
Global identification of mammalian host and nested gene pairs reveal tissue-specific transcriptional interplay
- 1Department of Medical and Molecular Genetics, King's College London, London SE1 9RT, United Kingdom;
- 2Department of Infectious Diseases, King's College London, London SE1 9RT, United Kingdom
-
↵3 These authors are joint first authors and contributed equally to this work.
Abstract
Nucleotide sequences along a gene provide instructions to transcriptional and cotranscriptional machinery allowing genome expansion into the transcriptome. Nucleotide sequence can often be shared between two genes and in some occurrences, a gene is located completely within a different gene; these are known as host/nested gene pairs. In these instances, if both genes are transcribed, overlap can result in a transcriptional crosstalk where genes regulate each other. Despite this, a comprehensive annotation of where such genes are located and their expression patterns is lacking. To address this, we provide an up-to-date catalog of host/nested gene pairs in mouse and human, showing that over a tenth of all genes contain a nested gene. We discovered that transcriptional co-occurrence is often tissue specific. This coexpression was especially prevalent within the transcriptionally permissive tissue, testis. We use this developmental system and scRNA-seq analysis to demonstrate that the coexpression of pairs can occur in single cells and transcription in the same place at the same time can enhance the transcript diversity of the host gene. In agreement, host genes are more transcript-diverse than the rest of the transcriptome. Host/nested gene configurations are common in both human and mouse, suggesting that interplay between gene pairs is a feature of the mammalian genome. This highlights the relevance of transcriptional crosstalk between genes which share nucleic acid sequence. The results and analysis are available on an Rshiny application (https://hngeneviewer.sites.er.kcl.ac.uk/hn_viewer/).
Classically genes are represented one after another along the chromosome's genomic sequence separated by intergenic stretches of DNA which are devoid of genes, in a strand-specific manner. However, in practice, the genomic organization is more complex, and genes can in reality occupy the same genomic space as each other and are considered as overlapping genes. The first overlapping pairs of genes were identified in the genomes of viruses (Weisbeek et al. 1977). Viruses are rapidly evolving organisms, with a small genome that must be packaged into a small capsid, and still maintain genetic novelty (Feiss et al. 1977; Wu et al. 2010). Thus, overlapping their genes allows them to deal with these constraints while enhancing their evolutionary stability because a mutation in an overlapping pair would affect more than one gene (Simon-Loriere et al. 2013). Higher-order organisms, on the other hand, contain vast amount of intergenic space for genes to occupy, do not have the requirement of rapid evolution, and yet, still contain genomic overlaps (Veeramachaneni et al. 2004). In eukaryotes, the first of these was identified in 1986 in Drosophila and mouse (Henikoff et al. 1986; Spencer et al. 1986; Williams and Fried 1986).
In 2019, a study in humans estimated that ∼26% of the protein-coding genes overlapped with at least one other protein-coding gene (Chen et al. 2019). Most of the time, the overlap involved a noncoding sequence (5′ untranslated region or UTR, 3′ UTR or intronic sequence) and sometimes only a few nucleotides were involved at the 3′ or 5′ end of the genes. However, larger genomic overlaps exist and can result in a gene being fully contained within a completely different gene. This configuration of genes contained one within the other is known as a host/nested gene pair. Genes organized this way are likely to cooperatively regulate one another because they occupy the same genomic space.
The steric hindrance observed when two RNA polymerases transcribe two different genes in the same genomic region (Billingsley et al. 2012) and transcriptional collision observed at convergent genes (Prescott and Proudfoot 2002) suggest that host and nested genes may prevent one another's expression and may be mutually exclusively expressed. Furthermore, a repressive chromatin environment that prevents instances of intragenic spurious initiation of transcription is established in gene bodies by transcription itself (Latos et al. 2012; Neri et al. 2017). This is likely to repress nested gene expression when the host gene is expressed. On the other hand, nested gene expression has been shown to impact host gene pre-mRNA processing mechanisms, and, as a consequence, can generate host RNA isoforms with different stability and coding capacity (Licatalosi and Darnell 2010). This was shown for example at two independent imprinted loci, where the expression of a nested gene is associated with “premature” alternative polyadenylation of the host gene upstream of the nested gene, whereas its silencing is associated with polyadenylation at the canonical 3′ UTR (Wood et al. 2008; Cowley et al. 2012). This extends beyond the imprinted context and has been suggested to occur at other host genes harboring nested LINE1 retrotransposons, nested genes, or putative intragenic promoters (Kaer et al. 2011; Amante et al. 2020). Growing evidence indicates that the short isoforms generated through “premature” polyadenylation are key for cell functions (Singh et al. 2018).
Thus, even though the role and the consequences of the host/nested gene organization are starting to be uncovered, an updated genome-wide analysis is urgently needed. As such, characterization and identification of all possible host/nested genes are crucial for the understanding of gene regulation both in terms of the level of expression and isoform regulation.
Previous compilations of host/nested genes are now outdated. Two previous analyses using NCBI RefSeq and microarray data, at the time, identified 373 and 128 host/nested gene pairs in human (Yu et al. 2005; Assis et al. 2008). Not only was the number of pairs identified different between the two analyses, but also conclusions regarding the coregulation of the pairs were different. Yu et al. (2005) demonstrated that most pairs were anticorrelated, suggesting mainly a mutually exclusive expression pattern while some showed a positive correlation and were expressed at the same time. Assis et al. (2008) did not identify any significant correlation between the pairs, suggesting that host and nested genes are not influencing each other's expression and that this organization is “neutral.”
These are the most recent collations of host–nested genes in human from 2005 and 2008, respectively (Yu et al. 2005; Assis et al. 2008). Genomic annotation is now much more comprehensive and advances in measurement technologies detect transcript isoforms in a more systematic way, as such, these previous lists underestimate the actual numbers (Frankish et al. 2019). Here, we identify and characterize the host/nested gene pairs in mouse and human using contemporary genomic annotations, RNA sequencing, and single-cell RNA sequencing (scRNA-seq) data, and collate this analysis into an accessible shiny app for use by the scientific community (https://hngeneviewer.sites.er.kcl.ac.uk/hn_viewer/).
Results
About a sixth of human (17%) and a tenth of mouse (12%) genes contain at least one nested gene
To obtain the most accurate and exhaustive list of host and nested genes in mouse and human, we decided to use the GENCODE consortium genomic annotation. We used the “comprehensive” set which includes the highest number of transcripts associated with protein-coding genes, pseudogenes, long noncoding RNAs and small noncoding RNAs genes (Frankish et al. 2019). Transcripts with “to be experimentally confirmed (TEC)” and “immunoglobulin (IG) variable chain or T cell receptor (TR) genes” biotypes were removed. In addition, complex loci where alternative promoter or recombined segments are annotated with different gene names but cannot be considered as independent genes were also removed (Jung et al. 2006; Strassburg et al. 2008; Jia and Wu 2020) (see Methods; Fig. 1) to ensure that a gene was defined as a validated transcriptional unit. Coordinate information for all the remaining genes was overlapped, and the result was filtered according to the extent of the overlap. Only pairs where all the transcripts of one gene were fully contained inside another transcript and where this involved transcripts from genes with different names were retained. When different host transcripts of the same genes were involved in a pair, the longest was selected. After filtering 7560 and 13,088 host/nested gene pairs in mouse and human remained, respectively (Supplemental Tables S1, S2). This represented 4801 (∼12% of the genes) and 7661 (∼17% of the genes) genes with at least one nested gene in mouse and human, respectively (Fig. 1; Supplemental Tables S1, S2).
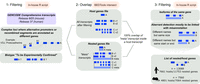
Host/nested gene pair identification. Schematic of the pipeline used to generate the list of host/nested gene pairs.
Hosts harbor nested genes preferentially inside a long intron and show no strong orientation bias
To identify any preferences of host/nested gene structure and location, we performed a suite of analyses based on their basic characteristics. Host genes were longer than their nested gene partner (Fig. 2A) and longer than the average size of the corresponding transcriptome (Supplemental Fig. S1A). However, Spearman's rank correlation analysis revealed that the positive relationship between host and nested gene sizes was weak (ρ < 0.3) (Fig. 2A). To minimize the impact to the transcriptome, we hypothesized that host genes would not contain a large number of nested genes and that they would preferentially locate within noncoding intronic host gene regions. Most of the host genes, indeed, harbored less than four nested genes (95th percentile, three in mouse and four in human) but in some extreme cases host genes could contain up to 77 nested genes in human and 186 in mouse (Fig. 2B). In addition, ∼75% of the nested genes are fully contained inside one intron of their host gene and ∼20% of nested genes span both an intron and an exon (Supplemental Fig. S1B). The remaining overlap with exonic sequences and 3%–5% of nested genes are completely embedded within exons (Supplemental Fig. S1B). In this case, however, nested genes predominantly localized to the last exon of host genes (Supplemental Fig. S1C). Within introns, nested genes were preferentially inside the largest host intron (Fig. 2C), as observed before (Yu et al. 2005). Previous studies reported that the largest introns of genes were usually located at the beginning of the gene (Smith 1988; Bradnam and Korf 2008). In line with this, we observed that nested genes were often located inside the first intron of their host (Supplemental Fig. S1D), and when looking in detail at the hosts containing up to 20 introns, nested genes were preferentially located inside the most 5′ introns of their host (Supplemental Fig. S1E). Assessment of pair orientation did reveal a slight bias toward being in the opposite orientation between pairs, but the distribution between opposing and same orientation was broadly similar (Fig. 2D). This pattern was similar for nested genes fully contained in introns and a stronger bias for opposite orientation was observed when nested genes spanned both exons and introns (Supplemental Fig. S1B–D).
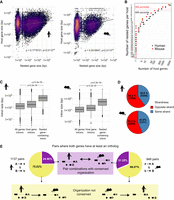
Host/nested gene pairs in mouse and human show similar characteristics. (A) Scatter plot showing the correspondence between the sizes of host and nested genes. The color gradient indicates the density. A base-10 log scale was used for the gene size. Spearman's correlation coefficient was determined which is shown on the graph alongside the resultant P-value. Linear regression analysis was computed and the regression line was added on the graph alongside the calculated 95% confidence intervals. (B) Scatter plots showing the number of nested genes per host. The dotted line represents the 95th percentile of the distribution. A base-10 log scale was used for the number of host genes. (C) Distribution of the intron size for all introns in all the genes, all introns in host genes, or the intron of the host containing a nested gene. A base-10 log scale was used for the intron size. The significance of the difference was tested using a Welch two sample t-test. The resulting P-value is mentioned on the graph. (D) The proportion of host/nested gene pairs with both genes in the same orientation or in the opposite direction. (E) Overlap and conservation between the pairs having an ortholog for both host and nested in the other species.
Given that host/nested gene pairs had a surprising coverage of over a tenth of genes across the genome, we asked if these pairs were conserved between mouse and human. We observed that more host genes had at least an ortholog in the other species than nested genes (64.01% vs. 15.61% in human and 75% vs. 20.99% in mouse) (Supplemental Fig. S2A). Considering only the pairs where both genes have an identified ortholog in the other species (1137 pairs in human and 948 pairs in mouse), we found that 24.98% of the human pairs and 31.33% of the mouse pairs were conserved in a similar host/nested gene configuration in the other species (Fig. 2E), as exemplified by Mcph1/Angpt2 and MCPH1/ANGPT2 (Supplemental Fig. S2B).
Host and nested genes are enriched for different functional classes
To better characterize the genes involved in host/nested gene pairs, we took advantage of the broad functional classes of biotypes available in GENCODE (Frankish et al. 2019). By comparison to the distribution of biotypes among all the genes, we found that host genes were enriched for protein-coding genes (P < 0.001, hypergeometric test) (Fig. 3A,B). The nested genes were enriched for small RNAs in general, whereas host genes were depleted for these transcripts in both species (P < 0.001, hypergeometric test) (Fig. 3A,B). When looking at the number of instances for each biotype, we observed that most of the host genes, but also a high proportion of the nested genes were protein-coding genes (Supplemental Fig. S3A). As expected from the enrichments, we also detected a high number of noncoding RNA in the nested genes list (lncRNAs, antisense RNAs, and miRNAs) (Supplemental Fig. S3A). Finally, when considering the correspondence of biotypes between the host and nested genes per pair, no trend was observed for host and nested genes biotype association, other than the expected higher proportion of pairs of genes being organized in the opposite direction when one of the partners was annotated as an antisense transcript (Fig. 3C). Mammalian promoters can be broadly classified into CpG island (CGI) and non-CGI promoters involving different gene regulatory mechanisms. We classified host, nested, and all gene promoters for their association with a CGI and found a slight bias for CGI promoter association to host genes versus nested genes (Supplemental Fig. S3B). However, this was mainly due to differences in biotype distribution, as, for example, protein-coding genes that are enriched in host genes, are more likely to be CGI associated (Supplemental Fig. S3C).
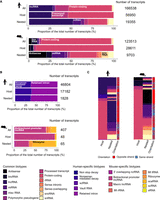
Host and nested gene biotypes. (A) Bar chart representing the proportions of biotypes which are common across both species. Proportions are displayed for all genes, host genes, and nested genes in human and mouse genomes. (B) Bar chart representing proportions of biotypes unique to human and mouse genomes across all genes, host genes, and nested genes. (C) Heatmap showing the correspondence between host and nested gene biotypes per pair and their orientation to each other.
Expression profile of host and nested genes in adult tissues
Host and nested genes were enriched for distinct functional classes and so we sought to annotate their biological function via Gene Ontology and expression analysis. Ontology analysis shows that host genes are enriched for terms associated with neural differentiation and synapse development (Supplemental Fig. S4A) and nested genes for terms associated with negative regulation of translation and gene silencing as expected given that a large proportion of nested genes were small RNAs (Supplemental Fig. S4B).
The Gene Ontology analysis of host genes suggested that they could be enriched for genes important for tissue specification (Supplemental Fig. S4A). Therefore, we decided to investigate whether the host genes were expressed in a tissue-specific way using bulk tissue ENCODE RNA sequencing data (Supplemental Tables S4, S5; Davis et al. 2018). As a first approach, we calculated the standard deviation of host and nested gene expression. It showed that host genes had a greater variance in expression level (Supplemental Fig. S5A), suggesting that these genes are expressed in a limited set of tissues. As the standard deviation is influenced by the level of expression, we investigated the global level of expression across each tissue for host and nested genes in comparison to all genes. Higher expression level of the host genes and lower expression of the nested genes were observed for protein-coding genes and some other biotypes (Fig. 4A). The higher expression of host protein-coding genes was detected in all tissues tested, whereas nested protein-coding genes had lower expression levels (Supplemental Fig. S5B,C). This suggests that the differences between standard deviations could be due to differences in basal expression levels. To explore this, we computed the τ (Tau) index which is a tissue-specificity metric (Yanai et al. 2005). The value of τ varies between 0 for housekeeping genes (or broadly expressed genes) and 1 for tissue-specific genes. In both mouse and human, we observed that host genes are more broadly expressed and nested genes are more tissue specific than the rest of the genome (Fig. 4B), suggesting that host/nested organization could play a role in tissue-specific regulation of expression.
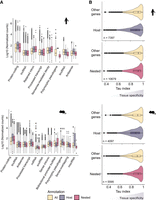
Host and nested gene expression profiles and tissue specificity. (A) Distribution of the expression of all, host, and nested genes for the biotypes associated with the three categories in human and mouse transcriptomes (B) Distribution of τ index calculated using the normalized expression level detected in RNA sequencing data sets. The higher the value of τ the more the gene exhibits tissue-specific expression. Across 1000 matched-size random samples, the application of a Kolmogorov–Smirnov test to these distributions always resulted in a P < 0.05. The figure shows one representative matched sample comparison.
Coexpression of host and nested gene pairs are linked to tissue specificity
As intragenic elements have been previously associated with tissue specificity (Amante et al. 2020), we hypothesized that coexpression could influence tissue-specific profiles and tested this by focusing on the correlation between host and nested genes expression. This also tested the model whereby there is a steric hindrance between two RNA polymerases transcribing in the same genomic region (Billingsley et al. 2012) and host gene expression represses nested gene expression (Neri et al. 2017). In this case, host and nested genes should be expressed in a mutually exclusive way. A Spearman's rank correlation coefficient (ρ) was calculated for each pair between the expression profile of the host and the nested gene across tissues (as exemplified for the conserved pair Mcph1/Angpt2 in Fig. 5A). The distribution of ρ showed a peak between 0 and 0.5, indicating that, for most pairs, there is no positive or negative relationship between the expression level of the host and the nested genes in both species. This distribution was independent of host/nested gene pair orientation (Fig. 5B; Supplemental Fig. S6A). Furthermore, conserved host/nested gene pairs between species showed a less variable correlation in expression when compared to correlations between pairs of genes that were conserved but were only host/nested in one species (Supplemental Fig. S6C,D).
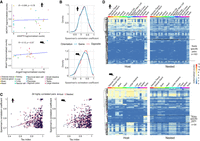
Tissue-specific coexpression of host/nested gene pairs. (A) Examples of correlation plots used to calculate Spearman's rank correlation coefficient for the conserved pair Mcph1/Angpt2. (B) Distribution of the Spearman's rank correlation coefficient for all host/nested gene pairs depending on their orientation to each other. (C) Correlation between the Spearman's rank correlation coefficient value and the τ index for all the pairs exhibiting a Spearman's rank correlation coefficient higher than 0.7. (D) Heatmap showing the normalized expression level detected in RNA sequencing data sets for host and nested gene pairs with a Spearman's rank correlation coefficient higher than 0.7. Host genes were clustered using hierarchical clustering according to their expression profile and their nested counterpart were maintained in the same order. The dotted lines are flanking the groups of pairs specifically coexpressed in testis.
In addition, anticorrelated expression was rare (ρ = <−0.7), while highly correlated pairs were more common (ρ = >0.7) (Fig. 5B). To determine whether this correlation was linked to the host and nested gene expression profile, the relationship between the correlation and the tissue specificity was evaluated. The higher the correlation, the more the host and nested genes were expressed in a tissue-specific manner as determined by the τ (Fig. 5C; Supplemental Fig. S6B). Examination of the expression profile of these highly correlated pairs across tissues reveals a large group of pairs with coexpression specific to the testis (163 pairs) (Fig. 5D). This was in line with the previous report of the testis showing both widespread gene activity and high transcriptome diversity (Soumillon et al. 2013). In addition, vast changes in gene expression occur, in the testis during the process of spermatogenesis (Jan et al. 2017). Thus, the testis is both an excellent developmental and mechanistic model tissue to begin to understand the functional role of host and nested coexpression.
Single-cell RNA sequencing reveals patterns of expression of host/nested gene pairs
Because, testis is a complex tissue, consisting of both somatic and germline cells at different stages of differentiation, bulk RNA sequencing does not offer the necessary resolution to determine whether pairs of genes are expressed in the same cells or in different cells or are mutually exclusive. To assess this, a human testis scRNA-seq data set was leveraged (Di Persio et al. 2021). Expression of 64 out of 163 testis-specific pairs was detected in this data set, which we attributed to the limited depth of single-cell sequencing approaches. A Spearman's rank correlation analysis was performed between host and nested gene expression per cell on the subset of testis-specific pairs identified by the ENCODE analysis (Fig. 5D). Most of the pairs demonstrated a low absolute value of the Spearman's rank correlation coefficient (Supplemental Fig. S7A). However, as Spearman's rank correlation is skewed by the low and zero counts of scRNA-seq data, it was not suitable to assess coexpression, nor, mutually exclusive expression of genes (Pollen et al. 2014; Sanchez-Taltavull et al. 2020; Li and Li 2021). Indeed, host/nested gene pairs demonstrate different profiles of coexpression despite similar Spearman's rank correlation coefficients. (AL109954.1/CST8, R = 0.29 and RNASE11/AL163195.2, R = 0.25) (Supplemental Fig. S7B). To overcome this, we classified the single cells according to the host and nested gene expression based on a quarter of the maximum expression value for each gene (Supplemental Fig. S7C). Expression above the threshold was indicative of robust detection and allows cell classification. Because we wanted to investigate the relationship between host and nested genes, we applied a filter of a minimum of 1% of the cells of the data set which had to have robust expression for both the host and the nested gene, selecting 34 pairs. Finally, expression above the threshold for both the host and the nested gene allows the identification of cells which coexpress the pair. Expression above one threshold but below the other indicates expression of only one member of the pair, whereas expression below both thresholds classified the cells as nonexpressing the pair (Supplemental Fig. S7C). This generated an accurate representation of mutual exclusive expression and coexpression, e.g., for LINC02253/AC020704.1 (0.06% of cells where both genes are expressed but 2.13% and 4.32% of cells with exclusive expression of the host and the nested gene, respectively) and CASC16/AC026462.3 (16.64% of cells where both genes are expressed), respectively (Fig. 6A).
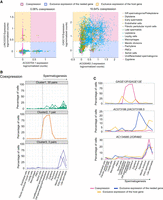
Host and nested genes with high coexpression in the testis can exhibit dynamic coexpression patterns during spermatogenesis at the single-cell level. (A) Scatter plots showing host and nested genes normalized expression level in testis single cells for two example pairs: LINC02253/AC020704.1 and CASC16/AC026462.3. (B) The proportion of cells which coexpress host and nested gene pairs across spermatogenesis. Three distinct coexpression profiles were determined by k-means clustering. (C) Line plot showing the profiles of expression calculated using the method detailed in Supplemental Figure S7C for three different pairs of host and nested genes. GAGE12F/GAGE12E is an example of a pair where host and nested genes are mainly coexpressed; AC073188.2/AC073188.5 is an example of a pair with mutually exclusive expression; and AC1349980.2/OR4M2 is a pair with a complex expression profile in testis.
Transcriptional interplay between host and nested genes during spermatogenesis
Coexpression or exclusive expression was limited to some of the cell types, as such we hypothesized that they are tightly regulated during development. For example, AL163195.3/AL163195.2 was coexpressed mainly during the late stages of spermatogenesis (meiotic division, spermatids), and expression of AL163195.3 alone was also observed at earlier stages (spermatogonia, leptotene, zygotene, pachytene) and in some of the somatic cells (Supplemental Fig. S7D). Globally, the interplay between host and nested gene expression was defined for each pair based on the percentage of cells defined as coexpressing or mutually expressing in every cell type (Supplemental Fig. S7E). We performed k-mean clustering (Fig. 6B; Supplemental Fig. S8A) and identified distinct profiles of coexpression or exclusive expression, indicating that host/nested gene expression and interplay are regulated according to cell types and during the process of spermatogenesis. However, a profile with a simple relationship (i.e., mostly coexpressed or host expressed in some cell types and nested gene in others) was rare. For example, GAGE12E/GAGE12F was mainly coexpressed and AC073188.2/AC073188.5 exhibited mutually exclusive expression (Fig. 6C). Most of the pairs showed a complex profile sometimes with coexpression and sometimes exclusive expression, as exemplified by AC134980.2/OR4M2 and IGSF11/IGSF11-AS1 (Fig. 6C; Supplemental Fig. S8B). For some pairs presenting this more complex expression profile, we observed that while the host gene was broadly expressed, the nested gene expression was limited to the testis (Fig 7A). IGSF11 is a known regulator of meiosis during spermatogenesis (Chen et al. 2021), suggesting that the coexpression of host and nested genes and the changes in isoforms expression could be key for its function in testis.
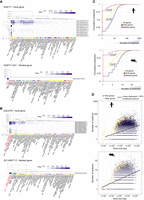
Host and nested gene coexpression is correlated with regulation of isoform diversity. (A) Profile of expression of the different isoforms (rows) of the IGSF11/IGSF11-AS1 pair in different tissues (columns) from the GTEx portal data on isoforms expression (https://gtexportal.org/home/). Isoform and tissue were ordered by hierarchal clustering using the Euclidean distance and average linkages. Tissues where coexpression is happening are in red. (B) Profile of expression of the different isoforms (rows) of the GALNT9/AC148477.2 pair in different tissues (columns) from the GTEx portal data on isoforms expression (https://gtexportal.org/home/). Isoform and tissue were ordered by hierarchal clustering using the Euclidean distance and average linkages. Tissues where coexpression is happening are in red. (C) Cumulative proportion of the number of isoforms for host genes, nested genes, and all genes. A base-10 log scale was used for the number of isoforms. The significance of the difference was tested using a DTS statistic test to compare the empirical cumulative distribution functions (ECDFs) (Dowd 2020). The resulting P-values are mentioned on the graph. (D) Scatter plot showing the isoform number according to gene size (of the longest isoform) for host genes and nonhost genes. A base-10 log scale was used for the number of isoforms and the gene size. Linear regression analysis was computed and the regression line was added on the graph alongside the calculated 95% confidence intervals.
Host genes show a greater number of isoforms and coexpression correlates with changes in transcript diversity
Using the GTEx portal data on isoform expression (Lonsdale et al. 2013), we observed that the nested gene expression restricted to testis was associated for the IGSF11/IGSF11-AS1 gene pair, with a greater diversity of host isoforms expressed in testis (Fig. 7A; Supplemental Fig. S8B). The correlation between host-specific isoforms and expression of the nested gene in the testis was also observed for pairs which were not detected in the single-cell RNA-seq data set, such as MGAM/OR9A4 and HMX1/AC116612.1 (Supplemental Fig. S9A,B). Beyond testis, AC1484477.2 nested gene high expression in two parts of the brain was associated with the expression of an increased number of isoforms of its host GALNT9 (Fig. 7B), whereas AL117382.2 nested gene exclusive expression in the liver was associated with the expression of five different isoforms of HNF4A one of which is unique to this tissue (ENST00000372920.1) (Supplemental Fig. S10A). Given the correlation between coexpression and isoform regulation observed here and in previous studies (Wood et al. 2008; Cowley et al. 2012; Amante et al. 2020), we hypothesized that host genes would be more transcript diverse. Evaluation of isoform enrichment identified that in comparison to the transcriptome, host genes are isoform rich, in the GENCODE annotation (P < 0.05, two-sample Kolmogorov–Smirnov test) (Fig. 7C). Previous work has identified that gene length is positively correlated with isoform number (Kopelman et al. 2005; McCoy and Fire 2024). As host genes were enriched for longer genes (Supplemental Fig. S1A), and we also observed a positive correlation between gene size and number of isoforms (Supplemental Fig. S10B), we asked if the enrichment for higher number of isoforms was associated with host genes being longer. To address this, we performed a linear regression analysis on the relationship between gene length and isoform number for host genes and nonhost genes (Fig. 7D). The absence of overlap between the 95% confidence intervals suggested that host genes contained more isoforms independently of their length. This was supported by subtracting the effect of length on isoform number by an ANCOVA analysis and a Tukey post-hoc test (mouse: P < 0.0001, human: P < 0.0001).
Discussion
Global compilation of host/nested gene pairs in mammals illustrates their transcriptional profile during tissue specification, and interplay with RNA processing mechanisms and isoform regulation. Compared to the most exhaustive previously known list (Yu et al. 2005), we identified ∼50 times more pairs likely due to the continuous improvement of sequencing technologies allowing the annotation of more genes and transcripts. For example, between 2003 and 2013, the number of transcripts annotated in the RefSeq database increased by 23% (Pruitt et al. 2014). In addition, the comprehensive GENCODE annotation provides an exhaustive annotation for transcripts (Frankish et al. 2015, 2019). The resulting collection is the most comprehensive list of host/nested gene pairs in mouse and human which can be explored through the web application developed for this study (https://hngeneviewer.sites.er.kcl.ac.uk/hn_viewer/). Ultimately, the accuracy of the pair identification is dependent on how precise the current genome annotations are and on the development of new technologies, such as long-read sequencing which will iteratively advance the detection of all possible genomic transcripts (Leung et al. 2021).
Contrary to the previous studies, a lower conservation of the host/nested genes was observed between mouse and human (between 21% and 34% of the pairs with known orthologs). This suggests that these events are mainly species-specific and the difference to previous studies can be explained by the updated annotation and inclusion of more diverse transcript types (multiple types vs. protein-coding only). Furthermore, the conservation analysis is highly dependent on the annotation of the ortholog gene list, and it cannot be excluded that some conserved pairs were missed or excluded due to annotation defaults. A more detailed conservation study, including other species, could improve the conservation level of host–nested gene pairs. This would also be useful for investigating the origins of host/nested gene pairs and for identifying the mechanisms involved, such as de novo promoter generation inside a gene, transfer of a gene inside another through genomic rearrangement or (retro)transposition and extension of the host gene by the acquisition of new exons which internalize an adjacent gene (Wright et al. 2022). These data indicate that most of the nested genes are fully contained within large introns of their hosts which suggest that there is a bias toward the acquisition of nested genes in these regions or a selection against events that interfere with the coding sequences of the host gene. This is also an indication that the impact of such a configuration would mainly be at the transcriptional level.
To test the transcriptional impact of one gene on the other, we conducted a correlation analysis between host and nested gene expression across multiple tissues. Previous analysis suggested gene pairs were anticorrelated or that there was no relationship regarding expression (Yu et al. 2005; Assis et al. 2008). Our results agree with the latter with a lack of direct correlation or anticorrelation between host and nested gene expression for most of the pairs. We identified that host genes were more likely to be broadly expressed and nested genes were more likely to be tissue specific. When coexpression was considered, the pairs showed patterns related to tissue specificity, suggesting that the nested gene is leading to a tissue specificity interplay with its host gene.
A high degree of coexpression of host and nested genes was observed in the testis which correlates with reports of widespread transcription in this tissue (Melé et al. 2015; Xia et al. 2020). Permissive transcription in the testis has been proposed to give rise to novel genes (Kaessmann 2010) and we observed that nested genes were less conserved than their host counterpart. This suggests that nested genes are more recent in evolutionary time and could indicate that nested genes are acquired through evolution during spermatogenesis. Because testis is a complex tissue, we leveraged scRNA-seq data where the different somatic and germ cell types are identifiable. In addition, we developed a novel analysis method to assess true coexpression in an individual cell-dependent manner. Thus, we investigated the coexpression and mutual exclusive behavior of host/nested genes in individual cells and showed that host and nested genes can be coexpressed in the same cells. We expect that the coexpression of pairs detected in single cells is an underestimate, given the limited depth of single-cell sequencing and the poly(A) selection associated with most RNA sequencing data sets. For most of the pairs, the profile was complex, and for some of them, such as IGSF11/IGSF11-AS1, changes in isoform diversity at the host are associated with the expression of the nested gene only in testis. Previous work has demonstrated that the testis exhibits widespread transcription, in particular for smaller genes (McCoy and Fire 2024), alongside a higher transcriptome diversity compared to other tissues (Soumillon et al. 2013). We propose that one mechanism to provide this observed diversity is through the widespread activity of nested genes that subsequently impact isoform regulation of their corresponding host.
Even if more common in testis, we also observed a correlation between nested gene tissue-specific expression and host isoform regulation in the brain and liver. More globally, host genes were found to exhibit more transcript isoforms than other genes, suggesting an important role for the host/nested genes’ genomic organization in the regulation of transcript diversity. Previous works indicate that this can happen through the process of alternative polyadenylation (Wood et al. 2008; Kaer et al. 2011; Cowley et al. 2012), but the other mechanisms leading to transcript diversity, such as the use of alternative promoters or alternative splicing could be also regulated by the interplay between host and nested genes. The dissection of mechanisms involved would require the improvement of technical approaches, such as long-read sequencing, capturing precisely the transcription start site, end site, and splicing profile of individual RNA molecules at a sufficient depth.
Methods
Identification of host–nested gene pairs in human (hg19) and mouse (mm10) genomes
To generate a list of all host/nested genes, comprehensive GENCODE annotations of the human (GRCh37/hg19, [LiftOver from Release 37 GRCh38/hg38]) and mouse (mm10, Release M25 [GRCm38.p6]) genomes were downloaded as a BED file from the UCSC Table Browser (https://genome.ucsc.edu/cgi-bin/hgTables). Transcripts denoted as TEC, IG variable chain, and TR genes, as well as complex loci (Protocadherin's and UDP-glucuronosyltransferase), were removed from the list. The resulting list was used as a reference for transcripts for the hosts. To only select pairs where all the transcripts of the nested gene were included inside the host, the filtered list was also used to generate a “metagene” list consisting of the most extreme coordinates of all possible transcripts of a single gene. BEDTools 2.29.2 (Quinlan and Hall 2010) intersect function was used to overlap the metagenes list with a reference list of transcripts for the hosts. The option -f 1.0 was used, meaning that the minimum overlap required was 100% of a metagene overlapping with another transcript. To keep only the overlap involving different genes, using an in-house R script (R Core Team 2023), genes which contained the same name, the same size, and where the start or end were the same were removed. When multiple transcripts from the same gene were involved in the same pair of genes, the longest transcript was kept. Annotation of opposite and same strand-oriented pairs was determined by an in-house R script, checking if the strands of the pairs were: same (+:+/−:−), or, opposite (+:−/−:+). A full list of host and nested gene pairs is available in Supplemental Tables S1 and S2.
Characterization of the host/nested gene pairs
Information about transcript biotypes, strands, number of isoforms, and gene sizes was retrieved from GTF files downloaded from the GENCODE website (https://www.gencodegenes.org/, Release M25 (GRCm38.p6) for mouse and Release 37 (GRCh38) for human). The GTF files can also be downloaded at (https://figshare.com/articles/dataset/GTF_files_to_accompany_Montibus_et_al_2024/26349091). The functional enrichment analysis (Gene Ontology) was performed using g:Profiler (version e109_eg56_p17_1d3191d) with the Bonferroni correction method applying a significance threshold of 0.05 (Raudvere et al. 2019). The complete results are available in Supplemental Table S3.
Annotation of the nested gene location
ChIPseeker (version 1.24.0) (Wang et al. 2022) was used to annotate the location of the nested genes inside their host. A custom GTF file containing only the information about the host transcripts involved in host/nested gene pairs was used as a reference and a BED file containing the start and end coordinates of the nested genes was used as a query. The genomicAnnotationPriority was defined as “Exon,” “Intron,” “Promoter,” “5UTR,” “3UTR,” “Downstream,” and “Intergenic.” The pairs with successful nested gene annotation inside their host were retained and further rounds of annotation were used when necessary (after removal of the already annotated pairs) to annotate the missing pairs (e.g., when a gene is nested inside two different genes). Finally, for the last pairs with missing annotation, manual annotation was performed. The table containing the annotation and exon/intron numbers was retrieved and cross referenced with the GENCODE GTF file for intron/exon characteristics.
Annotation of CGI location
A region of ±1 bp was taken from the TSS of all transcripts and the BEDTools 2.29.2 (Quinlan and Hall 2010) intersect with the -f 1.0, for 100% overlap, with CGI coordinates from Illingworth et al. (2010). Extraction of either host or nested genes associated with a CGI was performed by filtering the gene symbols with the CGI annotation from the BEDTools output. The genes not present in this list were denoted as having a “Non-CGI” promoter. Specific annotation of transcripts and their biotype was performed by filtering the BEDTools output using transcript IDs as opposed to gene symbols.
Conservation analysis
The list of all the human–mouse orthologs was downloaded using the Ensembl BioMart software suite (Cunningham et al. 2022) with the following filter “Homolog filters” option “Orthologous Mouse Genes” from the human gene list. The table was next downloaded with the following attributes: Gene stable ID, Transcript stable ID, Mouse gene stable ID, Mouse gene name, Mouse protein, or transcript stable ID. The orthologs information was next overlapped with the lists of host/nested genes using an in-house R script (R Core Team 2023) to identify the pairs where both the host and the nested genes were conserved in the two species and from them, the 349 combinations of pairs where the genes were in a host/nested configuration in both species.
ENCODE RNA sequencing analysis
Paired-end RNA sequencing data from multiple tissues was used in both human and mouse (for accession numbers, see Supplemental Tables S4, S5). Reads were aligned using kallisto (Bray et al. 2016) and a read count table per sample was generated using tximport (Soneson et al. 2016); reads were then normalized using DESeq2 (Love et al. 2014). Pairs of host/nested genes were processed with a pair_id which was used to sort normalized read matrix data based on the corresponding host and nested gene ENST_ID using a custom R script (R Core Team 2023). Correlations of host/nested gene expression across all samples were performed using Spearman's rank correlation coefficient. Spearman's rank correlation coefficient was also used for the comparison of correlations between pairs with conserved organization within species and pairs with host/nested gene organization specific to one species and their nonhost/nested genes counterpart. Highly correlated (coefficient >0.7) host/nested gene pairs were hierarchically clustered using the complete-linkage clustering method using the pheatmap package. Tissue specificity of the host, nested, and all genes were evaluated using the standard deviation of expression across all tissues, and, calculation of τ index (Yanai et al. 2005) using the package tspex (Camargo et al. 2020) on the normalized count matrix. All expression analysis was performed on all types of gene biotypes. We acknowledge that there is an enrichment for protein-coding gene biotypes given that the ENCODE RNA sequencing data are poly(A) selected.
scRNA sequencing analysis
A table with Integrated, normalized counts (GSE153947_Normal_integrated_data.tsv.gz) per cell from the scRNA-seq data set during normal spermatogenesis was retrieved from the NCBI Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/) under accession number GSE153947 (Di Persio et al. 2021). The data set was filtered to only include host and nested genes which were highly correlated and expressed in testis from the ENCODE RNA sequencing analysis. The expression of a gene was reduced to a binary signal whereby if expression reached above the threshold (a quarter of the maximum expression), a value of 1 was given, and a value of 0 if below this threshold. Host/nested gene pairs were coexpressed in a single cell if the sum of this value was 2. If the sum equal to 1, then single cells were only expressing either the host or nested gene. If the sum equaled 0, the single cells were not expressing either gene. This threshold was empirically determined by manually scanning gene pairs until it was clear that the segregation of the bimodal distribution occurred between expressing and nonexpressing cells. Multiple thresholds were attempted, and manual inspection of several scatter plots allowed us to decide on the quarter threshold. A schematic illustrating this method is available in Supplemental Figure S7C. For further analysis, pairs where over 1% of cells were expressing both transcripts were considered. The proportion of cells coexpressing each host/nested gene pair was clustered via k-means clustering using the factoextra package (Version 1.0.7.999).
Software availability
Codes and tables containing the information needed to repeat the analysis are available at https://github.com/Bertille-Montibus/host_nested_genes. The code is also available as a file (Supplemental Code) associated with this paper. Analysis of data and generation of figures de novo is also within an RShiny application (https://hngeneviewer.sites.er.kcl.ac.uk/hn_viewer/).
Competing interest statement
The authors declare no competing interests.
Acknowledgments
We thank Hannah Mischo for advice and useful discussions. We thank Lukasz Zalewski and Stuart Morrison for their assistance with the R Shiny app. We thank Hallgerdur Kolbeinsdottir for the critical reading of the manuscript. The Genotype-Tissue Expression (GTEx) Project was supported by the Common Fund of the Office of the Director of the National Institutes of Health, and by National Cancer Institute, National Human Genome Research Institute, National Heart, Lung, and Blood Institute, National Institute on Drug Abuse, National Institute of Mental Health, and National Institute of Neurological Disorders and Stroke. The data used for the analyses described in this manuscript were obtained from: the GTEx Portal on February 13, 2024. J.A.C. is supported by the UK Medical Research Council (MR/N013700/1), and King's College London and is a member of the MRC Doctoral Training Partnership in Biomedical Sciences. B.M. is supported by funds from King's College London (to R.J.O.).
Author contributions: B.M. and J.A.C. conceived the idea, performed the analysis, and wrote the manuscript. J.A.C. developed the shiny app with support and guidance from R.T.M.-N. R.J.O. provided project supervision, discussed the work with B.M. and J.A.C., and critically reviewed and contributed to the final manuscript.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.279430.124.
-
Freely available online through the Genome Research Open Access option.
- Received April 3, 2024.
- Accepted October 17, 2024.
This article, published in Genome Research, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








