
Haplotype-resolved 3D chromatin architecture of the hybrid pig
- Yu Lin1,9,
- Jing Li1,9,
- Yiren Gu2,3,9,
- Long Jin1,9,
- Jingyi Bai1,
- Jiaman Zhang1,
- Yujie Wang1,
- Pengliang Liu1,
- Keren Long1,
- Mengnan He1,
- Diyan Li4,
- Can Liu1,
- Ziyin Han1,
- Yu Zhang1,
- Xiaokai Li1,
- Bo Zeng1,
- Lu Lu1,
- Fanli Kong1,
- Ying Sun1,5,
- Yongliang Fan1,
- Xun Wang1,
- Tao Wang4,
- An'an Jiang1,
- Jideng Ma1,
- Linyuan Shen1,
- Li Zhu1,
- Yanzhi Jiang1,
- Guoqing Tang1,
- Xiaolan Fan1,
- Qingyou Liu6,
- Hua Li6,
- Jinyong Wang7,8,
- Li Chen7,8,
- Liangpeng Ge7,8,
- Xuewei Li1,
- Qianzi Tang1 and
- Mingzhou Li1
- 1State Key Laboratory of Swine and Poultry Breeding Industry, College of Animal Science and Technology, Sichuan Agricultural University, Chengdu 611130, China;
- 2College of Animal and Veterinary Sciences, Southwest Minzu University, Chengdu 610041, China;
- 3Animal Breeding and Genetics Key Laboratory of Sichuan Province, Sichuan Animal Science Academy, Chengdu 610066, China;
- 4School of Pharmacy, Chengdu University, Chengdu 610106, China;
- 5Institute of Geriatric Health, Sichuan Provincial People's Hospital, University of Electronic Science and Technology of China, Chengdu 610072, China;
- 6Animal Molecular Design and Precise Breeding Key Laboratory of Guangdong Province, School of Life Science and Engineering, Foshan University, Foshan 528225, China;
- 7Pig Industry Sciences Key Laboratory of Ministry of Agriculture and Rural Affairs, Chongqing Academy of Animal Sciences, Chongqing 402460, China;
- 8National Center of Technology Innovation for Pigs, Chongqing 402460, China
-
↵9 These authors contributed equally to this work.
Abstract
In diploid mammals, allele-specific three-dimensional (3D) genome architecture may lead to imbalanced gene expression. Through ultradeep in situ Hi-C sequencing of three representative somatic tissues (liver, skeletal muscle, and brain) from hybrid pigs generated by reciprocal crosses of phenotypically and physiologically divergent Berkshire and Tibetan pigs, we uncover extensive chromatin reorganization between homologous chromosomes across multiple scales. Haplotype-based interrogation of multi-omic data revealed the tissue dependence of 3D chromatin conformation, suggesting that parent-of-origin-specific conformation may drive gene imprinting. We quantify the effects of genetic variations and histone modifications on allelic differences of long-range promoter–enhancer contacts, which likely contribute to the phenotypic differences between the parental pig breeds. We also observe the fine structure of somatically paired homologous chromosomes in the pig genome, which has a functional implication genome-wide. This work illustrates how allele-specific chromatin architecture facilitates concomitant shifts in allele-biased gene expression, as well as the possible consequential phenotypic changes in mammals.
It is well understood that the vast majority of somatic cells in mammals inherit two haploid genomes that unequally contribute to cellular function. Substantial differences have been identified between homologous chromosomes (homologs) in gene transcription (Reinius and Sandberg 2015), DNA methylation (Xie et al. 2012), and chromatin accessibility (Lindsly et al. 2021) and replicating time (Rivera-Mulia et al. 2018). In addition, higher-order chromatin structure is now emerging as an important regulator of gene expression (Oudelaar and Higgs 2021). Advances in high-throughput chromatin conformation capture (Hi-C) and its derivatives have led to revelations in the full scope of three-dimensional (3D) chromatin dynamics during mammalian development and lineage specification (Akgol Oksuz et al. 2021). However, conventional Hi-C data cannot generally distinguish between different allelic copies, and most previous studies infer an average chromatin architecture that incorporates the diploid genome without considering possible differences between homologs (Li et al. 2021). Variability in multilevel chromatin architecture between homologs remains largely unexplored; characterizing the 3D genome organization of a mammalian diploid genome remains challenging (Du et al. 2017; Han et al. 2020; Tan et al. 2021).
Sus scrofa (i.e., pig or swine) is of enormous agricultural significance and informative biomedical model for human developmental processes (Liu et al. 2021; Zhu et al. 2021) and complex diseases (Yan et al. 2018; Moretti et al. 2020) in addition to its utility in vaccine (Decaro and Lorusso 2020) and drug design (Schelstraete et al. 2020) owing to the relatively close anatomical (Jin et al. 2021; Lunney et al. 2021), physiological (Sjöstedt et al. 2020; Karlsson et al. 2022), immunological (Dawson et al. 2013), and genetic (Groenen et al. 2012; Warr et al. 2020; Zhao et al. 2021) similarities with humans. The ability to generate genome-editing mutations in the pig combined with somatic nuclear cloning procedures has resulted in a number of new models for various human diseases (Zhi et al. 2022) and suggests the potential use of pigs as a source for xenotransplantation (Yue et al. 2021). Extensive genomic divergence is known to have occurred between European and Asian pigs (Groenen et al. 2012). Since animal breeding became more organized in the eighteenth century, and especially since the strong genetic selection of inbred commercial lines within the past 70 years (Groenen et al. 2012), many of these breeds show remarkable phenotypic diversity and genetic adaptations that provide a sufficient number of segregating sites to distinguish allele-specific information (Li et al. 2017).
To characterize the 3D spatial organization of the diploid genome in mammals and to investigate a possible role of allelic bias in chromatin architecture in transcriptional regulation, we introduce a pig model generated by a reciprocal cross of highly selected European Berkshire and Asian aboriginal Tibetan pigs. Using the F1 hybrid progeny, we then examine how allelic differences in 3D chromatin architecture broadly affect gene expression, and eventually phenotype, at multiple levels.
Results
Construction of high-resolution diploid Hi-C contact maps
To generate chromosome-span haplotypes for use in assigning “omic” data from diploid genomes to their parental origins, we performed reciprocal crosses of two genetically distinct pig breeds (three families of Tibetan [♂] × Berkshire [♀], and three families of Berkshire [♂] × Tibetan [♀]) and sequenced the whole genomes of six parent–child trios (∼122.25× coverage for each individual, n = 18) (Fig. 1A).
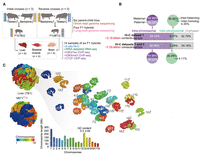
Three-dimensional (3D) structures of diploid genomes in hybrid pigs. (A) Schematic representation of reciprocal crosses between genetically distinct Berkshire and Tibetan pig breeds. Liver, skeletal muscle, and brain of F1 hybrids were collected for multi-omic assays (for details, see Supplemental Fig. S1). F1i and F1r denote the F1 hybrids of initial and reverse crosses, respectively. (B) Allele assignment of in situ Hi-C contacts in F1 hybrids. A total of about 48.95 billion valid Hi-C contacts were generated, including about 3.15 billion autosomal contacts for each of the 14 samples (six liver, four muscle, and four brain samples; data set 1). To reliably identify PEIs throughout the diploid genome, we performed additional in situ Hi-C assays for 12 samples (data set 2), resulting in a final total of about 97.53 billion valid Hi-C contacts (about 7.29 billion autosomal contacts for each of the 12 samples, including four liver, four muscle, and four brain samples) in the combined data sets 1 and 2 (for details, see Supplemental Fig. S1). (C) 3D genome structure of a representative liver sample (see Supplemental Methods). (Left) Whole genome; (right) the 18 autosome pairs visualized separately.
To understand the 3D structural basis of transcriptional control in a diploid genome, we performed in situ Hi-C for three tissues from newborn female filial 1 (F1), representing the three germ layers: the liver (endoderm, n = 6), the brain (ectoderm, n = 4), and skeletal muscle (mesoderm, n = 4). In total, approximately 48.95 billion valid Hi-C contacts (data set 1: about 3.50 billion for each sample, n = 14) were generated, and the transcriptional profiles of corresponding samples were obtained using RNA-seq (Fig. 1A; Supplemental Figs. S1, S2).
Combining trio-based genomic and Hi-C data from F1 hybrids enabled construction of chromosome-span haplotypes that included 99.33% of all heterozygous single-nucleotide variants (SNVs; about 4.54 per kb) for each F1 hybrid (see Methods) (Supplemental Fig. S3). Through SNV phasing combined with local imputation (Methods) (Supplemental Fig. S4), we built diploid Hi-C maps for 14 samples based on the unambiguous maternal and paternal contacts, each containing about 1.04 billion intra-chromosomal contacts (with maximum resolution of 2-kb), and about 100.13 million inter-chromosomal contacts (∼4.37% occurring between homologs) (Fig. 1B; Supplemental Fig. S5A–D). The allelic expression of about 11,430 protein-coding genes (59.14% of the autosomal genes) (Supplemental Fig. S5E–H) was estimated using Allelome.PRO (Andergassen et al. 2015).
After efficient Hi-C data normalization (Supplemental Figs. S6, S7), we reconstructed the 3D genome structures of the 14 diploid samples from their haplotype-resolved Hi-C maps at 20-kb resolution (Fig. 1C; see Supplemental Methods). The pig genome contained full sets of functional condensin II subunits (a determinant of architecture type) (Supplemental Fig. S8A; Hoencamp et al. 2021); its 3D structures had characteristically uneven mass distribution (i.e., nucleotides were concentrated in the nucleus interior, except in the hollow nucleolus) (Supplemental Fig. S8B) and displayed distinct chromosomal territories (Supplemental Fig. S8C). Chromosomes preferred the centromere-facing-out (no Rabl-like) configuration (Supplemental Fig. S8D) and sequence-dependent spatial organization of chromatin (Supplemental Fig. S8E–I). These findings agreed with human and rodent studies (Du et al. 2017; Stevens et al. 2017; Tan et al. 2021). Additionally, homolog pairs were nearly equidistant from the center of the 3D nucleus (Supplemental Fig. S8J) and in closer spatial proximity (Supplemental Fig. S8K–M), with highly similar intra- and inter-chromosomal contact patterns (indicated by multiple-chromosomal intermingling intensity) (Supplemental Fig. S8H), compared with nonhomologous chromosomes (heterologs).
Haplotype-resolved analyses of Hi-C maps revealed that different tissues displayed more distinct differences in 3D genome architecture than that between the same tissue in either parent of origin or between parental breeds (Table 1; Supplemental Fig. S8N–S), which was recapitulated by allelic expression data (Supplemental Fig. S8T,U).
Summary of allelic chromatin reorganization at multiple scales
Allelically compartmental rearrangements
In total, ∼9.91% of the genome (∼224.51-Mb regions) was found to have different compartmental status between tissues (i.e., A/B switches) (Fig. 2A; Table 1; Supplemental Figs. S9A–E, S10A; Supplemental Table S1) using the method described by Rowley et al. (2017), which was highly consistent with the A/B compartment switches detected using HOMER (overlapping ratios > 92%) (Supplemental Figs. S10B,C; Lin et al. 2012). Genes located in tissue-restricted A compartment regions showed generally higher expression than the same genes in the B compartment in other tissues (Supplemental Fig. S10D,E). These tissue-dependent transcriptionally active genes were mainly involved in specialized functions of their respective tissue (Supplemental Fig. S10F).
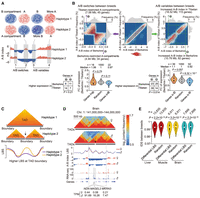
Compartmental rearrangements and variable TAD boundaries among haploid genomes. (A) Identification of compartmental rearrangements. Regions with distinct compartment status were defined (i.e., from B [negative value of A-B index] to A [positive value of A-B index], or from A to B; scenarios 1 and 2, respectively) as A/B switched regions. Additionally, compartment scores (i.e., the A-B index values) for regions with the same status were compared between haploid genomes (not between those with distinct switches in compartment status) and identified regions with significantly higher compartment scores as A/B variable regions (P < 0.05, paired Student's t-test, and |ΔA-B index| > 0.5; scenarios 3 and 4). (B) Allelic compartmental rearrangements in muscle. We identified compartment A regions exclusively found in the Tibetan (7.58 Mb) or Berkshire (4.94 Mb) alleles (top left) and regions with significantly elevated compartment scores in the Tibetan (18.52 Mb) and Berkshire (10.78 Mb) alleles (top right). Genes with testable differences in allele (i.e., with informative SNVs and TPM ≥ 0.5 in at least one allele) tended to be located in the more accessible chromatin regions and generally showed increased expression; violin plots show changes in allelic expression (Wilcoxon rank-sum test) and counts (Fisher's exact test) of genes located in regions with allelic differences in compartmentalization (i.e., A/B switches, left; A/B variables, right). (C) Identification of shifted TAD boundaries (see Supplemental Methods). (D) Paternal-specific TAD boundaries adjacent to three paternally expressed imprinted genes (NDN, MAGEL2, and MKRN3). (E) In comparison to genomic background, regions with allelic differences in boundary strength (LBS, P < 0.05, paired Student's t-test) showed greater allelic divergence in sequence divergence (reflected by the lower pairwise haplotype similarities between two parental breeds determined by identity score [IDS]) (see Supplemental Methods). P-values are from a Wilcoxon rank-sum test. Hi-C maps at 20-kb resolution were used to generate these graphs.
Given the rarity of regions that show compartment switching between parent of origin (∼0.35% of the genome; ∼7.97 Mb) and between parental breeds (∼0.71% of the genome; ∼16.19 Mb), we compared scores (i.e., A-B index values) between alleles with regions in same compartment and found that these regions had significantly different compartment scores (termed as A/B variables; P < 0.05, paired Student's t-test, and |ΔA-B index| > 0.5) (Fig. 2A; Supplemental Fig. S11A; Supplemental Table S2), which indicate altered chromatin activity and were highly correlated with varying gene expression levels and, therefore, deserve consideration. This analysis showed that ∼0.55% (∼12.51 Mb) of the genome regions displayed allelic differences in compartmentalization (including A/B switches and A/B variables) between parents of origin (Table 1; Supplemental Fig. S11A; Supplemental Table S2), which were enriched with 126 known, imprinted mammalian genes (P < 0.002, χ2-test) (Supplemental Figs. S11B,C, S12). Genomic regions containing known, imprinted genes also had subtle but significantly higher variability in compartment scores between alleles (Supplemental Fig. S11D). These results indicated that parent-of-origin-specific compartmentalization could serve as an underlying mechanism for genome imprinting (Rao et al. 2014; Dixon et al. 2015).
After comparing compartment switching in parents, we examined compartment switching between breeds and found that ∼2.27% (∼51.37 Mb) of the genome had allele-specific variable or switched compartments between the parental breeds (Table 1; Supplemental Table S3). These regions had a characteristically higher sequence divergence between alleles (i.e., higher density of SNVs and short indels and, thus, lower pairwise similarity between haplotype identity score [IDS]) (see Supplemental Methods; Supplemental Fig. S11E). Genes located in these regions showed agreement in their allelic bias between compartmentalization and expression (Fig. 2B). Although separating sequence variations from other confounding factors affecting compartmentalization remains challenging, genes located in breed-restricted A compartment regions potentially reflected some of the phenotypic differences between the parental breeds (Supplemental Fig. S11F). Notably, genes located in the Tibetan-restricted A compartment regions (more accessible and relatively active) were enriched for annotations associated with the nonshivering thermogenesis to resist cold stress (“brown fat cell differentiation,” three genes), disease resistance (“adaptive immune response,” 13 genes; “immunoglobulin production,” four genes), and resistance to intensive solar ultraviolet radiation–induced DNA damage (“regulation of DNA-dependent DNA replication,” four genes) (Li et al. 2013; Witt and Huerta-Sánchez 2019). These results suggest that the compartmentations in hybrid pigs are separately inherited from their parents which showed the different adaption of parental breeds to their local environments.
Allelically variable TAD boundaries
To identify allelic differences in 3D genome architecture that led to imbalanced expression at a finer scale than compartments, we partitioned the haploid genomes into approximately 3956 topologically associating domains (TADs) (Supplemental Fig. S9F–L). Pairwise comparison between tissues identified ∼3.93% (about 184) that had shifted boundaries between tissues (indicated by changes in boundary position with significantly different local boundary scores [LBSs]) (see Fig. 2C; Table 1; Supplemental Table S4; Supplemental Methods; Han et al. 2020). In addition, tissue-restricted boundaries showed substantial differences in compartmentalization, whereas boundaries that were the same between tissues did not (Supplemental Fig. S13A,B). Genes near these tissue-restricted boundaries also showed greater differences in expression than those near boundaries that were the same between tissue, and included critical marker genes associated with core functions of that tissue (Supplemental Fig. S13C–H). These results highlight the function of TADs in facilitating local interactions and establishing regulatory context for their respective genes (Beagan and Phillips-Cremins 2020).
In total, three TAD boundaries were found to be altered between parent of origin across tissues that contained or were near to known imprinted genes (Table 1; Supplemental Table S5), which were well supported by the changes in TAD boundary scores calculated using a dedicated tool for TAD boundary analysis, TADCompare (Supplemental Fig. S11G–J), showing the reliability of our results. Two of these were paternal-specific boundaries, including one in brain and muscle that may affect three imprinted genes (NDN, MAGEL2, and MKRN3) (Fig. 2D; Supplemental Fig. S11G) and another in brain and liver affecting two imprinted genes (CASD1 and SGCE) (Supplemental Fig. S11H,I). One maternal-specific boundary was identified in muscle that most likely influenced seven imprinted genes (ASCL2, CD81, H19, IGF2, INS, TH, and TSSC4) (Supplemental Fig. S11J). The imprinted genes around allele-specific boundaries generally showed increased transcriptional activity, supporting the possibility that parent-of-origin-specific TAD organization provides a structural context conducive to activation of imprinted genes (Monk et al. 2019).
Although no altered boundary positions were detected between haplotypes of the two parental breeds, ∼190.97 Mb (∼8.43%) of genomic regions had allelic differences in boundary strength (LBS; P < 0.05, paired Student's t-test) (Table 1; Supplemental Table S6) and showed significantly higher sequence divergence among alleles than other regions (P ≤ 3.2 × 10–12, Wilcoxon rank-sum test) (Fig. 2E).
Haplotype-resolved interrogation of tissue- and imprinting-specific PEI organizations in gene expression control
The information necessary for regulating transcription is conveyed through long-range physical interactions between promoters and enhancers (Song et al. 2019; Oh et al. 2021). To compile an extensive but reliable genome-wide catalog of PEIs in the diploid pig genome, we conducted additional in situ Hi-C assays (data set 2) for 12 of the 14 aforementioned samples (three tissues from four F1 hybrids, two F1 from the initial and two F1 from reciprocal crosses) (Fig. 1A,B; Supplemental Fig. S1). By combining both Hi-C data sets, we built diploid Hi-C maps for each of the 12 samples, including about 2.28 billion intra-chromosomal contacts (a maximum resolution of 1-kb), and about 239.91 million inter-chromosomal contacts (4.11% between homologs) per map (Fig. 1B; Supplemental Figs. S2C, S5I–K). We identified 29,352 enhancers assigned to 7399 promoters (38.28% of the autosomal genes) across 24 haploid genomes at 5-kb resolution (Supplemental Fig. S14A–G).
To elucidate how this observed extensive differences of PEIs may contribute to the transcriptomic divergence across haploid genomes, we calculated the regulatory potential score (RPS; a spatial proximity-based index of the combined regulatory effects of multiple enhancers for a given gene) for each promoter using a unified set of PEIs from all haplotypes of a given tissue (see Supplemental Methods; Li et al. 2022; Zhi et al. 2022). As expected, genes with a larger RPS had higher expression within haplotypes (Supplemental Fig. S14H). Beyond characterizing the spatial proximity of enhancers and promoters, we also measured allelic imbalances in the activity of enhancers and promoters by generating H3K27ac and H3K4me3 ChIP-seq signal enrichment profiles (Supplemental Figs. S1, S2G, S14I–P; Villar et al. 2015; Pérez-Rico et al. 2017).
Examination of the haplotype-resolved Hi-C maps and ChIP-seq signals revealed extensive differences of PEIs between tissues (Supplemental Fig. S14Q–S). A total of 4865 genes showed a significantly different RPS between pairs of tissues (Table 1; Supplemental Fig. S15A; Supplemental Table S7), the majority of which were also differentially transcribed between tissues (Supplemental Fig. S15B). These sets of tissue-specific active genes were enriched in annotations related to their respective core functions (Supplemental Fig. S15C). Representative examples are shown in Supplemental Figure S16. Notably, examination of individual tissue-specific markers indicated that a high RPS was a prevalent feature of germ layer markers and that their expression was elevated in tissues originating from their respective germ layers (Supplemental Fig. S15D–F), which supported the hypothesis that transcriptional programming (as well as spatial proximity of genes and their enhancers) retains lineage- or differentiation-specific memory (Hon et al. 2013; Javierre et al. 2016). Representative examples are shown in Supplemental Figure S17.
Genome imprinting represents a phenotypically consequential outcome of allele-specific expression (Tucci et al. 2019). By comparing data from F1 hybrids produced by reciprocal crosses, we were able to survey genome-wide allelic differences in chromatin conformation for 126 known imprinted genes (Supplemental Fig. S12), nearly half of which are located adjacently to each other as previously reported (Santini et al. 2021). This analysis identified a set of 101 testable imprinted genes with informative SNVs and enhancer interactions in at least one tissue (Fig. 3A; Supplemental Table S8), among which 12 had strongly significant parent-of-origin-specific PEI organization (highly consistent between replicates) and generally corresponding parental expression bias in two or three tissues (Fig. 3B; Supplemental Fig. S18). Visual examination of a representative case showed the lower deviations of parent-of-origin-specific 3D structure for the region around the promoter (paternally expressed MAGEL2) across replicates and different tissues (Fig. 3C). These results highlighted the possible mechanistic contribution of chromatin architecture in imprinting phenomenon (Monk et al. 2019; Tan et al. 2021).

Parent-of-origin-specific PEI organization of imprinted genes. (A) Quantification of PEIs in 126 previously described imprinted genes identified 101 genes with informative SNVs that interacted with enhancers in at least one tissue. Plots of differential RPSs between parents of origin in at least two tissues are shown for 12 representative imprinted genes (|ΔRPS| > 0.3 and P < 0.05, paired Student's t-test). (B) Allelic differences in RPS and expression of 12 imprinted genes with differential RPS listed in A. The similarity (Pearson's r) of interactions in promoter-centered regions (200 kb upstream of and downstream from the promoter bin) among eight haplotypes is shown for each tissue. (C) Allelic promoter-centered interactions in the representative imprinted gene MAGEL2. Haplotype-resolved Hi-C maps (left), 3D structural models (middle), and interaction metaplots (right) of promoter-centered regions across replicates and tissues. Enhanced paternal allele-specific interactions between the promoter and its 200-kb upstream regions. Black lines in metaplots show interactions of the other parental allele. Hi-C maps at 5-kb resolution were used to generate these graphs.
Effects of genetic variations and histone modifications on allelic PEI differences
Because both genetic and epigenetic regulation shape gene expression patterns, we next explored whether and how inherited sequence variation and histone modifications (the environmentally induced transgenerational epigenetic inheritance) (Fitz-James and Cavalli 2022) impacted allelic PEI differences in F1 hybrids. Considering the functional impacts of sequence variations on the genome, we comprehensively surveyed the SNVs and short and large indels to determine their potential effects on allelic PEI formation. For this analysis, we generated long-read sequence data for the four F1 hybrids (∼125.22× coverage per individual) using the Oxford Nanopore Technology (ONT) platform and identified their sequence-resolved large indel alleles (Supplemental Fig. S19; see Supplemental Methods). As expected, higher divergence between the alleles of promoters or enhancers was linked to more differences in PEI intensity between parental alleles (Fig. 4A,B). We then estimated the probability of long-range interactions for each PEI in light of their respective, specific variants using the PEP algorithm (Yang et al. 2017). We found that PEIs with higher intensity had a higher probability of interaction within alleles (Supplemental Fig. S20A) and between alleles (Supplemental Fig. S20B), supporting that genetic variations can affect dynamic PEI formation (Spielmann et al. 2018; Kvon et al. 2020). A representative example of the potential for PEI disruption by multiple SNVs and indels is shown for SMAD4 in Supplemental Figure S20, C through E.
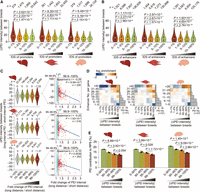
Influence of allelic sequence variations on PEIs. (A,B) Promoters (A) and enhancers (B), with higher allelic sequence divergence (especially those with the lowest 1% of IDS between parental breeds) (see Supplemental Methods), showed greater differences in PEI intensity. P-values are from a Wilcoxon rank-sum test. (C) Effects on PEI intensity caused by allelic variation-induced differences in PEI bridging distances. PEIs with the top 1% of increases in bridging distance showed significantly decreased PEI intensity (0%–99% vs. 99%–100%: P = 5.03 × 10–12 in liver, P = 2.21 × 10–6 in muscle, and P = 3.26 × 10–10 in brain; Wilcoxon rank-sum test; left). This trend is more prominent in PEIs with the greatest extension (allelic fold change in linear bridging distance of the top 0.1%: 1.17–1.21), which showed strongly negative correlations between bridging distance and PEI intensity of PEIs (Spearman's r = –0.15 to –0.29, P ≤ 0.0036; right). (D) PEIs with enrichment for differential allelic intensity (x-axis) among genes that contact a variable number of enhancers (y-axis) in the three tissues. (E) PEIs showing greater differential allelic intensity tend to contribute less to RPS (including the combined regulatory effects of multiple enhancers for a given gene) and thus exert the least impact on gene expression. P-values are from a paired Student's t-test. Hi-C maps at 5-kb resolution were used to generate these graphs.
Additionally, comparison of allelic differences in the bridging distance between the promoter and enhancer supported our general hypothesis that closer linear chromosomal proximity between a promoter and enhancer is associated with closer proximity in 3D nuclear space and thus, with higher PEI intensity and vice versa (Fig. 4C). Simulation of PEI intensities based on the allelic variations in bridging distances using Huynh's algorithm (Huynh and Hormozdiari 2019) further supported this likelihood (Supplemental Fig. S20F,G). Moreover, the significantly greater activity of H3K4me3-enriched promoters or H3K27ac-enriched enhancers generally resulted in allele-specific increases in PEI intensity (Supplemental Fig. S20H–K).
We found that allele-specific PEIs occurred more frequently in genes that contacted multiple enhancers (Fig. 4D), providing validation for the idea that enhancers function in a redundant manner; that is, the ubiquitous presence of multiple enhancers could serve as a mechanism to reinforce local regulatory circuitry necessary to buffer environmental stresses and genetic disruption, ultimately resulting in phenotypical robustness (Kvon et al. 2021; Laverré et al. 2022). Importantly, these allele-specific PEIs exert relatively mild effects on RPS between parental alleles (Fig. 4E). In addition, we identified the allele-specific loss of two CCCTC-binding factor (CTCF)-mediated loops in a Tibetan allele in the diploid Hi-C maps of liver. In these cases, the CTCF binding motifs of the loop anchor were effectively disrupted by a 1-bp deletion (“G,” Chromosome 2: 578,22,013) and a SNV (“G” > “T,” Chromosome 8: 53,792,054), respectively, but only resulted in subtle impacts on the expression of genes within these loops (Supplemental Fig. S21). These results are analogous with the findings in human cancers that 3D genome organization is disrupted by somatic genomic rearrangements, typically resulting in the fusion of discrete TADs, the gain or loss of CTCF-mediated loops, and enhancer “hijacking” (Weischenfeldt et al. 2017; Huynh and Hormozdiari 2019), whereas known SV polymorphisms in healthy human populations have significantly less impact on 3D organization (Akdemir et al. 2020).
Allelic differences in PEIs associated with phenotypic differences between parental breeds
We next asked whether the allelic differences of PEIs in F1 hybrids could contribute to phenotypic divergence between the parental breeds (Li et al. 2013, 2014; Jeong et al. 2015). In total, about 520 genes were identified that showed a differential RPS between the Berkshire and Tibetan alleles across tissues (Fig. 5A; Table 1; Supplemental Table S9), which potentially could be an effect of breed-specific genetic variations. Population-level analysis of the 82 purebred diploid pig genomes (Berkshire, n = 21; Tibetan, n = 61) further suggested that variants in promoter and enhancers were almost fixed in the more genetically homogeneous Berkshire pigs (Fig. 5A), which could reflect an effect of rigorous selection during breeding (Li et al. 2014; Jeong et al. 2015; Fu et al. 2020), although functional analyses are required to validate the nonneutrality of these genes. Below, we examine some genes with highly significant differential RPS between parental breeds that are likely candidates responsible for functional divergence in different tissues between parental breeds.

Allelic differences of PEIs in F1 hybrids. (A, left) Volcano plots of genes with a differential RPS (|ΔRPS| > 0.3 and P < 0.05, paired Student's t-test) between Berkshire and Tibetan alleles in three tissues. (Middle) Differential PEI intensities, distributions of differential PEI intensities, and estimated probabilities of PEIs based on their sequence variants (see Supplemental Methods) are shown. (Right) Degree of sequence similarity for promoters and enhancers in pairwise comparisons among eight haplotypes (Tibetan [n = 4] and Berkshire [n = 4]; measured by IDS) and among 82 diploid genomes in population-level analysis of purebred pigs (measured by pairwise IBS distances; see Supplemental Methods). Among the 82 diploids,12 from the six trios in this study (Berkshire, n = 6; Tibetan, n = 6) and 70 are publicly available (Berkshire, n = 15; Tibetan, n = 55); SNVs were retrieved from the ISwine database (http://iswine.iomics.pro/pig-iqgs/iqgs/index). Representative functional genes are labeled. (B) Significantly enriched GO-BP terms or KEGG pathways (performed using Metascape) for genes showing differential RPS between Berkshire and Tibetan haplotypes in the liver (left), muscle (middle), and brain (right).
Liver is the metabolic hub for nutrient homeostasis (Ben-Moshe and Itzkovitz 2019); its function may contribute to major phenotypic differences between Tibetan (long-standing survived in highland at a free-range farming state) (Li et al. 2013) and Berkshire pigs (enhanced, high efficiency energy storage under high calorie diets in captivity) (Li et al. 2014; Jeong et al. 2015). As expected, a large suite of genes (n = 254) with a higher RPS in the Berkshire allele showed enrichment for “maintenance of location” (six genes) (Fig. 5B), which is necessary for proper subcellular localization of protein complexes or organelles and is therefore essential for lipid transport (Wong et al. 2019). Notably, we found that the Berkshire allele had a coenhanced RPS along with the expression of C12ORF29 (an anti-obesity factor) (Wang et al. 2020) and CAV2 (a protective factor against nonalcoholic fatty liver disease; Yang et al. 2020) compared with that in Tibetan liver tissue (Supplemental Fig. S22A–H). These results suggested a possible protective mechanism for Berkshire pigs against the deleterious effects of a “diabetogenic” environment (Gerstein and Waltman 2006). Conversely, Tibetan pigs are known to show relatively high sensitivity to induction of obesity-related comorbidities when given excessive dietary energy in pig farms (Zhu et al. 2009; Li et al. 2013). Our analysis identified 278 Tibetan alleles with higher RPS than the Berkshire alleles that could contribute to inflammatory processes, including “chemokine signaling pathway” (seven genes) and “positive regulation of cell-substrate adhesion” (six genes) (Fig. 5B). In addition, two typical inflammatory markers (ABI3 and ITGA6) also displayed greater RPS and expression levels in the Tibetan allele compared with those of the Berkshire allele (Supplemental Fig. S22I–P; Verma et al. 2018; Richter et al. 2021).
In muscle tissue, our analysis uncovered 265 Berkshire alleles with a larger RPS than their Tibetan counterpart alleles that are involved in energy metabolism and muscle growth (e.g., six genes related to “insulin secretion”) (Fig. 5B). Nonetheless, 273 genes were found with a greater RPS in the Tibetan allele that are involved in “regulation of mitochondrial fission” (five genes) (Fig. 5B), defects which may cause muscle wasting in adult mammals (Kraus et al. 2021). These findings most likely reflected a consequence of artificial selection for faster growth in Berkshire pigs (daily weight gain during 2–6 mo of age, ∼594.67 g/d) and greater meat yield (myofiber cross-sectional area, ∼5231.84 μm2) in contrast to Tibetan pigs (∼269.35 g/d and ∼2574.05 μm2) (Supplemental Fig. S23A; Wang et al. 2011), which only recently attracted focused breeding attention for pork production at high altitudes.
The plasticity of skeletal muscle in mammals is largely attributable to myofiber heterogeneity (Jin et al. 2021). In comparison to glycolytic type II, oxidative type I myofibers are more resistant to fatigue and are rich in triglyceride and fatty droplets (Malenfant et al. 2001). In the muscle of our F1 hybrids, we observed that genes involved in lipid metabolism, such as “regulation of fat cell differentiation” (six genes) were more structurally active (i.e., greater PEI intensity) in the Tibetan allele compared with that in the Berkshire allele (Fig. 5B). This finding was consistent with a marked increase in the proportion of lipid-rich type I myofibers in muscle of Tibetan pigs (∼26.26%) compared with that in Berkshire pigs (∼12.05%) (Supplemental Fig. S23B,C). Consistent with these data, DDIT4L, a specific regulator of type II myofiber (Pisani et al. 2005) had lower RPS and expression in the Tibetan allele (Supplemental Fig. S23D–F). The triglycerides accumulating between or within myofibers represent a substantial energy source (Roepstorff et al. 2005). The higher proportion of type I myofibers in Tibetan pigs compared with that in Berkshire pigs may be both an effect of selection and husbandry practices as well as a contributing factor in the enhanced athletic performance of grazing Tibetan pigs compared with that of Berkshire pigs raised in limited space for activity in pig farms.
We also identified genes with a differential RPS in the brain that are involved in functional and behavioral differences between Berkshire and Tibetan pigs. In total, we identified 253 genes with a differentially higher RPS in the Berkshire allele that were notably enriched in “cellular response to nutrient” (three genes) (Fig. 5B), indicating a functional role in the control of appetite and food intake. In the Tibetan allele, 236 genes were found with a significantly higher RPS that were enriched in “response to carbohydrate” (seven genes) (Fig. 5). Among these, Pfkl reportedly evokes higher rates of glucose utilization in the mice brain (Supplemental Fig. S22Q–T; Dienel 2019). These findings together show the distinct effects on central nervous system control of metabolism accomplished through breeding selection (i.e., Berkshire) versus the effects of natural selection in the Tibetan pig in the absence of artificial selection (Li et al. 2013). To protect the Berkshire against the deleterious effects of an extremely high-calorie concentrated diet, the central nervous system triggers a reduction in food intake upon sensing dietary excess (Mancini and Horvath 2018). In contrast, Tibetan pigs require high efficiency utilization of low-calorie carbohydrates to adapt to the limited nutritional resources typical of highland grazing (Zhu et al. 2009; Li et al. 2013). This adaptive response potentially facilitates maintenance of brain homeostasis through a “competent brain-pull” mechanism (Herhaus et al. 2020), which is indispensable to maintain high energy levels in the brain despite food deprivation.
These collective observations of differential RPS between Tibetan and Berkshire pigs in genes that are well established to participate in metabolism, muscle development, and behavior provide strong evidence linking phenotype in pigs with regulatory control of development mediated by 3D chromatin architecture.
Structured homolog pairing in pig genome
Experimental evidence from flies and mammals suggests communication between homologs, which can contribute to transcriptional regulation (Koeman et al. 2008; Joyce et al. 2016; Child et al. 2021). Using ultradeep Hi-C data obtained from 12 diploid samples, each containing about 9.86 million inter-homolog contacts (Fig. 1B), we assessed the extent of homolog proximity in pig genomes. The Hi-C maps revealed apparent inter-homolog interactions across the entire length of each autosome at 1-Mb resolution (Fig. 6A), as well as observed structurally coordinated inter-homolog and intra-chromosome interactions (Supplemental Fig. S24A–D). These results were consistent with findings in flies (AlHaj Abed et al. 2019; Erceg et al. 2019), suggesting similarity or conservation in the mechanisms of contact formation. Further, enhanced allelic pairing was evident in active A compartments, implying a likely functional role for homolog pairing in compartmentalization (Supplemental Fig. S24E–G). However, no significant correlation was found between HPS and LBS (a parameter used to define TAD boundary strength) (Supplemental Fig. S24H). More experimental evidence is necessary to determine the relationship between homolog pairing and TAD formation. These results were recapitulated by analyses at 100-kb resolution that showed higher density of contacts (a 5.92-fold increase in inter-homolog contacts in genomic bins from 20-kb to 100-kb), which suggested greater reliability (Supplemental Fig. S25).
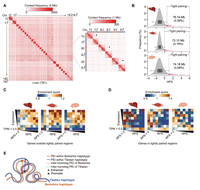
Highly structured homolog pairing in F1 hybrids. (A) Hi-C maps showing signals of genome-wide homolog pairing (arrows) in representative somatic tissue (liver) of a hybrid pig. Magnified Hi-C maps of Chromosomes 7, 8, and 9 are shown on the right. (B) Identification of tightly paired loci in all three somatic tissues of hybrid pig based on distribution of homolog pairing score (HPS) values. In the boxplot, the internal line indicates the median; the box limits indicate the 25th and 75th quartiles; and the whiskers extend to 1.5 × IQR from the quartiles. Rare tightly paired loci (liver, 76.74 Mb; muscle, 72.12 Mb; brain, 74.18 Mb) are defined as those with a HPS above Q3 + 1.5 × IQR. (C,D) Enrichment of RPSs (x-axis) from gene expression (y-axis) indicates the increased covariations between allelic expression and intra-chromosomal-based RPSs (i.e., genes with a larger RPS had higher expression) for genes outside the tightly paired regions (C) compared with genes located in the tightly paired regions (D). (E) Schematic representation of intra-chromosomal (solid lines) and inter-homolog (dashed lines) promoter–enhancer interactions in tightly paired loci; the formers are generally much stronger than the latter.
We next investigated the infrequent homologous interactions in which an enhancer may act on the homolog of its corresponding promoter in an inter-chromosomal trans-manner (Galouzis and Prud'homme 2021). Examination of PEIs between enhancers and promoters from homologs showed that covariation between allelic expression and RPS decreased for genes located in tightly paired loci with their enhancers (∼74.35 Mb, or ∼3.28% of the genome; see Methods) (Fig. 6B–D; Supplemental Fig. S24I,J), indicating that the RPS only reflects intra-chromosomal regulatory effects of PEI. This result suggests that although rare, tightly organized homolog pairing may result in an additional layer of transcriptional regulation (Fig. 6E), and further foundational assays are required to validate the impact of inter-homolog interactions in modulating gene transcription.
Discussion
Many long-standing, fundamental biological questions are linked to diploid 3D genome architecture in mammals (Jerkovic and Cavalli 2021; Zhang et al. 2021). Here, we generated ultra-high-resolution Hi-C diploid maps (at 1-kb maximum resolution) for three porcine tissues that represent distinct embryonic germ layers. These Hi-C maps were then used to characterize the 3D nuclear organization of the diploid genomes and highlight features of the spatial organization of homologs. In depth, genome-wide, functional examination of haplotypic changes in 3D chromatin organization were then conducted at multiple scales using these data, including between tissues, parents of origin, and parental breeds.
We found that 3D organization was different between tissues and was highly correlated with the expression of genes involved in the specialized functions of each tissue and in the development of their respective germ layers. This study provides an allele-specific survey of 126 known imprinted genes and confirms a parent-of-origin-specific 3D conformation for at least 12 of them. The results of this work show that homologs are differentially organized in 3D structures and that this organization is strongly correlated with widespread allelic imbalance in gene expression. Additionally, genetic variations and histone modifications were found to affect the allelic reorganization of promoter–enhancer interactions (PEIs) linked to allelic imbalances in gene expression in these hybrid pigs. It should be noted that although relatively stringent criteria and rigorous analyses were used to screen differential A/B compartmentalization, shifts in TAD boundaries, and rewired PEIs, the advanced statistical methods used for these large-scale Hi-C data do not include P-value adjustment for possible false positives, which will require further experimental validation in future work.
Somatic homolog pairing (i.e., colocalization of homologs) has been extensively investigated in fruit flies (AlHaj Abed et al. 2019; Erceg et al. 2019) but has been rarely explored in mammals, mainly owing to inadequate chromosomal contact data or technical difficulties. Here, using a similar analytical pipeline to that used in fruit fly, with our unprecedented (∼1 kb) high-resolution diploid Hi-C maps, we could observe homolog pairing in porcine somatic tissues, even only using the heterozygous SNVs of the highest confidence to phasing Hi-C data (Supplemental Fig. S26). This homolog pairing correlated with genomic compartmentalization and might represent an additional layer of transcriptional regulation between alleles. However, two previous studies that reported finding similar signals in humans (Hoencamp et al. 2021) and mice (Selvaraj et al. 2013) were unable to verify whether these data are evidence of extremely rare inter-homolog interactions or are artifacts arising from misalignment (Hoencamp et al. 2021) and noise (Selvaraj et al. 2013). Further investigation is needed to either exclude such possible technical biases in sequencing, mapping, and assignment that interfere with recognition of inter-homolog interactions or, alternatively, experimentally validate the occurrence of homolog pairing phenomena in mammals.
Considering that our bulk Hi-C data were obtained from cell population-scale assays, we cannot rule out that cellular heterogeneity may, to some extent, contribute to the observed allelic differences in chromatin features. Thus, single-cell analyses may be used to validate these scenarios in future studies (Tan et al. 2021; Zhou et al. 2021).
In recent decades, pigs, and miniature pig breeds in particular, have recently emerged as an attractive biomedical model for humans (Lunney et al. 2021). More than 730 genetically distinct pig breeds have been recognized worldwide, and thus, understanding the control of phenotype between genetically similar breeds is of enormous significance to animal husbandry (Chen et al. 2007; Li et al. 2017). Indeed, the F1 progenies of these breeding lines are known to show remarkable phenotypic changes (Chen et al. 2007; Groenen et al. 2012). These progenies also contain a large number of segregating sites that can be used to distinguish parental haploids; the heterozygous SNP density of F1 hybrids in our pig model is about 4.54 per kilobase but is about one per kilobase in humans, about five per kilobase in fruit flies, and about 7.69 per kilobase in mice (de Wit 2017; AlHaj Abed et al. 2019).
Our haplotype-based interrogation of PEI organization greatly expands the annotation of regulatory DNA elements (enhancers) currently available in the pig reference genome. Using the NHGRI-EBI GWAS catalog (Buniello et al. 2019) and the liftOver tool (https://genome.ucsc.edu/cgi-bin/hgLiftOver), we found that DNA sequence variations (human noncoding SNPs) associated with specific traits or diseases were enriched in enhancers in the pig genome (mean SNP enrichment scores in enhancers vs. nonenhancer regions: 1.34 vs. 0.97, P = 0.002, paired Student's t-test) (Supplemental Fig. S27A), confirming the functional importance of enhancers (Maurano et al. 2012; Hnisz et al. 2013). Supporting our findings of allele-specific PEIs between tissues associated with tissue-specific gene regulation related to core functions, we observed that SNPs associated with cholesterol level are enriched in metabolically active liver, SNPs linked to oxygen supply are enriched in high oxygen-consuming muscle, and Alzheimer's disease–associated SNPs are enriched in neuron-rich brain tissue (Supplemental Fig. S27B). Further, analysis of pig QTLs recapitulated our findings from human GWAS data (mean enrichment scores for enhancers vs. nonenhancer regions: 1.44 vs. 0.87, P = 0.01, paired Student's t-test) (Supplemental Fig. S27C). That is, we detected notable enrichment for meat-related QTLs in muscle tissue and hormone-related QTLs in liver and brain (Supplemental Fig. S27D). These results, combined with the preponderance of physiological similarities between pigs and humans, strongly support the use of pigs as a model for human disease (Jin et al. 2021; Pan et al. 2021; Zhao et al. 2021).
Recent studies in humans have revealed that most disease- and phenotype-associated variants do not affect protein-coding sequences but instead lie in noncoding regulatory regions (∼98% of the genome) (Aganezov et al. 2022; Nurk et al. 2022). Nonetheless, establishing causal links between phenotypes in humans and the rapidly expanding list of noncoding variants poses an increasingly large challenge (Spielmann et al. 2018; Kvon et al. 2020; Nasser et al. 2021). Our allele-specific survey of the impacts of (epi-)genetic variations on PEI differences between parental breeds in the F1 hybrid progeny greatly expands our knowledge of how 3D chromatin organization affects phenotype through transcriptional programming and assigns previously unrecognized function to large parts of the noncoding genome.
Methods
Pig breed crosses
We performed reciprocal crosses (parent–child trios, n = 6) of two pig breeds and generated trios of Tibetan (♂) × Berkshire (♀; n = 3) and Berkshire (♂) × Tibetan (♀; n = 3). Ear tissues from the two parental pigs of the six trios were collected (n = 12). A total of 14 samples comprising liver (n = 6), skeletal muscle (n = 4), and brain (n = 4) of newborn F1 females were collected and snap-frozen in liquid nitrogen for subsequent assays (for details, see Fig. 1A; Supplemental Fig. S1). All research involving living pigs were conducted according to the regulations for the administration of affairs concerning experimental animals (Ministry of Science and Technology, China, revised in March 2017) and approved by the animal ethical and welfare committee of Sichuan Agricultural University under permit no. 20220194. The animals were humanely euthanized as necessary to prevent suffering.
Whole genome sequencing of parent–child trios and in situ Hi-C assays of hybrids
We sequenced the whole genomes of six parent–child trios (n = 18, including 12 parents and six F1 hybrids) (see Supplemental Methods), and constructed eight to 15 in situ Hi-C libraries (technical replicates) for each of the 14 samples of newborn F1 females (see Supplemental Methods). The heterozygous SNVs of each F1 hybrid were masked by converting them to “N” bases to accurately distinguish allele-specific Hi-C contacts (Du et al. 2017). High-quality Hi-C reads were aligned to the variant masked pig reference genome (Sscrofa 11.1), which is the only chromosome-level assembly and therefore provides the best assembly continuity, and sequences representing experimental Hi-C artifacts and other uninformative di-tags were removed, using the standard pipeline provided by HiC-Pro (v 2.9.0) (Servant et al. 2015). Consequently, we obtained about 7.72 billion contacts for each of the 14 samples (data set 1) and an additional approximately 9.27 billion contacts for each of 12 samples (data set 2) (Supplemental Fig. S1).
Construction of chromosomal-span haplotypes
We generated two parental haplotypes for each autosome by combining the information of heterozygous SNVs obtained from whole genome sequencing (see Supplemental Methods) and the Hi-C contacts (see Supplemental Methods). For each trio, we first phased the heterozygous SNVs of F1 hybrids when at least one parental genotype was homozygous (Supplemental Fig. S3E). We also phased heterozygous variants of unknown parental origin based on Hi-C contacts using HapCUT2 (v 1.3.1) (Supplemental Fig. S3F; Edge et al. 2017). We constructed chromosomal-span haplotypes for each F1 hybrid by combining the results of the two strategies, incorporating the vast majority of all heterozygous SNVs. This generated two haplotypes for each chromosome, one for the maternal and another for the paternal alleles.
Reconstruction of haplotype-resolved Hi-C maps
We reconstructed haplotype-resolved Hi-C maps for 14 diploid F1 pig samples using SNPsplit (v 0.3.4) (Krueger and Andrews 2016), HaploHiC (v 0.32) (Lindsly et al. 2021), and the Juicer Tools (Supplemental Fig. S3; see Supplemental Methods; Durand et al. 2016). The reconstruction analysis was separately applied to data set 1 (a maximum resolution of 2-kb resolution for a total of 14 F1 samples) and the aggregation of data sets 1 and 2 (a maximum 1-kb resolution for 12 F1 samples) (Supplemental Fig. S1). The Juicer Tools (Durand et al. 2016) software was used to generate Knight–Ruiz (KR) matrix balancing normalized (Knight and Ruiz 2013) contact matrices for haplotype-resolved intra-chromosomal (at 20-kb and 100-kb resolution) and inter-chromosomal (at 1-Mb resolution) Hi-C maps. Notably, normalized intra- and inter-chromosomal contact matrices (36 × 36) were generated for 18 pairs of homologous autosomes for each sample. After this, the intra- and inter-chromosomal matrices were further normalized across haplotypes using the quantile method (Fletez-Brant et al. 2017) and across samples using the counts per million (CPM; normalized to the average abundance across all samples) strategy. The normalization efficiency was further validated by the cyclic loess method (Stansfield et al. 2019).
Haplotype-resolved interrogation of chromatin architecture, gene expression, and histone modifications
We reconstructed the 3D genome architectures for each sample using the normalized contact matrices from haplotype-resolved Hi-C data (see Supplemental Methods). We identified A/B compartments by performing PCA and using the A-B index (Rowley et al. 2017). Differential compartments, including A/B switched and A/B variable compartments, were identified between haplotypes (see Supplemental Methods). TADs at a resolution of 20 kb were determined (Rowley et al. 2017). TAD boundary shifts were identified between haplotypes (see Supplemental Methods). We identified potential PEIs using the PSYCHIC algorithm (Ron et al. 2017) with default parameters. To investigate the regulatory effects of multiple enhancers on a gene, we calculated a RPS for each gene using high-confidence PEIs, as previously reported (Zhi et al. 2022). Genes with differential RPS were identified between haplotypes.
Total RNA-seq data from 14 samples used for an in situ Hi-C assay were also generated. We assess allelic expression, using the phased exonic SNVs (see Supplemental Methods) as implemented in Allelome.PRO (Andergassen et al. 2015). We performed ChIP-seq using antibodies against H3K27ac (a canonical histone mark of enhancers) and H3K4me3 (an active histone mark of promoters) for 12 samples (liver, skeletal muscle, and brain from each of four F1 hybrids) and qualified the allele-specific activity of promoters and enhancers for each sample (see Supplemental Methods).
Calculation of inter-homolog pairing scores
To measure the strength of interactions between homologs, we generated inter-chromosomal normalized Hi-C maps at 20-kb resolution
and calculated inter-homolog pairing scores (HPSs) for each 20-kb bin. The HPS of a 20-kb bin is the log2-transformed average contact frequency between homologs within a window of W bins upstream of and downstream from the specific 20-kb bin (AlHaj Abed et al. 2019). HPS is defined as
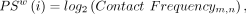 where m and n correspond to the i − Wth and i + Wth bins. The intra-chromosomal score (ICS) was calculated using the intra-chromosomal Hi-C maps. This analysis was applied
to aggregation of the Hi-C data sets 1 and 2 (Supplemental Fig. S1).
where m and n correspond to the i − Wth and i + Wth bins. The intra-chromosomal score (ICS) was calculated using the intra-chromosomal Hi-C maps. This analysis was applied
to aggregation of the Hi-C data sets 1 and 2 (Supplemental Fig. S1).
Data access
All raw sequencing data generated in this study have been submitted to the Genome Sequence Archive of China National Center for Bioinformation (GSA; https://ngdc.cncb.ac.cn/gsa) under accession number CRA006472. Processed files generated in this study have been submitted to the Open Archive for Miscellaneous Data (OMIX; https://ngdc.cncb.ac.cn/omix) under accession number OMIX004825. The spatial transcriptomic data were submitted to the NCBI Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/) under accession number GSE203094. All of the scripts and pipelines used for the analyses in this study are available at GitHub (https://github.com/QianZiTang/linyu) and as Supplemental Scripts.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
We thank Dr. Isaac V. Greenhut for valuable discussion and feedback on the manuscript. This work was supported by the National Key R & D Program of China (2020YFA0509500 and 2023YFD1301302 to M.L., 2021YFA0805903 and 2022YFD1301200 to L.J., 2021YFD1300800 and 2023YFD1300400 to L.L., 2021YFD1301102 to L.C., and 2022YFF1000100 to J.L. and Q.T.), the National Natural Science Foundation of China (32225046 to M.L., 32102507 and U22A20507 to J.L., 32072687 to L.G., 32341050 and 32272837 to L.J., 32102512 and 32372846 to K.L., and 32202630 and 32341051 to F.K.), the Major Program for Science & Technology lnnovation (2023ZD0404404 and 2023ZD0406906 to X.W.), the Tackling Project for Agricultural Key Core Technologies of China (NK2022110602 to L.J.), the Sichuan Science and Technology Program (2021ZDZX0008 and 2022JDJQ0054 to L.J. and Q.T., 2021YFYZ0009 to M.L., 2021YFYZ0030 to Y.G., 2021YFH0033 to J.M., 2022NSFSC1618 to J.L., 2022NSFSC1781 to F.K., 2023NSFSC0169 to X.F., and 2022NSFSC0056 to K.L.), the Major Science & Technology Projects of Tibet Autonomous Region (XZ202101ZD0005N to J.M.), and the Foundation of Key Laboratory of Pig Industry Sciences (22519C to F.K.).
Author contributions: Conceptualization was by M.L. and Y.G. Methodology was by Y.L. and Q.T. Validation was by Y.L., J.L., L.J., Y.Z., and X.K.L. Formal analysis was by Y.L., J.L., L.J., J.B., and J.Z. Investigation was by Y.W., P.L., K.L., M.H., D.L., C.L., Z.H., B.Z., L.L., F.K., and X.F. Resources were by Y.G., Y.S., Y.F., X.W., T.W., A.J., J.M., J.W., L.C., and L.G. Data curation was by L.S., L.Z., Y.J., and G.T. Writing was by M.L., J.L, L.J., and Q.T. Visualization was by Q.L., H.L., and X.W.L. Supervision was by M.L., Q.T., and J.L. Project administration was by M.L. Funding acquisition was by M.L.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.278101.123.
-
Freely available online through the Genome Research Open Access option.
- Received May 16, 2023.
- Accepted February 15, 2024.
This article, published in Genome Research, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








