
Pangenome-genotyped structural variation improves molecular phenotype mapping in cattle
Abstract
Expression and splicing quantitative trait loci (e/sQTL) are large contributors to phenotypic variability. Achieving sufficient statistical power for e/sQTL mapping requires large cohorts with both genotypes and molecular phenotypes, and so, the genomic variation is often called from short-read alignments, which are unable to comprehensively resolve structural variation. Here we build a pangenome from 16 HiFi haplotype-resolved cattle assemblies to identify small and structural variation and genotype them with PanGenie in 307 short-read samples. We find high (>90%) concordance of PanGenie-genotyped and DeepVariant-called small variation and confidently genotype close to 21 million small and 43,000 structural variants in the larger population. We validate 85% of these structural variants (with MAF > 0.1) directly with a subset of 25 short-read samples that also have medium coverage HiFi reads. We then conduct e/sQTL mapping with this comprehensive variant set in a subset of 117 cattle that have testis transcriptome data, and find 92 structural variants as causal candidates for eQTL and 73 for sQTL. We find that roughly half of the top associated structural variants affecting expression or splicing are transposable elements, such as SV-eQTL for STN1 and MYH7 and SV-sQTL for CEP89 and ASAH2. Extensive linkage disequilibrium between small and structural variation results in only 28 additional eQTL and 17 sQTL discovered when including SVs, although many top associated SVs are compelling candidates.
Assigning functional information to genetic variants is challenging. Genome-wide association studies (GWAS) have revealed many quantitative trait loci (QTL) in cattle (Fang and Pausch 2019; Freebern et al. 2020), as well as other species (Filiault and Maloof 2012; Yengo et al. 2022), but require substantial a priori knowledge of phenotypes or traits of interest. Alternatively, expression QTL (eQTL) mapping can use “molecular phenotypes,” such as RNA abundance, to identify regulatory variants, which may contribute to inherited trait variation (e.g., carcass yield [Leal-Gutiérrez et al. 2020; Wang et al. 2022], male fertility [Mapel et al. 2024], and female fertility [Forutan et al. 2023]). Similarly, variants that are associated with alternative splicing or differential isoform usage can be identified through splicing QTL (sQTL) mapping (e.g., milk production [Xiang et al. 2018] and male fertility [Mapel et al. 2024]). In particular, sQTL have been suggested as a leading candidate for explaining a substantial portion of complex trait and disease heritability (Xiang et al. 2023). Alternative splicing can also affect gene expression, and associated variants may also appear as eQTL (Yamaguchi et al. 2022).
Detecting e/sQTL relies on both accurate and complete quantification of RNA abundance, as well as the availability of matched genotypes from the same samples. Recently, several long-read cohorts have shown the importance of including structural variants (SVs) in explaining phenotypic variation in human (Beyter et al. 2021), tomato (Alonge et al. 2020), and rice (Shang et al. 2022). However, most e/sQTL studies, particularly those in livestock, primarily rely on short-read sequencing (Littlejohn et al. 2016) or genotyping arrays (Liu et al. 2020; Cai et al. 2023) to assess genomic variants in enough samples to ensure sufficient statistical power to detect associations with molecular phenotypes. SVs, such as indels >50 bp, have thus been predominantly neglected in GWAS and e/sQTL studies, despite contributing substantially to phenotype variation (Alonge et al. 2020; Scott et al. 2021). Some recent work has used short reads from various cattle breeds to call SVs (Zhou et al. 2022a; Bhati et al. 2023; Lee et al. 2023) but was primarily restricted to deletions and duplications and required extreme filtering to remove false positives. Long and accurate sequencing reads, like Pacific Biosciences (PacBio) HiFi and those produced with Oxford Nanopore Technologies (ONT) r10 chemistries, have the potential to access both small variants (including SNPs and indels <50 bp) and SVs but are costly when sequencing entire mapping cohorts.
A recent intermediate approach, PanGenie (Ebler et al. 2022), produces a “pangenome” variation panel from high-quality, haplotype-resolved assemblies (which can access all scales of variation). Additional samples can then be efficiently genotyped against this panel using k-mers. Crucially, short-read sequencing can be used to produce these k-mers, enabling genotyping of both small variants and SVs in existing biobank-sized short-read cohorts. Earlier pangenome genotyping methods (Chen et al. 2019; Sirén et al. 2021) relied on more laborious sequence alignment to genotype samples, impeding scaling to larger cohorts.
Separate domestication events, adaptation to various environments, and selection for different phenotypic characteristics led to the emergence of several hundred breeds of taurine and indicine cattle (Loftus et al. 1994). The genetic diversity across breeds is huge, but the genetic diversity within typical taurine breeds is low because of the widespread use of few sires in artificial insemination, resulting in a 50-fold lower effective population size in cattle than in human populations (Tenesa et al. 2007; Hall 2016). Fewer than 20 animals typically explain more than half of the genetic diversity of current taurine populations, which suggests that even a limited number of cattle genomes will represent a large fraction of SVs that segregate within breeds (Jansen et al. 2013; Daetwyler et al. 2014).
Here, we created a pangenome variation panel of small variants and SVs from 16 haplotype-resolved cattle assemblies and 307 short-read samples of predominant Brown Swiss (BSW) and Original Braunvieh (OBV) ancestry. We leverage this pangenome variation panel to investigate how eQTL and sQTL mapping in testis tissue benefits from including SVs.
Results
Pangenome genotyping of small variants and SVs
We created the pangenome variant panel using 16 HiFi-based haplotype-resolved cattle assemblies, including four previously published assemblies (two Original Braunvieh, one Brown Swiss, and one Piedmontese) (Crysnanto et al. 2021; Leonard et al. 2022), eight newly generated assemblies from previously published data (four Original Braunvieh and four Brown Swiss) (Leonard et al. 2023), and four assemblies from new data (four Brown Swiss). All assemblies were aligned to the cattle reference genome, ARS-UCD1.2 (Rosen et al. 2020), followed by calling variants from the alignments in confident regions (as described by PanGenie). Through this approach, we identified 12,918,792 SNPs, 3,123,739 indels <50 bp, and 53,297 SVs ≥50 bp, comparable to studies with human samples (Ebert et al. 2021). The three OBV individuals (six haplotypes) were a sire–dam–offspring trio, allowing us to estimate the SNP and SV Mendelian inconsistency rate in the pangenome as 1.06% and 2.32%, respectively.
We compared this set of assembly-derived SVs against SVs called directly from the HiFi reads with Sniffles using Jasmine (Kirsche et al. 2023), requiring two SVs to be the same event type and within 100 bp to be considered as overlapping. As expected, we confirmed a high level of overlap with 86% of assembly SVs recovered in the Sniffles SVs (Fig. 1A). However, there was some disagreement, particularly for large insertions exceeding the average HiFi read size (Fig. 1B). In these circumstances, the read alignments end in soft or hard clips on both ends of the insertion, and the SV cannot be directly detected (Supplemental Fig. 1). Because the assemblies are effectively a single read with megabase-scale length, they can cleanly resolve larger insertion SVs. Deletions larger than the read length can generally still be directly detected. There were spikes of SV frequency for both insertions and deletions approximately of size 1.3 kb, largely confirmed by RepeatMasker to be endogenous retrovirus (ERV) sequence.
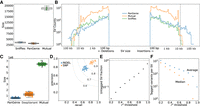
Concordance of variants genotyped by PanGenie. (A) SV overlap between PanGenie and Sniffles for the eight individuals used to create the pangenome variant panel. (B) SV size distribution for the groups in A. The gray dashed lines indicate 15 kb, the average read length for the HiFi reads used by Sniffles. (C) Small variant overlap between PanGenie-genotyped variants and DeepVariant-called variants for the 307 short-read samples. (D) Precision and recall for the 307 samples from C. The gray lines are the F-score boundaries for the indicated values. (E) Fraction of all SVs tagged by small variants at different thresholds of r2 within a linkage window of 1000 kb across the 307 samples. (F) Average and median number of variants that tag each SV across different r2 thresholds.
With PanGenie, we genotyped all the pangenome variation for 307 Braunvieh samples (consisting of Brown Swiss/Original Braunvieh/mixed breeds originating from a common ancestral population) using short sequencing reads. We further supplemented this genotyped variation by directly calling variants with DeepVariant on the 307 samples and merged the PanGenie-called and DeepVariant-called variation into a “PanGenie+” set. The larger sample size for DeepVariant (307 samples vs. eight individuals with haplotype-resolved assemblies) meant more small variants were called, although the majority were mutually present (Fig. 1C). The overwhelming majority of SNPs and SVs in the pangenome variation panel were present in the larger cohort, 98.8% and 96.2%, respectively, whereas small indels (<50 bp) were more frequently missing (80.8% present). The genotype concordance of the calls was also high, with mean F-scores of 0.90 and 0.72 for SNPs and indels, respectively, across the 307 samples (Fig. 1D). There were also four distinct sire–dam–offspring trios in the 307 samples, which we used to validate the genotyping accuracy. The Mendelian inconsistency rate was 1.15% and 4.46% for SNPs and SVs, respectively. We also confirmed the genotyped small variants and SVs were both independently able to recover the expected population structure (in addition to establishing the representativeness of the PanGenie assemblies for their respective breeds) through principal component analyses, although the more complete small variant set explained slightly more of the structure (Supplemental Fig. 2).
We also examined the linkage disequilibrium (LD) between small variants and SVs, finding that ∼70% of SVs are strongly tagged (r2 > 0.8) by small variants within a 1-Mb cis-window, whereas only ∼5% of SVs are poorly tagged (r2 < 0.2) (Fig. 1E). SVs were tagged by an average of 116 variants (median, 15 variants) within that window above the strongly tagged threshold (Fig. 1F).
Variant discovery in a cohort subset with long-reads
We also collected moderate coverage (12.9 ± 1.4-fold) of PacBio HiFi reads on 25 samples of predominant Braunvieh ancestry and called SVs from the long-read alignments using Sniffles. We find that even a small number of samples captures a large portion of SVs present in a given population (Fig. 2A), and we estimate that roughly 100 samples would likely capture nearly all SVs that segregate in a typical taurine cattle breed such as Braunvieh, finding only approximately 100 new SVs per additional sample beyond this population size (Supplemental Fig. 3). Using Jasmine again with a 100-bp distance threshold, we identified that 69% of SVs discovered through the 25 long-read alignments were already present in the PanGenie variant set and genotyped into the larger population (Fig. 2B), rising to 85% when considering only SVs with allele frequency >10% (Fig. 2C). There were also 15,930 SVs that were not in the PanGenie variant set; however, these are likely singleton or rare SVs present in the 25 samples unrelated to those used in constructing the pangenome panel. As such, there is a nonnegligible portion of SVs that could only be discovered through including additional assemblies into the PanGenie variant set or directly calling SVs with long reads on each sample in the e/sQTL set.
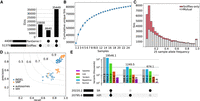
Comparison of variant calling with a small long-read cohort. (A) SV intersection between PanGenie (called from eight individuals with haplotype-resolved assemblies) and Sniffles (called from 25 HiFi read samples). (B) SV saturation for 25 HiFi read samples. Markers indicate the mean value of unique SVs over 10 random shuffles of sample order, and error bars represent the standard deviation. The dotted line is a fitted curve of the form f(x) = ax−b + c, predicting saturation at approximately 175,000 SVs. (C) SV overlap for different allele frequency (based on the 25 samples) bins. (D) Small variant accuracy of HiFi-based and short-read-based calls, taking the short-read data as truth, stratified by autosomes and sex chromosomes for SNPs and indels. Large markers indicate the mean over the 25 samples. (E) Small variant intersections between HiFi-based and short-read-based calls in genomic regions identified as centromeric satellites, low mappability, tandem repeats, repetitive, and “normal” (all other regions). A large proportion of variants called in the challenging regions were unique to HiFi-based alignment and calling.
We were also able to compare small variant accuracy between HiFi and short reads in the 25 samples with about 10-fold coverage of both sequencing approaches. Notably, although there are minor differences for autosome-wide alignments between HiFi and short reads, with HiFi read alignments covering only 0.3% more of the autosomal bases than the short-read alignments, there is a moderate and large effect for the X and Y Chromosomes, respectively: 3.5% and 31.5%. The improved alignments in the sex chromosomes contributed most of the additional variants called by HiFi reads over short reads (Supplemental Fig. 4). Taking the short-read variants as truth, the mean SNP and indel F-score was 0.92 and 0.82, respectively (Fig. 2D), where the higher recall than precision is largely owing to the additional variants called by HiFi reads. Similarly, we observed that HiFi-based alignments (at comparable coverage) called substantially more variants in regions annotated as centromeric satellites, low mappability, tandem repeats, and repetitive, resulting from inconsistent and lower-quality short-read alignments, whereas the number of variants in “normal” regions was comparable (Fig. 2E).
cis-eQTL mapping
After splitting multiallelic variants and filtering at 1% minor allele frequency in the PanGenie+ set, 20,931,316 variants remained for downstream analyses, including 17,439,736 SNPs, 3,449,049 small indels, and 42,531 SVs (Table 1). There were 8355 SVs >1 kb, of which 2103 were >5 kb. Because the SVs were only genotyped through PanGenie and small variants were also called directly, there were fewer rare SVs filtered out compared with SNPs (Supplemental Fig. 5).
Breakdown of variants for SNPs, small insertions and deletions (<50 bp), and SV insertion and deletions (≥50 bp) for the total merged PanGenie+ variant set and the MAF-filtered variant set
We then investigated the impact of SVs on gene expression in a subset of 117 mature bulls for which we also had deep total RNA sequencing from testis tissue, with 257 ± 35 million paired-end reads per sample. After aligning to the cattle reference genome and annotation (Ensembl release 104), followed by quantifying expression as transcripts per million (TPM), we retained 19,440 genes for cis-eQTL mapping. We ran a permutation analysis to determine the significance thresholds, followed by a conditional analysis, finding 3,677,218 associated variants for 15,406 eQTL (11,030 expressed genes [eGenes]). Of those variants, 6985 were SVs (including 1412 and 97 SVs >1 kb and >10 kb, respectively). Association testing in a relatively small cohort of animals with widespread LD often produces identical test statistics for multiple nearby variants. As the most significantly associated variant is not necessarily the causative variant, we also considered variants with conditional significance within 1.5× of the top variant (adapted from Sanchez et al. 2017) as candidate causal variants. We find 92 SV-eQTL in which 25 have eSVs as the unique-top variant (Fig. 3A) and 58 eQTL in which the top variant is an SV that is in near-perfect LD with a small variant (Fig. 3B).
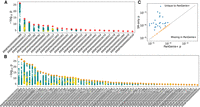
cis-QTL mapping. (A) Twenty-five independent eGene signals with red diamonds denoting SVs as uniquely top hits. Other SVs are shown as yellow diamonds, and small variations are shown as teal circles. (B) Fifty-eight independent eGene signals with SVs as top hits in LD, with small variants denoted as orange diamonds and with yellow diamonds and teal circles as described in A. (C) eGenes that are present in only the PanGenie+ data set or the short-read-only DeepVariant data set. The dashed line indicates equal significance thresholds between the two conditional analyses.
We also performed the permutation and conditional analyses using small variants combined with 52,221 SVs directly discovered and genotyped through the cohort short reads with DELLY and INSurVeyor. There were 3,615,699 variants associated with the expression of 11,061 eGenes. All eGenes found uniquely with the short-read data set were just missed by the significance threshold in the PanGenie+ data set, suggesting they are of marginal importance (Fig. 3C). On the other hand, there were 26 eGenes found only with the PanGenie+ data set, including four for which the top eVariant was an SV (and the remaining were typically small indels within tandem repeats). Nearly half of the PanGenie+ SV-eQTL were not discovered through the short reads alone, whereas a further quarter were discovered but poorly genotyped and were not significant eQTL (Supplemental Table 1; Supplemental Fig. 6).
We further examined in more detail several eGenes that are affected by SVs identified uniquely with the PanGenie+ set (Supplemental Table 2). For example, we identified a strong cis-eQTL ∼14 kb downstream from the annotated translation termination codon of STN1 (ENSBTAG00000015019) encoding STN1 subunit of the CST complex (Fig. 4A). This cis-eQTL was significantly associated with 672 variants, although one of the top variants (P = 1.99 × 10−22) was a 5.9-kb deletion containing 3.9 kb of DNA transposons, RTEs, and ERV-LTR elements, occurring with a frequency of 32% in the 117 animals. The deletion is in high LD (r2 = 0.923) with the top SNP (Chr 26: 24,452,023 bp) and the association signal only slightly lower. STN1 is moderately expressed (13.59 ± 1.92 TPM) in testis, but the deletion reduces STN1 mRNA abundance (effect size [β] of −1.11). Closer inspection of the eQTL also revealed limitations of the current functional annotation of the bovine reference genome. The Ensembl annotation of STN1 contains five transcripts, whereas the RefSeq annotation suggests 10 isoforms, of which eight are expressed in testis, including one (XM_024985601.1) that has an intron overlapping the deletion (Supplemental Fig. 7A). Although the deletion reduces the expression of three isoforms, including the canonical isoform (NM_001076849.1), it increases the abundance of five other isoforms (Supplemental Fig. 7B). Because the canonical isoform is more abundant than all other isoforms, the deletion overall reduces STN1 expression. We corroborated that the SV-eQTL impacts not only the overall expression of STN1 but also its relative isoform abundance, as this SV is also strongly associated with a sQTL (P = 3.96 × 10−22) (Supplemental Fig. 7C), although it was not the top candidate.
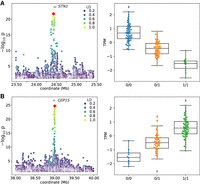
Nominal eQTL association significance (left) and normalized TPM values for the expressed gene (right) for STN1 (A) and CEP15 (B). The red diamond represents the top-associated SV. Linkage disequilibrium (LD) between the SV and all other variants within the cis-window is indicated with the color gradient.
A 118-bp deletion was strongly associated with CEP15 (ENSBTAG00000001889, encoding centrosomal protein 15) mRNA abundance (Fig. 4B). The deleted sequence is a short interspersed nuclear element (SINE). The SV-eQTL was located 8 kb downstream from the transcription start site of CEP15 and was 1.4× as significant (P = 6.60 × 10−27) as the closest SNP. The deletion was associated with increased (β = 1.23) CEP15 expression.
We also examined two prominent insertion SV-eQTL. The expression of MYH7 (ENSBTAG00000009703) encoding myosin heavy chain 7 was associated with a 388-bp insertion (P = 1.27 × 10−22) consisting almost entirely of LINE sequence. The LINE sequence was inserted 8.3 kb downstream from MYH7 and increased mRNA abundance (β = 1.37). The expression of LOC112443864 (ENSBTAG00000053433) encoding MHC class I polypeptide-related sequence B-like was associated with an 11.6-kb insertion (P = 2.18 × 10−25) containing 2.3 kb of SINE, LINE, and ERV-LTR elements 7.4 kb upstream (β = 1.09) (Supplemental Fig. 8). Given its location nearby the bovine leukocyte antigen (BoLA) complex, this SV potentially could contribute to eQTL in immune-related tissues.
We realized that the 11.6-kb insertion affecting LOC112443864 expression also highlights difficulties in association testing with large SVs. The original pangenome variant panel constructed from the 16 haplotypes contained three near-identical (>99.9% sequence identity) insertion alleles, differing by only several SNPs. Each allele, when considered individually for molQTL mapping after PanGenie-based genotyping, was below the significance threshold for LOC112443864, but curating and merging the alleles before genotyping and conducting the eQTL analysis revealed a highly significant peak (Supplemental Fig. 9).
cis-sQTL mapping
We performed a similar analysis for sQTL, now using intron excision ratios as the phenotypes. We tested for associations in 14,243 genes with 46,417 splicing clusters. With the PanGenie+ variant set, we find 3,613,475 associated variants for 16,893 sQTL (7064 spliced genes [sGenes] and 10,629 splicing clusters), of which 5366 were SVs and 1061 were SVs >1 kb. Again, we found only 11 additional sGenes compared with using the short-read-only data set, but we did find 73 SV-sQTL with 15 sSVs as the unique-top variant (Fig. 5A) and 58 sQTL for which the top variant is an SV that is in near-perfect LD with a small variant (Fig. 5B; Supplemental Table 2). Similar to our observations for SV-eQTL, over half of the PanGenie+ SV-sQTL were undetected by short reads with a further quarter with low genotyping accuracy (Supplemental Table 1).
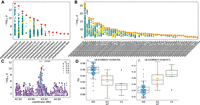
cis-sQTL mapping. (A) Fifteen independent sGene cluster signals with SVs as the unique-top variant and (B) 58 SVs as top variants in LD with small variants, with the color and marker meanings as described in Figure 4. (C) Nominal association significance for CEP89, where the two red diamonds indicate the same variant affecting two separate junction splicings within the sQTL cluster. (D) Percentage spliced in (PSI) across the two significantly associated junctions (indicated by number from C) within the sQTL cluster.
We examined an sQTL for CEP89 (ENSBTAG00000004864) encoding centrosomal protein 89 in more detail, noting that a 1.3-kb insertion at Chr 18: 43,395,289 3 kb downstream from the transcription start site and 420 bp upstream of the 3′ splice site of the second intron was the top associated variant for a splice cluster containing two splice junctions. This 1.3-kb insertion was approximately six times more significant (P = 6.0 × 10−11) than the next highest SNP, with β = −0.86 (Fig. 5C). The inserted sequence was almost entirely an LTR retrotransposon and was present in ∼30% of samples, contributing to alternative splicing (Fig. 5D). The two associated splice junctions span the second and third exon of CEP89 (Supplemental Fig. 10). However, the annotation of three CEP89 transcripts in Ensembl again appears incomplete as RefSeq indicates seven CEP89 isoforms. Although the SV does not affect the overall CEP89 expression (i.e., CEP89 was not an eGene), it is associated with the abundance of two isoforms, suggesting that this sQTL promotes alternative isoform usage and so impacts the relative abundance of distinct CEP89 isoforms (Supplemental Fig. 10C).
We also examined an sQTL for ASAH2 (ENSBTAG00000003529) encoding N-acylsphingosine amidohydrolase 2. The top associated variant was 34 kb downstream and was a 3.6-kb insertion and was approximately 150 times more significant (P = 1.60 × 10−11) than the next most significant SNP, causing alternative splicing (Supplemental Fig. 11) with β = 1.24. The inserted sequence contained ∼2 kb of BovB repeats, another transposable element.
The inserted sequence was located within a putative duplication, causing potential misalignments of short reads and thus erroneously calling a C-to-T transition at 26:8694035. This variant has previously been reported (EVA: rs385128608). However, long-read alignments more strongly support the 2-kb insertion detected through the assemblies, although even these were complicated to validate (Supplemental Fig. 12). Although the SV genotyped through PanGenie appears to be the most accurate, the limited LD observed with adjacent small variations may also indicate imperfect genotyping owing to limited unique k-mers in the region. More generally, there are 4338 instances in which a SNP and SV share a starting genomic coordinate, of which 1851 (42.7%) occur in regions identified as VNTRs (Leonard et al. 2023), in which short reads can easily misalign and appear as motif variation rather than insertions/deletions of additional repeats. There are 599 and 425 SV-e/sVariants, respectively, overlapped by SNPs, of which 61 and 85, respectively, are highly significant (P < 1 × 10−10).
Discussion
Genotyping pangenome-discovered variation, including both small variants and SVs, with short reads improves eQTL and sQTL mapping over just using the short reads alone. Compared with the long-read truth set, we detected too few deletions (10,000 vs. 32,000) and too many insertions (43,000 vs. 37,000) with short reads, and of the discovered SVs, many were poorly genotyped. Many putative short-read SVs were filtered out, like the 388-bp insertion affecting MYH7, which could not be confidently clustered as per-sample alleles erroneously ranged from 130 to 458 bp. Furthermore, ∼20% of discovered insertions were incompletely assembled, which would hinder downstream characterization of the sequence and prevent differentiation between multiple alleles, which occur frequently at large SVs. From the same set of short reads and assemblies from eight individuals, PanGenie was able to accurately genotype variation and discover several new e/sGenes, as well as compelling new SV causal candidates.
Given the relatively small effective population size (about 100) and high relatedness of the Original Braunvieh and Brown Swiss breeds, even 16 haplotypes were sufficient to capture 69% of SVs in 25 unrelated Original Braunvieh or Brown Swiss HiFi samples. The assemblies also could detect larger insertions compared with using the samples’ raw sequencing directly with read-based approaches, as recently observed (Harvey et al. 2023). Furthermore, we identify cases like an SV-sQTL for ASAH2, in which SNPs (including some reported in public databases) may actually be SVs. PanGenie can resolve such cis-e/sQTL associations with the genotyped SVs correctly called from high-quality assemblies. Creating haplotype-resolved assemblies is currently the biggest bottleneck in this approach, but given a larger number of initial assemblies, association mapping may further improve by only using PanGenie genotyped variation and not supplementing with occasionally erroneous short-read-called variation.
We observe extensive LD between SNPs and SVs, as reported elsewhere (Zhou et al. 2022b; Lee et al. 2023), which limits the number of e/sGenes that were uniquely discovered by the PanGenie variant set. However, including SVs revealed equally or more significant top associations and, in some cases, more compelling causal candidates (e.g., deletion of an entire exon) than the tagging SNPs. This is particularly true for 53 and 41 insertion SV-e/sQTL, respectively, which can only comprehensively be interrogated through long reads or assembly-based approaches. Still, the variant set lacks a moderate fraction of SVs segregating in this cattle population, particularly rare alleles that might have an especially strong impact on gene expression and splicing (Li et al. 2017; Wagner et al. 2023), and so, a full long-read cohort may provide greater power to find untagged SV-QTL.
Several of the SV-QTL examined in detail (e.g., STN1, CEP89, and ASAH2) contain inserted or deleted sequences that are largely composed of transposable elements, like ERV-LTR, BovB, and hobo transposons. More generally, we found 51 out of 92 (55.4%) SV-eQTL and 37 out of 73 (50.7%) SV-sQTL contained transposable elements (Supplemental Table 3), matching previous observations of SVs widely containing mobile genetic elements (Chaisson et al. 2019; Ebert et al. 2021). Although many of these e/sQTL were also associated with SNPs that were in LD with SVs, transposable elements are widely reported to be able to mediate expression (Almeida et al. 2007; Elbarbary et al. 2016; Platt et al. 2018; Kelly et al. 2022), and so are strong candidates for being the causal variants. Given the SV size distribution spike around the size of LTR elements, it is likely such transposable elements will increasingly be identified as a driving force behind bovine phenotypic diversity.
Association mapping with SVs is not just a simple extension to using SNPs, owing to SVs’ greater proclivity of having highly similar but distinct alleles. Larger SVs (e.g., >1 kb) are likely to appear multiallelic across older assemblies or individual high-quality reads (typically quality value of about 30, or one error expected per one kb). Distinguishing technical noise (errors in reads/assemblies) from meaningless biological variation (differences in allele have no functional consequence) or from meaningful biological variation (differences in allele may functionally impact gene regulation) is an open and challenging question. Addressing this question is particularly critical for pangenomes containing diverse (sub)species, as multiallelic but similar SVs become increasingly common, which can dilute significant associations below their thresholds.
We also confirm recent results that at moderate coverages (about 10-fold), HiFi reads can replace short reads for small variant calling, while accurately calling SVs (Harvey et al. 2023; Kucuk et al. 2023). The former is especially true in highly repetitive regions like centromeric satellites or tandem repeats, which have largely been challenging to assess with short reads even using dedicated tools. As such, future large efforts, like the Bovine Long Read Consortium (BovLRC) (Nguyen et al. 2023), will likely be able to assess nearly all genomic variation from only a single data source of accurate long reads, as well as providing sufficient samples for statistically significant QTL mapping of rare SVs and trans-QTL. However, in the intermediate future, although solely long-read cohorts are prohibitively costly, we show that several assemblies and pangenome genotyping of SVs can greatly improve our ability to detect additional e/sQTL as well as identify more compelling causal candidates.
Methods
HiFi sequencing
We extracted high-molecular-weight DNA from blood of two animals with the Qiagen MagAttract HMW kit, following the manufacturer's protocols. PacBio HiFi libraries were generated and sequenced on three SMRT cells each by the Functional Genomic Center Zurich (FGCZ).
Testis tissue from 25 additional BSW/OBV individuals was sampled from a commercial abattoir in Zürich, Switzerland. We extracted high-molecular-weight DNA with the Monarch HMW extraction kit for tissue (New England BioLabs) and followed the manufacturer's recommendations. DNA fragment length and quality were assessed by the FGCZ with the Femto pulse system (Agilent). PacBio HiFi libraries were produced and sequenced on one SMRT cell per individual with a Sequel IIe.
Genome assembly
Four Original Braunvieh and four Brown Swiss haplotypes were assembled from publicly available data (under NCBI BioProject [https://www.ncbi.nlm.nih.gov/bioproject/] accession number PRJEB42335). In addition, we assembled four Brown Swiss haplotypes from new HiFi data (accession codes ERS15606279 and ERS15606280) from two F1s. We used hifiasm (v0.19.4-r575) (Cheng et al. 2021) to generate the haplotype-resolved assemblies, using default parameters and providing parental k-mers of size 31 counted by yak (v0.1-r66-dirty, https://github.com/lh3/yak) for the two trios. We scaffolded the resulting contigs to ARS-UCD1.2 using RagTag (v2.1.0) (Alonge et al. 2022) with the additional parameters “-cx asm20.”
PanGenie genotyping
We created the variant panel from the 16 cattle assemblies following the approach laid out by PanGenie (Ebler et al. 2022). Briefly, we aligned each assembly to ARS-UCD1.2 with minimap2 (v2.24-r1122) (Li 2018) with the parameters “-ax asm20 -m 10,000 -z 10,000,50 -r 50,000 ‐‐end-bonus=100 -O 5,56 -E 4,1 -B 5,” followed by calling haploid variants for each haplotype with paftools.js. Variants were merged into diploid calls and filtered according to PanGenie. We additionally modified the merging step to consider SVs with >98% sequence identity to be part of the same cluster and take the first SV of the cluster as the allele.
We genotyped 307 short-read samples using PanGenie (v.2.1.1) (Ebler et al. 2022) with the pangenome variant panel using default parameters. Each VCF was then merged using BCFtools (v1.17) merge (Danecek et al. 2021).
Small variant calling
We aligned short-read samples (available from NCBI BioProject accession no. PRJEB28191) to the ARS-UCD1.2 reference using BWA-MEM (v0.717) (Li 2013) using the -M flag, followed by coordinate sorting and deduplicating with SAMtools (v1.17) (Danecek et al. 2021). Variants were called per-sample using DeepVariant (v1.5.0) (Poplin et al. 2018) with the “WGS” model and jointly genotyped and filtered using GLnexus (v1.4.1) (Yun et al. 2021) with the “DeepVariantWGS” configuration. Sporadically missing variants were then imputed using Beagle (v5.4) (Browning et al. 2018).
We aligned long-read samples to ARS-UCD1.2 using minimap2 with the parameters “-ax map-hifi” and converted to BAM files as described above. We called variants as above, except using the “PACBIO” model for DeepVariant.
Long-read SV calling
We called and jointly genotyped SVs using Sniffles (v2.0.7) (Smolka et al. 2024) on the aligned long-read files with the parameter “‐‐min_sv_len=50.” For the assembly haplotype samples, we additionally used the “‐‐phased” parameter. We filtered out BND-type variants as well as variants exceeding 100 kb with BCFtools view.
Short-read SV calling
We used DELLY (v1.1.8) (Rausch et al. 2012) to call SVs per sample from the aligned short-read BAM files, before using delly merge with the flags “‐‐minsize 50 ‐‐precise ‐‐pass” to create a list of SV sites. We then used delly call to force-genotype these SV sites. We also used INSurVeyor (v1.1.2) (Rajaby et al. 2023) to similarly discover SV sites, merged across samples with SurVClusterer (v1.0, https://github.com/Mesh89/SurVClusterer), and then force-genotype using SurVTyper (796e9d0; https://github.com/kensung-lab/SurVTyper). We removed all insertions from the DELLY SV calls before merging with the INSurVeyor SV calls using BCFtools concat to create a unified set of insertions and deletions from short reads.
Variant analyses
We used BCFtools to merge the DeepVariant short-read-called variants with the PanGenie genotyped variants for the 307 samples, using the concat command with the “-D” flag to remove duplicate variants (giving allele/genotype priority to DeepVariant). Indels were left-normalized with BCFtools norm.
We assessed genotype accuracy using hap.py (v0.3.15, https://github.com/Illumina/hap.py), using the short-read-called variants as truth and the HiFi-called variants as query. We determined the overlap of the two variant sets using BCFtools isec with parameters “-c some -n +1” to allow partial overlapping of multiallelic sites, followed by determination of the proportion in centromeric satellites using BEDTools (v2.30.0) (Quinlan and Hall 2010) intersect on those positions and annotated regions. We determined if multiples SVs were “the same” using Jasmine (v1.1.5) (Kirsche et al. 2023), allowing intersections up to the smaller of max_dist_linear = 1 (proportional to SV size) and max_dist = 1000 (1 kb).
We used the BCFtools mendelian2 plugin to assess Mendelian inconsistency rates.
RNA sequencing and alignment
RNA from 117 testis samples were sequenced from paired-end total RNA libraries, as described previously (Mapel et al. 2024), available from the NCBI BioProject accession number PRJEB46995. Briefly, the sequencing reads were trimmed using fastp (v0.23.4) (Chen et al. 2018) and aligned to ARS_UCD1.2 and the Ensembl gene annotation (release 104) with STAR (version 2.7.9a) (Dobin et al. 2013). We produced an additional set of alignments with the flag ‐‐waspOutputMode to account for allelic mapping bias for sQTL analyses.
QTL analyses
Gene quantification and covariate files were processed for e/sQTL analyses as previously described (Mapel et al. 2024). Briefly, to quantify gene-level expression in TPM, we used QTLtools quan (Delaneau et al. 2017), and to infer gene-level read counts, we used featureCounts (Liao et al. 2014). We removed lowly eGenes and only included genes with ≥0.1 TPM in ≥20% of samples and six or more reads in ≥20% of samples. Filtered expression values were quantile-normalized and inverse normal transformed for downstream analyses.
For splicing quantification, we considered intron-excision values from intron clusters identified. Specifically, we identified exon–exon junctions from WASP-filtered reads with RegTools (Cotto et al. 2023), followed by using Leafcutter (Li et al. 2018) to construct intron clusters and an altered “map_clusters_to_ genes.R” script to map clusters to the cattle gene annotation (Ensembl release 104). We filtered introns with read counts in <50% of samples, introns with low variability across samples, and introns with fewer than max(10, 0.1n) unique values (where n is sample size). We used the “prepare_phenotype_table.py” script from Leafcutter to normalize filtered counts and produce files for sQTL mapping.
We filtered variants with MAF < 0.01 and split multiallelic sites using BCFtools view and norm, respectively. We performed all association testing (for both e/sQTL) using QTLtools (v1.3.1) (Delaneau et al. 2017). Permutation analyses were performed using a 1-Mb cis-window 2000 times with a false-discovery rate of 0.05, which determined the significance thresholds for each gene in the conditional pass. Nominal association was performed using a significance threshold of 0.05. LD scores for specific variants were calculated using PLINK v1.9 (Chang et al. 2015).
The abundance of RefSeq (version 106, GCF_002263795.1) transcripts was quantified using kallisto (version 0.46.1) (Bray et al. 2016) and aggregated to the gene level using R (v4.2) (R Core Team 2022) with the package tximport (Soneson et al. 2016).
Data access
The HiFi data generated in this study have been submitted to the NCBI BioProject database (https://www.ncbi.nlm.nih.gov/bioproject/) under accession numbers PRJEB46995 (accessions SAMEA113612078, SAMEA113612079, SAMEA113612080, SAMEA113612081, SAMEA113612082, SAMEA113612083, SAMEA113612084, SAMEA113612085, SAMEA113612086, SAMEA113612087, SAMEA113612088, SAMEA113612089, SAMEA113612090, SAMEA113612091, SAMEA113612092, SAMEA113612093, SAMEA113612094, SAMEA113612095, SAMEA113612096, SAMEA113612097, SAMEA113612098, SAMEA113612099, SAMEA113612100, SAMEA113612101, SAMEA113612102) for the truth set long reads and PRJEB42335 (accessions SAMEA113612103 and SAMEA113612104) for the new assemblies. The F1 parental short-read data generated in this study have been submitted under accession number PRJEB18113 (accessions SAMEA8565028 [sire] & SAMEA8565098 [dam] and SAMEA32980918 [sire] & SAMEA32981668 [dam], respectively). All scripts are available at GitHub (https://github.com/AnimalGenomicsETH/pangenome_molQTL) and as Supplemental Code.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
We thank Dr. Anna Bratus-Neuenschwander and Dr. Catharine Aquino from the ETH Zurich technology platform FGCZ (https://fgcz.ch) for DNA fragment analysis and DNA and RNA sequencing. We also thank Eirini Lampraki from Pacific Biosciences for DNA sequencing. We thank Dr. Cord Drögemüller (University of Bern) for providing blood samples. This work was financially supported by the Swiss National Science Foundation (SNSF), an ETH Research Grant, and Swissgenetics. The funders had no role in study design, data collection and analysis, interpretation of the data, decision to publish, or preparation of the manuscript.
Author contributions: A.S.L. and H.P. conceived the study. X.M.M. performed the DNA and RNA sequencing. A.S.L. constructed the genome assemblies and created the small, structural, and PanGenie variant sets. A.S.L. ran the e/sQTL association analyses with input from X.M.M. A.S.L. and H.P. performed detailed analyses on specific QTL. A.S.L. and H.P. wrote the manuscript. All authors read and approved the final manuscript.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.278267.123.
- Received July 11, 2023.
- Accepted February 1, 2024.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see https://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








