strain using RepeatMasker-4.1.0 results (Smit 1999; Larkin et al. 2021). FlyBase TE families with at least three copies of >200 bp and <1% divergence were retained. This resulted in a reference library for the eight strains with 129 TE families. Next, RepeatModeler2 was run on the seven DSPR genomes and reference iso-1 strain followed by removal of non-TE repeats like tRNA, satellites, rRNA, etc., as well as TE subfamilies using bash scripts (Flynn et al. 2020). Putative novel TE family consensus was identified (see Supplemental Methods), and each genome was remasked with combined TE library (novel and reference TEs).
Nonreference TE insertion detection from long reads
Raw long reads used in SV detection were also used for unique TE insertion analyses. Reads (>1 kb) were mapped to iso-1 reference genome (without Y-linked contigs and all contigs <20 kb) using minimap2.1 default parameters, and the resulting SAM file was converted to BAM file, sorted, and indexed using SAMtools (Li et al. 2009; Li 2018). TLDR, a de novo TE detection program (Ewing et al. 2020), was run for each strain using the comprehensive D. melanogaster TE library curated in this study. High-confidence TLDR insertion calls for all eight strains are reported in Supplemental Table S7.
Data access
All raw and processed sequencing data generated in this study have been submitted to the NCBI BioProject database (https://www.ncbi.nlm.nih.gov/bioproject/) under accession number PRJNA1013381. Relevant source data and methods for figures are available as Supplemental Material. Additional raw data files, TE consensus sequences, and scripts are available as Supplemental Code and at GitHub (https://github.com/kerogens101/Dmel_piC).
Competing interest statement
The authors declare no competing interests.
Acknowledgments
This work was supported by National Institutes of Health grants R01-GM119125 to A.G.C. and R35-GM122550 to C.F. S.P.S. is supported by the Distinguished Scholar Award from the Cornell Center for Vertebrate Genomics. We thank Jullien Flynn for her assistance in RepeatModeler2 runs and consensus filtering and Michelle Stitzer for recommendations on structural variant analysis. We also thank Dan Barbash, Justin Blumenstiel, and Andrew Grimson for helpful discussions on the analyses and members of the Clark and Feschotte laboratories for valuable feedback on the manuscript.
Author contributions: S.P.S. performed all experiments. S.P.S. performed all computational analysis and created all figures with input from A.G.C. and C.F. S.P.S., A.G.C., and C.F. wrote the manuscript. Initial project conception was by S.P.S. and A.G.C.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.278062.123.
- Received May 8, 2023.
- Accepted May 7, 2024.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see https://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








