
Long-read RNA sequencing of archival tissues reveals novel genes and transcripts associated with clear cell renal cell carcinoma recurrence and immune evasion
- Joshua Lee1,2,3,4,
- Elizabeth A. Snell4,
- Joanne Brown5,
- Charlotte E. Booth1,2,
- Rosamonde E. Banks5,
- Daniel J. Turner4,6,
- Naveen S. Vasudev5 and
- Dimitris Lagos1,2
- 1Hull York Medical School, University of York, York YO10 5DD, United Kingdom;
- 2York Biomedical Research Institute, University of York, York YO10 5DD, United Kingdom;
- 3Department of Biology, University of York, York YO10 5DD, United Kingdom;
- 4Oxford Nanopore Technologies Plc, Oxford OX4 4DQ, United Kingdom;
- 5Leeds Institute of Medical Research at St James's, University of Leeds, St James's University Hospital, Leeds LS9 7TF, United Kingdom
Abstract
The use of long-read direct RNA sequencing (DRS) and PCR cDNA sequencing (PCS) in clinical oncology remains limited, with no direct comparison between the two methods. We used DRS and PCS to study clear cell renal cell carcinoma (ccRCC), focusing on new transcript and gene discovery. Twelve primary ccRCC archival tumors, six from patients who went on to relapse, were analyzed. Results were validated in an independent cohort of 20 patients by qRT-PCR and compared to DRS analysis of RCC4 cells. In archival clinical samples and due to the long-term storage, the average read length was lower (400–500 nt) than that achieved through DRS of RCC4 cells (>1100 nt). Still, deconvolution analysis showed a loss of immune infiltrate in primary tumors of patients who relapse as reported by others. Differentially expressed genes in patients who went on to relapse were determined with good overlap between DRS and PCS, identifying LINC04216 and the T-cell exhaustion marker TOX as novel candidate recurrence-associated genes. Novel transcript analysis revealed over 10,000 candidate novel transcripts detected by both methods and in ccRCC cells in vitro, including a novel CD274 (PD-L1) transcript encoding for the soluble version of the protein with a longer 3′ UTR and lower stability than the annotated transcript. Both methods identified 414 novel genes, also detected in RCC4 cells, including a novel noncoding gene overexpressed in patients who relapse. Overall, we showcase the use of PCS and DRS in archival tumor samples to uncover unmapped features of cancer transcriptomes, linked to disease progression and immune evasion.
Kidney cancer contributes ∼2% of all newly diagnosed cancer cases worldwide (Sung et al. 2021). The most common form of kidney cancer is renal cell carcinoma (RCC) and the most frequent RCC type is clear cell RCC (ccRCC, ∼75% of all RCC cases [Ricketts et al. 2018]). Inactivation of the VHL gene function is an almost universal hallmark of ccRCC. Secondary mutations are required in hotspot genes, including PBRM1, SETD2, and BAP1, as well as copy number changes in Chromosomes 9p and 14q (Hsieh et al. 2018). Of note, ccRCC tumors contain one of the highest percentages of tumor-infiltrating immune cells among all cancer types at ∼30% of all cells (Aran et al. 2015; Rooney et al. 2015; Ricketts et al. 2018). Treatment of localized ccRCC typically involves the removal of part or all of the kidney (radical/partial nephrectomy). Approximately one-third of patients have metastases detected at preoperative screening and 30%–50% develop metastases after the removal of the primary tumor (Rini et al. 2009). Several approaches have been proposed for assessing the risk of disease recurrence following surgery (Cotta et al. 2023). Scores based on gene expression signatures have also been proposed to refine risk prediction (Brannon et al. 2010; Rini et al. 2015; Morgan et al. 2018). However, despite a recognized need (Correa et al. 2019; Vasudev et al. 2020), so far, no set of biomarkers has reached routine clinical practice.
Aberrant co- and posttranscriptional events (e.g. alternative splicing/polyadenylation, posttranscriptional modifications, etc.), drive oncogenesis but also tumor immunogenicity (Sveen et al. 2014; Smith et al. 2019). Our understanding of cancer transcriptomes is nearly exclusively based on short-read sequencing platforms. Given that the average length of an mRNA is 1.5–2 kb in mammals this approach requires high depth of sequencing to confidently call transcript variants and is limited with regard to reconstruction of full-length novel transcripts. Often the reliance on reference genomes/transcriptomes means that this approach misses or discards novel transcripts. Furthermore, it is extremely difficult, if not impossible, to confidently establish transcriptional codependencies, i.e. coexistence of distinct features (e.g. specific splice junctions and untranslated regions—UTRs) on the same transcript. Long-read direct RNA sequencing (DRS) and PCR cDNA sequencing (PCS) have emerged as transformative methodological alternatives to overcome these limitations (Nature Methods Editors 2023). Yet, in cancer, there are only a handful of reports using long-read sequencing in tumor samples from patients with solid (Qu et al. 2022; Veiga et al. 2022; Mock et al. 2023) or blood cancers (Tang et al. 2020; Pratanwanich et al. 2021; Cortés-López et al. 2023), with only one example of using DRS (in three myeloma patient samples [Pratanwanich et al. 2021]). Currently, there are no reports directly comparing PCS to DRS in clinical samples and long-read sequencing has not been applied to kidney cancer.
Here, we aimed to explore whether archival surgical fresh frozen nephrectomy tissue samples (typically stored for over 10 years) could be used for Oxford Nanopore Technologies (ONT) DRS (RNA002 kit) and PCS (PCS111 kit) analyses (Garalde et al. 2018) to explore ccRCC transcriptomes. We focused on differential gene expression analysis to identify novel candidate predictors of disease relapse and the discovery of novel genes and transcripts with evidence of cancer cell-intrinsic expression and potential association with disease relapse.
Results
To demonstrate the feasibility of utilizing long-read RNA sequencing technologies for characterizing ccRCC transcriptomes in archival surgical fresh frozen specimens, we sequenced 12 snap-frozen nephrectomy samples using ONT PCS and DRS on ONT PromethION flow cells, using 200 ng and 2 μg of total RNA, respectively. These samples consisted of six specimens from patients who later developed ccRCC recurrence and six nonrecurrent controls (see Methods and Supplemental Table S1). For each clinical specimen, the same RNA sample was used for DRS and PCS analysis. No significant differences in RNA quality (based on RNA integrity numbers [RINs]) were observed between recurrent and nonrecurrent controls (Supplemental Fig. S1A). An overview of the study design and data analysis pipeline is shown in Figure 1A.
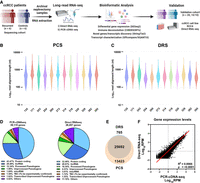
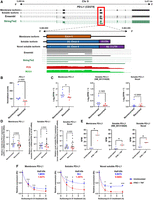
DRS and PCS of ccRCC nephrectomy samples. (A) Summary of study design and data analysis workflow—figure made with BioRender (https://www.biorender.com). (B) Violin plot showing Log10 transformed raw read lengths of passed reads generated by PCS. (C) as in (B), but for DRS. (D) Pie chart depicting the proportions of gene biotypes of all mapped genes from the reference genome (Ensembl release 105, GRCh38) mapped PCS and DRS reads of sequenced tumor samples. (E) Venn diagram showing the overlap between PCS and DRS mapped genes. (F) Correlation between gene expression levels (Log10 reads per million [RPM]) of all genes mapped by both PCS and DRS (n = 25,692). Diagonal line represents the line of best fit. R2 value was computed to measure goodness-of-fit and P-value was generated from F-test, with P < 0.05 considered statistically significant. Lowest expression values shown correspond to the minimum normalized abundance derived for genes detected only at one read in the sample with the highest total number of reads.
DRS and PCS of ccRCC nephrectomy samples
All nephrectomy specimens were successfully sequenced using both PCS and DRS. After 72 h of sequencing, PCS generated reads ranging from 50 million to 85 million (median = 56.6 million, total = 701 million), with ∼80% qualified as pass reads (median = 45.8 million, total = 561 million) having a minimum read Q score of 7. DRS generated between 2.4 million and 5.5 million reads (median = 4.6 million, total = 52.6 million), with ∼70% qualified as pass reads (median = 3.2 million, total = 37.4 million). Summary sequencing output statistics can be found in Table 1 and Supplemental Table S2.
Sequencing statistics of PCS and DRS of archival ccRCC tumor samples
Both PCS and DRS reads were next mapped to the human reference genome. The median alignment length for PCS and DRS reads were 517 and 405 nt, respectively, which is lower than that typically observed (Garalde et al. 2018). A range of 21%–37.1% (median = 25.95) of PCS- and 3.2%–18.1% (median = 7.6%) of DRS-aligned reads represent full-length transcripts (coverage of at least 95% of the mapped reference annotation) (Fig. 1B,C). As RNA molecules are sequenced from 3′ end, gene coverage is biased toward 3′ end (Supplemental Fig. S1B). Overall, PCS and DRS reads achieved median accuracies of 95.5% and 90.5%. The longest aligned reads for PCS and DRS were 27,854 and 7822 nt, respectively. This was likely due to the significantly higher sequencing depth achieved by PCS. These results demonstrated the capability of ONT long-read RNA sequencing to produce high depth, PCS and DRS sequencing data sets from flash-frozen historical clinical specimens. The modest average read length is likely due to the long-term storage of these samples.
To evaluate the ability of long-read sequencing to capture the diversity of the transcriptome, we examined the RNA biotypes of genes identified by PCS and DRS. PCS identified 39,115 genes across the 12 samples, with a median of 26,203 mapped genes per specimen (Supplemental Fig. S1C). Among all PCS-mapped genes, 45.47% were classified as protein-coding genes, 29.48% as long noncoding RNAs (lncRNA) and 12.34% as processed pseudogenes (Fig. 1D). In comparison, DRS identified 26,457 genes across the specimens (median = 18,057 per sample) (Supplemental Fig. S1C), with 32.48% classified as protein-coding genes, 28.81% as lncRNAs and 16.50% as processed pseudogenes (Fig. 1D); 25,692 genes were mapped by both methods (Fig. 1E); 13,423 genes were exclusively mapped by PCS, likely due to higher sequencing depth compared to DRS. Notably, 765 genes were exclusively mapped by DRS.
We observed that the majority of expressed genes for both PCS and DRS were protein-coding (89.4% and 91.7%, respectively), followed by mitochondrial rRNAs (mt-rRNAs) (5.35% and 4.66%), processed pseudogenes (2.57% and 1.74%) and lncRNA (1.71% and 1.10%) (Fig. 1D; Supplemental Fig. S1D). The observed bias toward the detection of protein-coding genes was likely due to both PCS and DRS using poly(A)-targeting probes for library construction, also meaning all sequenced transcripts are polyadenylated. Some read-through events were observed but we did not analyze them systematically as we were mindful of the relatively low percentage of full-length reads in our analyses of archival tissue. Despite using total RNA as input for PCS and DRS library preparation, highly abundant rRNAs were sequenced at negligible levels. Distribution of gene expression levels for each biotype by PCS and DRS are illustrated by violin plots in Supplemental Figure S1E. Furthermore, among genes mapped by both PCS and DRS (n = 25,692), we found significant correlation in their gene expression levels (Fig. 1F; Supplemental Fig. S2). Overall, while PCS provided greater sequencing depth, our data demonstrated that both methods can capture a diverse range of transcripts from archival clinical samples, yielding highly concordant gene expression profiles.
Differential gene expression analysis reveals that ccRCC recurrence is associated with suppressed tumor immune infiltration
We then tested whether DRS and PCS can identify features associated with ccRCC recurrence. After alignment to the reference genome, PCA did not result in visually distinct gene expression clusters correlating with ccRCC recurrence status, sex, or the number of mutations on ccRCC prognostic markers for either PCS or DRS (Supplemental Fig. S3A,B). We explored the effect of number of mutations in addition to VHL as we have previously shown poorer outcomes for tumors with VHL + 2 or more mutations (Scelo et al. 2014; Vasudev et al. 2023). No sample separation was observed based on RNA integrity or number of pass sequencing reads (Supplemental Fig. S3C,D). However, differential gene expression analysis identified 159 and 68 genes with significantly differential expression (|log2FoldChange| ≥ 2, Padj ≤ 0.1) between recurrent and nonrecurrent tumors by PCS and DRS, respectively, with substantial overlap (Fig. 2A,B; Supplemental Tables S3, S4). The directionality of gene expression among these differentially expressed genes (DEGs) showed strong correlation (Fig. 2C). We note that we did not observe any outliers with regard to the time to relapse within the recurrent disease group.
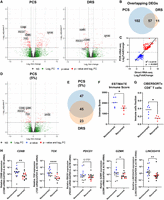
ccRCC recurrence is associated with suppressed tumor immune infiltration. (A) Volcano plots showing DEGs (red) between recurrent and nonrecurrent ccRCC tumors from PCS and DRS data using Ensembl genome reference (Ensembl release 105). (B) Venn diagram showing overlaps of DEGs identified by both PCS and DRS. (C) Correlation between log2FoldChange of DEGs identified by either or both PCS and DRS (recurrent vs. nonrecurrent ccRCC tumors). Diagonal line represents the line of best fit. R2 value was computed to measure goodness-of-fit and P-value was generated from F-test, with P ≤ 0.05 considered statistically significant. (D) Volcano plots showing DEGs (red) between recurrent and nonrecurrent ccRCC tumors from 5% subsampled PCS data using Ensembl genome reference (Ensembl release 105). (E) Venn diagram showing overlaps of DEGs identified by both 5% subsampled PCS and DRS. (F) Grouped dot plot showing an estimated immune score of nonrecurrent (blue) and recurrent (red) ccRCC tumor by the ESTIMATE algorithm, using PCS gene expression data. (G) Grouped dot plot showing the relative population of CD8+ T cells within immune infiltrates of nonrecurrent (blue) and recurrent (red) ccRCC tumors estimated by CIBERSORTx using PCS gene expression data. (H) CD8B, TOX, PD-1, GZMK, and LINC02416 mRNA levels measured by qRT-PCR in recurrent and nonrecurrent tumors from the sequenced cohort (blue and red, n = 12) and validation cohort (black, n = 20), relative to average mRNA levels in nonrecurrent tumors. mRNA levels were normalized to GAPDH and ACTB. For (A) and (D), blue and red dots represent significantly down and upregulated genes by either or both PCS and DRS. Dotted lines indicate the significance threshold (|log2FoldChange| ≥ 2, Padj ≤ 0.1). Names of genes that were validated by qRT-PCR with validation cohort are shown. For (F)–(H), two-tailed Mann–Whitney U tests were used with P ≤ 0.05 considered significant. (*) P < 0.05, (**) P < 0.01, (****) P < 0.0001. Line represents the median for each group.
As PCS produced substantially higher number of sequencing reads, we further evaluated ccRCC gene expression patterns using randomly subsampled PCS reads (5%) compared to DRS. We selected 5% as this brings the PCS depth to similar levels of the range achieved by DRS (2–4 million passed reads). PCA did not reveal a distinct cluster correlating with ccRCC recurrence (Supplemental Fig. S3E). We observed similar proportions of RNA biotypes of mapped genes across all tumor samples using 5% subsampled PCS data (Supplemental Fig. S3F). Differential gene expression analysis of 5% subsampled PCS data identified 92 DEGs, with significant overlap with DRS. The number of commonly identified DEGs between 5% PCS and DRS increased as a percentage of total DEGs identified by PCS and decreased as a percentage of total DEGs identified by DRS (Fig. 2D,E; Supplemental Table S5).
Within the overlapping DEGs between PCS, 5% PCS, and DRS, several key adaptive immune genes, including CD8B, PDCD1, GZMK, and TOX, were significantly downregulated in recurrent samples (Fig. 2A,B). To evaluate variations in biological processes (BPs) and pathways between recurrent and nonrecurrent ccRCC tumors, we performed Gene Ontology (GO) analysis. The top 10 most significantly enriched (by Padj) GO BPs terms from PCS data were all associated with adaptive immunity (Supplemental Fig. S4A). This pattern was also found using DRS data, where enrichment plot for the top 5 enriched GO BP terms (by Padj) by DRS showed identified suppression of adaptive immune response-related pathways (i.e. positive regulation of cell killing, T-cell-mediated cytotoxicity) in ccRCC recurrent samples (Supplemental Fig. S4B).
To further explore the relationship between ccRCC recurrence and immune infiltrate populations, we used the ESTIMATE algorithm (Yoshihara et al. 2013), which uses gene expression signatures to infer tumor purity and immune cell abundance. Using PCS data, we found that recurrent ccRCC exhibited significantly lower immune scores and higher levels of tumor purity compared with nonrecurrent controls (Fig. 2F; Supplemental Fig. S4C). DRS data displayed a borderline nonsignificant trend toward decrease in immune scores in recurrent ccRCC tumors (P = 0.0881) and a high degree of concordance was found between DRS and PCS ESTIMATE immune scores (R2 = 0.87, P < 0.0001) (Supplemental Fig. S4D,E). Both PCS and DRS data sets also had significantly lower immune scores for recurrent samples according to xCell analysis (Supplemental Fig. S4F).
Immune infiltrate profiles were further analyzed using another cell-type deconvolution algorithm, CIBERSORTx (Newman et al. 2019). Both PCS and DRS exhibited a significant reduction in the fraction of CD8+ T cell within recurrent ccRCC tumors when compared to nonrecurrent controls (Fig. 2G; Supplemental Fig. S4G,H). Similarly, EPIC, another immune cell-type deconvolution method (Racle and Gfeller 2020), also indicated suppression of CD8+ T cell within the immune infiltrates among the recurrent ccRCC tumors (Supplemental Fig. S4I). These findings agreed with a previously reported, qRT-PCR based, ccRCC recurrence prediction assay, which also linked lower expression levels of immune response genes with an increased likelihood of disease recurrence (Rini et al. 2015). Among the 11 recurrence-related gene makers examined in that study, our PCS and DRS analyses also identified that levels of NOS3 and CCL5 were significantly decreased in the recurrent tumors (Supplemental Fig. S4J).
Critically, we sought to validate our long-read sequencing results by an independent method (qRT-PCR) for CD8B, PDCD1, GZMK, and TOX using samples from both the sequenced cohort but also an independent validation cohort (n = 20, 10 from recurrent ccRCC patients and 10 nonrecurrent controls). This analysis confirmed significant downregulation of the CD8+ T-cell marker CD8B, the activation marker GZMK, and the T-cell exhaustion marker TOX in the recurrent tumors (Fig. 2H; Supplemental Fig. S5A–C). We note that for TOX the effect was statistically significant both in the pooled data and when analyzing the sequenced and validation cohorts separately. PDCD1 (also known as PD-1) levels were not statistically different when assessed by qRT-PCR between the two groups (Fig. 2H; Supplemental Fig. S5D). To explore if the additional sequencing depth achieved by PCS could lead to the identification of more candidate gene correlates of disease recurrence we used qRT-PCR to measure levels of three DEGs (LINC04216, LINC04217, and POU4F1) that were only significantly differentially expressed according to PCS. We found significant downregulation of LINC04216 using the sequenced cohort and when both cohorts were pooled (Fig. 2H; Supplemental Fig. S5E). No significant changes were found for LINC04217 and POU4F1 by qRT-PCR (Supplemental Fig. S5F,G).
Collectively, these findings demonstrated that both PCS and DRS can identify differential expression signatures associated with disease relapse. PCS and DRS showed a significant suppression of immune infiltration, particularly CD8+ T cells, in tumors of patients who later experience disease recurrence, and identified the exhaustion marker TOX and the LINC04216 noncoding RNA as novel candidate recurrence-associated genes.
Differential transcript usage analysis identifies candidate isoform switching events associated with ccRCC recurrence
One of the advantages of the long-read sequencing approach lies in its ability to identify and quantify transcript isoforms. By aligning sequencing reads against the reference transcriptome, isoform-level expression data can be used to detect differential transcript usage (DTU) events. We first compared reference genome- and reference transcriptome-based methods in gene expression and differential gene expression analysis. For both PCS and DRS, the reference transcriptome alignment method detected similar number of genes compared to reference genome alignment, with substantial overlap (Fig. 3A). Gene expression levels of the PCS and DRS of nephrectomy samples also displayed strong correlation between the two alignment methods (r = 0.7685 for PCS, r = 0.7087 for DRS) (Fig. 3B). Differential gene expression analysis using PCS and DRS reference transcriptome alignment data identified 197 and 34 significant DEGs between recurrent and nonrecurrent controls, respectively, with good overlap with the reference genome-alignment method (Fig. 3C; Supplemental Fig. 6A; Supplemental Tables S6, S7). The directionality of gene expression among these DEGs showed a strong correlation (Supplemental Fig. 6B).
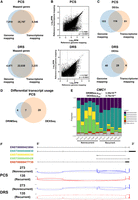
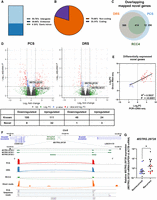
DTU events associated with ccRCC recurrence. (A) Venn diagram showing overlaps between reference genome- and reference transcriptome-alignment method mapped genes in PCS and DRS of nephrectomy samples. (B) Correlation between gene expression levels (Log10 RPM) of all genes mapped by both reference genome-alignment method and reference transcriptome alignment method in PCS (n = 35,797) and DRS (n = 22,038). Diagonal line represents the line of best fit. r value denotes Pearson's correlation coefficient and P-value was generated from F-test, with P < 0.05 considered statistically significant. (C) Venn diagram showing the overlaps of DEGs identified by both between reference genome- and reference transcriptome-alignment method in PCS and DRS of nephrectomy samples. (D) Venn diagram showing the overlaps of genes that displayed significant DTU by DRIMSeq and DEXSeq in PCS nephrectomy samples. (E) Stack bar graphs representing proportions of CMC1 isoforms in ccRCC tumors using PCS data. DRIMSeq and DEXSeq Padj values for DTU of CMC1 are indicated in the graph. (F) Graphical representation of CMC1 isoforms Ensembl reference annotations in Integrative Genomics Viewer (IGV), with black boxes representing exons. Sashimi plots of CMC1 from PCS and DRS recurrent (135) and nonrecurrent (273) ccRCC samples. Junction lines are shown for junction coverages with at least 5% of total CMC1 reads.
DTU analysis was carried out on both PCS and DRS of ccRCC tumor samples using DRIMSeq (Nowicka and Robinson 2016) and DEXSeq (Anders et al. 2012). Analysis of the PCS data identified 31 genes that displayed isoform switching in recurrent ccRCC tumors compared to nonrecurrent controls (Fig. 3D; Supplemental Table S8). These included CMC1 that showed statistically significant DTU by both DRIMSeq and DEXSeq and was also identified by DRS to display DTU (Supplemental Table S9). In PCS, most of the isoforms that are expressed in recurrent ccRCC samples are ENST000423894 and ENST00000466830 (Fig. 3E,F, labeled as purple and teal, respectively). In contrast to nonrecurrent counterparts, recurrent ccRCC specimens also expressed very low level of ENST00000468330 and ENST00000495428 (Fig. 3E,F, labeled as yellow and green, respectively). Overall, this analysis revealed a limited number of candidate disease recurrence-associated DTU events.
Long-read RNA sequencing enables the discovery of novel full-length transcripts expressed in ccRCC cells
A unique strength of long-read sequencing is the potential to discover novel transcript isoforms and genes, not currently included in the reference transcriptome. To identify novel transcript isoforms that are present in the ccRCC nephrectomy specimens, we applied StringTie2 to perform transcriptome assembly using PCS reads aligned to the reference genome. StringTie2 assembled isoforms were subsequently compared to the reference annotation (Ensembl release 105) with both SQANTI3 and GffCompare (see Methods). SQANTI3 classifies each assembled isoform into known or novel based on their splice junction matches. Known transcripts comprise full splice match (FSM) and incomplete splice match (ISM), whereas novel in catalog (NIC), novel not in catalog (NNC), antisense, fusion, genic, genic intron, and intergenic isoforms are classified as novel transcripts (Fig. 4A; Supplemental Table S10). Similarly, GffCompare assigns each StringTie2 assembled isoform with a transcript class code which corresponds to “Known” and “Novel” transcripts (Supplemental Fig. S7; Supplemental Table S10).
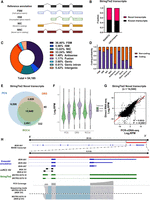
Long-read RNA sequencing enables the discovery of full-length novel transcripts. (A) Graphical representation of the major SQANTI3 isoform categories (antisense, genic intron, genic genomic, and intergenic not shown here). (B) Bar chart showing the proportion of Novel and known transcripts in StringTie2 assembly as curated by SQANTI3 and GffCompare. (C) Pie chart depicting the distribution of SQANTI3 isoform categories among StringTie2 assembled transcripts (n = 54,185). (D) Bar chart showing the proportion of coding and noncoding StringTie2 assembled transcripts by SQANTI3 isoform categories. (E) Venn diagram showing the number of overlapping mapped StringTie2 novel transcripts between PCS and DRS of ccRCC tumor samples, and DRS of ccRCC cell line RCC4. (F) Violin plot showing the expression levels (Log2 RPM) of known and novel transcripts in PCS and DRS of ccRCC tumor samples, and DRS of ccRCC cell line RCC4. The width of the violin plots represents the density of transcripts at different expression levels. Black dots represent mean expression levels. The top and bottom of box plots represent upper and lower quartiles, respectively. (G) Correlation between transcripts expression levels (Log2 RPM) of all StringTie2 novel transcripts mapped by both PCS and DRS (n = 14,544). Diagonal line represents the line of best fit. R2 value was computed to measure goodness-of-fit and P-value was generated from F-test, with P < 0.05 considered statistically significant. Lowest expression values shown correspond to the minimum normalized abundance. (H) IGV visualization of MVK reference annotations (blue), ccRCC-specific MVK splice junctions (black), StringTie2 assembled novel transcripts (green), PCS coverage track (gray) illustrating the depth of sequence coverage across the region of interest (red bar, hg38 Chr 12: 109,594,200–109,598,600) and PCS sequencing reads aligned to the reference genome in the region of interest.
Both SQANTI3 and GffCompare classifications revealed that novel transcripts constitute more than 50% of the assembled transcripts from the nephrectomy specimens (Fig. 4B). For SQANTI3 classification, the most prominent class of assembled transcripts were FSM (36.5%, n = 19,722 out of a total 54,185) (Fig. 4C). Within the FSM isoforms, 15.3% exhibited an alternative 3′ end (n = 3010), 11.0% contain an alternative 5′ end (n = 2170), and 4.9% of FSM transcripts display alternative 3′ and 5′ ends (n = 962) when compared to the reference annotation (Supplemental Table S11). Under a broader classification criterion, these isoforms could be considered as putative novel transcripts. Importantly, analysis by SQANTI3 also indicated that the large proportion of novel transcripts may possess coding potential (Fig. 4D).
Similarly, GffCompare analysis revealed that the predominant class of StringTie2 assembled transcripts was “j” (Novel, multiexon gene with at least one matched exon junction) (40.58%, n = 21,635), followed by “=” (Known, complete intron chain match) (28.32%, n = 15,099) (Supplemental Table S12). Applying the alternative long-read RNA-seq transcript assembler FLAIR on PCS reads, GffCompare characterization reaffirmed that the majority of assembled transcripts are novel isoforms (Supplemental Table S12). Detailed characterizations of novel StringTie2 assembled transcripts by SQANTI3 and GffCompare can be found in Supplemental Table S13.
Next, we asked whether the novel assembled transcripts can also be detected by DRS of nephrectomy samples and, ccRCC tumor cells in vitro. The latter was used to avoid artifacts associated with the modest read length and full-length transcript coverage achieved in clinical samples, and to indicate the cancer cell-intrinsic origin of these transcripts. To address this, we performed DRS analysis of the VHL-negative, ccRCC cell line RCC4 under both untreated and IFNG- and TNF-treated conditions using DRS. The cytokine treatment conditions aimed to simulate in part the transcriptomic response of tumor cells to immune cells. Workflow and sequencing statistics can be found in Supplemental Figure S8, and DEG analysis data can be found in Supplemental Table S14. The mean read length was over 1100 nt and the full-length transcript coverage over 45% for all samples. Out of the 26,834 novel isoforms that were mapped by PCS of nephrectomy samples, 14,544 were also mapped in at least one DRS of the nephrectomy samples, and 13,336 were also detected in the DRS of RCC4 samples, whereas 10,645 novel transcript isoforms were detected in all three data sets (Fig. 4E). Levels of novel isoforms that were detected in all three data sets showed comparable expression levels compared to known reference isoforms (Fig. 4F). Furthermore, expression levels of these novel isoforms exhibited strong concordance between PCS and DRS of nephrectomy samples (R2 = 0.6832, P = <0.0001) (Fig. 4G). Despite starting with a reference based on our PCS data set, a small number of StringTie2 transcripts were mapped exclusively in DRS of nephrectomy samples (n = 80) and RCC4 (n = 332). This may be due to the assignment of multimapping, ambiguous sequencing reads by the sequence alignment program (minimap2). These results reveal a plethora of previously uncharacterized and unmapped transcripts within the ccRCC transcriptome. Despite differences in sequencing depth, novel transcripts from PCS could also be mapped by DRS, with a substantial proportion of these novel transcripts also detected in ccRCC cells in vitro.
Long-read RNA sequencing reveals the full exonic structure of ccRCC splice variants
Taking advantage of the ability of long-read sequencing to reveal whole transcript exonic structures, we next examined recently reported novel, nonreference annotated splice variants (SVs) specific to ccRCC tumors, which were supported by proteomics data and associated with clinical outcomes (Chang et al. 2022). We found sequence read the evidence for all 16 reported SVs (15/16 for PCS of nephrectomies, 9/16 for DRS of nephrectomies, and 13/16 for DRS of RCC4). Moreover, PCS StringTie2 assembled transcripts spanning 11 of the unannotated SVs (Supplemental Table S15). For example, the StringTie2 assembled transcripts MSTRG.9279.18 and MSTRG.9269.19 accurately replicated two ccRCC-specific SVs from MVK (Fig. 4G). This was supported by reference genome-aligned reads from all three long-read RNA-seq data sets (Supplemental Fig. S9A). In addition, sequencing results showed that these ccRCC SVs adopt the 3′-UTR structure of MVK-002 (ENST00000392727) instead of the longer 3′ UTR from canonical MANE transcript MVK-001 (ENST00000228510) (Fig. 4H). Another example was HPCAL1, where two StringTie2 assembled transcripts (MSTRG.20400.11 and MSTRG.20400.12) were found to span the ccRCC-specific SV (Supplemental Fig. S9B). The two isoforms exhibit variation in the exon 3 usage, where evidence of exon 3 retention can be found in PCS as well as RCC4 sequencing results. Overall, three additional SVs (SYNPO, EGFR, and FAM107B) were found to be encompassed by two StringTie2 assembled transcripts (Supplemental Table S15). We also examined VHL isoform expression in PCS and DRS of nephrectomies, as well as DRS of RCC4. The 3′ UTR of the VHL mRNA is 4 kb-long. As such, even though full-length VHL transcripts were detected, most PCS and DRS of archival samples did not span the 5′ upstream exons (Supplemental Fig. S10). The longer sequencing read length achieved in the DRS analysis of RCC4 cells allowed us to capture mainly full-length VHL. The majority of expressed VHL transcripts in RCC4 correspond to ENST00000256474, but with a shorter 3′ UTR compared to the reference gene model. Collectively, using recently reported ccRCC-related SVs (Chang et al. 2022), we demonstrated the ability of long-read sequencing to reveal transcriptomic codependencies, in this case, the co-occurrence of specific SVs with specific UTRs, providing unparalleled insight into novel features of ccRCC.
Discovery of a novel soluble PD-L1 isoform expressed by ccRCC tumor cells
Having identified that a reduction in the immune infiltrate of ccRCC tumors was linked to disease recurrence and that the ccRCC transcriptome includes a high number of previously uncharacterized novel transcript isoforms we focused on transcripts of immune checkpoint proteins. Here, we focused on PD-L1 (official symbol CD274). While most studies on PD-L1 have focused on the membrane-bound isoform (mPD-L1), recent attention has been drawn to a soluble PD-L1 isoform (sPD-L1) lacking exon 5, 6, and 7. sPD-L1 is currently unannotated in the Ensembl gene annotation, but it has been described in the NCBI GenBank database (NM_001314029). An Ensembl annotated transcript (ENST00000474218) partially overlaps with the 3′ UTR of the GenBank sPD-L1 transcript, serving as a proxy for mapping sPD-L1. Upon closer inspection to the exon 4 and 3′-UTR region of sPD-L1 (Chr 5: 5,462,800–5,463,400), reference genome reads coverage and StringTie2 supported two distinct isoforms with varying 3′-UTR lengths (Fig. 5A). While the shorter sPD-L1 represents the GenBank transcript, the alternative sPD-L1 includes a 3′ UTR more than twice the length of GenBank annotation (61 nt vs. 154 nt) (Fig. 5A). StringTie2 assembly revealed this elongated 3′-UTR structure, with supporting evidence stemming from reference genome-aligned reads of PCS and DRS data from ccRCC tissues, as well as DRS data from RCC4 cells (Supplemental Fig. S11A–E). To further validate our findings, we performed short-read Illumina RNA sequencing analysis of RCC4 cells and we were able to detect reads corresponding to the novel sPD-L1 3′ UTR. Furthermore, analysis of publicly available short-read RNA-seq data of normal human kidney and lung tissues from the Genotype-Tissue Expression (GTEx) project (The GTEx Consortium 2013) also validated the existence of this PD-L1 isoform (Supplemental Fig. S11F,G).
Discovery of a novel sPD-L1 isoform expressed by ccRCC tumor cells. (A) IGV visualization of reference annotation of mPD-L1 isoform (black, ENST00000381577), sPD-L1 (black, NM_001314029), and StringTie2 reference annotation (green) (top tracks); graphical representation of membrane, soluble, and novel soluble PD-L1 exon 4; Ensembl (black) and StringTie2 reference annotations (green) and IGV coverage tracks for PCS of ccRCC tumors (red) and DRS of RCC4 (green). (B) Grouped dot plot showing reference DESeq2 normalized PD-L1 expression in nonrecurrent (blue) and recurrent (red) tumors' PCS data. DESeq2 Padj value is shown in the graph. Center line represents the median for each group. (C) Grouped dot plots showing normalized mPD-L1, sPD-L1, and novel sPD-L1 expression (log2(RPM + 1)) in nonrecurrent (blue) and recurrent (red) tumors’ PCS data. (D) Grouped dot plots showing mPD-L1, sPD-L1 (all isoforms), and novel sPD-L1 mRNA levels measured by qRT-PCR in recurrent and nonrecurrent tumors from sequenced cohort (blue and red, n = 12) and validation cohort (black, n = 20) relative to average mRNA levels in nonrecurrent tumors. (E) Stacked bar graphs representing proportions of mPD-L1, sPD-L1, and novel sPD-L1 isoforms in RCC4 cells based on DRS data. For (C)–(E), two-tailed Mann–Whitney U tests were used with P ≤ 0.05 considered significant. (*) P < 0.05. Center line represents the median for each group. (F) mRNA decay curves for mPD-L1, sPD-L1, and novel sPD-L1 in unstimulated (blue) and IFNG + TNF treated (red) RCC4 cells. Half-lives of isoforms are indicated in the graph (blue for unstimulated, red for IFNG + TNF treated RCC4). Comparisons were made using unpaired Student's t-test with P ≤ 0.05 considered significant. (n.s.) not significant, (*) P < 0.05, (**) P < 0.01, (***) P < 0.001.
Upon evaluating the expression of PD-L1 in ccRCC tumors, no significant disparity in gene-level expression was found between recurrent and nonrecurrent nephrectomy samples (Fig. 5B). However, at the isoform level, while mPD-L1 was suppressed in the recurrent samples, both sPD-L1 isoforms (NM_001314029 and novel sPD-L1) showed no significant differences (Fig. 5C). We note that in the clinical samples mPD-L1 is the most abundant PD-L1 transcript, whereas expression of the novel and annotated sPD-L1 isoforms is comparable (Fig. 5C). Subsequent expression validation via qRT-PCR with the sequenced and independent validation cohort displayed the same pattern of results, where mPD-L1 displayed a borderline nonsignificant (P = 0.09) downregulation, while sPD-L1 isoforms remained unchanged (Supplemental Fig. S12).
As all PD-L1 transcripts, including the novel sPD-L1 isoform, were detected in RCC4 cells by DRS, we sought to further explore their regulation in cancer cells. The expression of all PD-L1 isoforms increased in response by IFNG and TNF treatment (Fig. 5D). However, expression levels of mPD-L1 were profoundly more responsive to cytokine treatment than the soluble isoforms (∼30-fold induction of mPD-L1 as opposed to threefold to 10-fold induction of sPD-L1 isoforms) (Fig. 5D,E). mRNA stability assays revealed that cytokine treatment significantly reduced the stability of sPD-L1 but not mPD-L1 (Fig. 5F). Furthermore, the novel sPD-L1 isoform exhibited lower stability than the total sPD-L1 isoforms. Taken together, our findings revealed the existence of an up-to-now uncharacterized sPD-L1 isoform with a longer 3′ UTR and low stability, and key differences in the regulation of membrane and soluble PD-L1 isoforms in ccRCC tumors and in response to inflammatory cytokines in vitro.
Discovery of novel genes associated with ccRCC recurrence
In addition to the characterization of novel isoforms within known genes, long-read RNA-seq also enables the discovery of novel genes that are absent from the reference gene annotation. Using the PCS StringTie2 assembly, we identified 1350 novel genes (curated by SQANTI3) that were mapped in the PCS data set. The majority of these genes were classified as either intergenic (59.76%) or antisense (39.86%) transcripts (Fig. 6A). Most of these novel genes have a single isoform, with the majority being noncoding, multiexon isoforms featuring canonical splice sites (Fig. 6B; Supplemental Fig. S13A–C). The expression levels of these novel genes are similar to those of reference annotated genes, with the coding novel genes demonstrating higher expression levels than noncoding novel genes (Supplemental Fig. S13D). Importantly, of the 1350 novel genes that were mapped by PCS, 982 (72.7%) were also detected in the DRS data of tumor nephrectomies, and 414 (30.7%) novel genes were also mapped in the DRS data of RCC4 cells (Fig. 6C). This suggests that a large number of novel genes might be expressed in ccRCC tumor cells.
Discovery of ccRCC recurrence-associated novel genes by long-read RNA-seq. (A) Bar chart showing the isoform classifications of StringTie2 assembled transcripts from novel genes as classified by SQANTI3. (B) Pie chart illustrating the proportion of coding and noncoding StringTie2 assembled transcripts from novel genes as classified by SQANTI3. (C) Venn diagram showing the number of overlapping mapped novel genes between PCS and DRS of ccRCC tumor samples, and DRS of RCC4. (D) Volcano plots showing DEGs (red) between recurrent and nonrecurrent tumors from PCS and DRS data using StringTie2 assembled reference. Number of differentially expressed novel and known genes are shown in table below plots. Names of novel genes that were validated by qPCR with validation cohort are shown on plots. (E) Correlation between log2FoldChange of differentially expressed novel genes identified by either or both PCS and DRS between recurrent versus nonrecurrent tumors (n = 40). (F) IGV visualization of MSTRG.29728 isoforms StringTie2 reference annotation (green) and the closest neighboring genes (LINC01170 and CSNK1G3) in the Ensembl reference annotation (Ensembl release 105) at Chr 5: 123,500,000–124,300,000 (top track); Sashimi plot showing abundance of reference genome-aligned reads and splicing patterns along MSTRG.29728 (Chr 5: 123,859,000–123,868,000) for PCS (red) and DRS (blue) of ccRCC tumor samples, and DRS (green) and short-read Illumina sequencing (orange) of RCC4; representative PCS sequencing reads (gray) aligned to the reference genome in the region of interest. (G) MSTRG.29728 mRNA levels measured by qRT-PCR in recurrent and nonrecurrent tumors from sequenced cohort (blue and red, n = 12) and validation cohort (black, n = 20), relative to average mRNA levels in nonrecurrent tumors. mRNA levels were normalized to GAPDH and ACTB. Two-tailed Mann–Whitney U test was used with P ≤ 0.05 considered significant. (*) P < 0.05. Center line represents the median for each group.
Next, we performed DEG analysis with the PCS StringTie2 assembly and identified a set of significantly differentially expressed (|log2FoldChange| ≥ 2, Padj ≤ 0.1) novel genes (n = 40 for PCS, n = 4 for DRS) between recurrent and nonrecurrent samples (Fig. 6D; Supplemental Fig. S13E; Supplemental Tables S16–S19). The directionality of gene expression for these differentially expressed novel genes demonstrated strong concordance between PCS and DRS (Fig. 6E). Thirteen differentially expressed novel genes were also identified between untreated and IFNG and TNF treated RCC4 cells (Supplemental Fig. S13F).
To further validate our sequencing findings, we sought to experimentally measure the levels in recurrent and nonrecurrent ccRCC nephrectomies of two novel genes: MSTRG.29728 and MSTRG.38727 using qRT-PCR. MSTRG.29728, a StringTie2 assembled gene is located on Chromosome 5, with its nearest reference annotated genes (5′: CSNK1G3, 3′: LINC01170) situated more than 300 kb away (Fig. 6F). Notably, the presence of this novel gene was also supported by reference genome-aligned reads from the DRS and our short-read RNA sequencing analysis of RCC4 cells (Fig. 6F). Based on coverage data from all sequencing experiments, the most highly expressed MSTRG.29728 isoform consists of two exons (Supplemental Fig. S14A,B) and its levels of expression are intermediate (Supplemental Tables S16–S18). Analysis of publicly available cell line data further validated the existence of this gene and suggested that it was enriched in kidney cancer cell lines (Supplemental Fig. S14C). MSTRG.29728 was significantly upregulated in recurrent ccRCC tumors in both PCS and DRS data sets. This upregulation was confirmed by qRT-PCR in both sequenced and independent validation cohorts (Fig. 6G). The second tested novel gene, MSTRG.38727, is located on Chromosome X with read coverage from PCS and DRS of nephrectomy specimens, albeit absent in DRS data from RCC4 (Supplemental Fig. S15A–C). PCS sequencing results showed that MSTRG.38727 expression was highly elevated in three of the six recurrent ccRCC tumors (Supplemental Fig. S15D). This was corroborated through qRT-PCR validation within the sequenced cohort, but was not validated in the independent validation cohort (Supplemental Fig. S15E).
Overall, long-read sequencing revealed a high number of candidate novel genes present in ccRCC transcriptomes. Further testing for two such genes by orthogonal methods and in independent patient cohorts provided further support for their existence and, critically, identified MSTRG.29728 as a novel noncoding RNA gene associated with ccRCC recurrence in both study cohorts. We provisionally term MSTRG.29728 as RECART, for Renal Carcinoma Recurrence-Associated Transcript.
Discussion
Long-read sequencing technologies represent a new era in cancer genomics and RNA medicine (Sakamoto et al. 2020; Wang et al. 2023). We used DRS and PCS to explore transcriptomes of primary ccRCC tumors. Our study aimed to demonstrate the methodological application of long-read sequencing, both PCS and DRS, in cancer and specifically ccRCC, focusing on the use of archival fresh frozen tissue samples and new gene and transcript discovery, and using disease recurrence as a proof-of-principle context. Even though we analyzed a sequencing and an independent validation cohort, the relatively low total number of study participants (n = 32) is a limitation of our study that should be considered. In addition, the relatively modest read length acquired for clinical samples should also be considered as a limitation. As a mitigation for this, we used the RCC4 DRS data set as a high-quality reference, as well as further validation for selected isoforms and genes. Using this approach, we showcase how long-read RNA sequencing can lead to the discovery of novel disease-associated transcripts and genes, the existence of which is supported by multiple approaches including short-read Illumina sequencing, targeted qRT-PCR, and validation in independent cohorts. We opted not to perform more detailed analyses such as the estimation of poly(A) length per transcript or posttranscriptional RNA modification analyses (Krause et al. 2019). However, our work sets the foundation for follow-up studies using the new ONT DRS platform (RNA004) comparing the ability to detect changes in such features in fresh and archival samples.
From a methodological point of view, the distinguishing features of our study are (1) the use of long-term stored tissue, (2) the direct comparison between DRS and PCS of clinical samples, (3) the successful sequencing of archival fresh frozen tissue samples, and (4) the use of total RNA as starting material for DRS and PCS library preparation. In reference to the latter point, compared to the pg–ng range of total RNA input requirement for short-read RNA sequencing library preparation, previous studies using ONT DRS have typically used 50–500 ng of poly(A) enriched RNA, which is hugely demanding for clinical samples (Jain et al. 2022). Here, we used 2 μg and 200 ng total RNA for DRS and PCS from tissues, respectively, without poly(A) enrichment. Indeed, it has been suggested that poly(A) selection can introduce a potential bias toward mRNAs with longer poly(A) tails (Viscardi and Arribere 2022). PCS achieved a higher depth and, consequently, detected a higher number of transcripts and genes in all tested samples, and a higher number of DEGs in primary tumors of patients who experienced recurrence than DRS. We note that we did not multiplex samples for PCS, but used a subsampling approach (5% PCS) that identified similar gene expression patterns both with regard to DEGs and enriched pathways. The 5% subsampling level was chosen to reduce the PCS read depth within the range achieved by DRS (2–4 million passed reads). On the other hand, DRS does not include a PCR amplification step, providing further confidence in the overlapping gene sets between the two methods. With regard to read length, both methods produced long reads. On average, raw reads generated by PCS were longer than DRS reads likely because of the fact that raw reads from PCS have additional ligated reverse transcription, PCR amplification primer, and unique molecular identifier. Once aligned to the reference genome, both methods achieved similar read lengths, although PCS achieved a higher percentage of full-length transcripts likely due to the size selection step following PCR (Bayega et al. 2022). Alignment to reference transcriptome showed good correlation with genome mapping and DTU analysis identified candidate DTU events associated with recurrence, including changes in CMC1 transcript usage. It should be noted, however, that, when using historical samples and achieving relatively modest read lengths, there might be limitations in the ability to accurately measure ratios of different SVs of the same gene. We note that as we used archival tumor samples, our DTU analysis should be interpreted with caution as it is likely to be underestimating the number of disease relapse-associated DTU events. This is why we focused on gene-level comparisons and the discovery of novel transcripts and genes that could be validated by other methods including DRS of RCC4 cells that achieved higher quality measures and by qRT-PCR.
The primary biological objective of our study was to use DRS and PCS to explore ccRCC recurrence-associated transcriptome features including previously uncharacterized genes and transcripts. Our differential expression and deconvolution analyses identified a loss of immune infiltrate and specifically CD8+ T cells as a key feature of primary tumors that go on to relapse after surgery. This is reported by others (Ghatalia et al. 2019; Peng et al. 2022), providing a biological validation of our findings. Despite the low numbers of samples tested in our sequencing cohort, we were able to see similar expression patterns for NOS3 and CCL5, two previously reported recurrence markers (Rini et al. 2015), but also the novel finding of downregulation of TOX and LINC04216 linked to recurrence (note that TOX levels were not measured in the study that identified loss of NOS3 and CCL5 as recurrence markers [Rini et al. 2015]). In addition, our study also identified upregulation of a novel gene, MSTRG.29728 or RECART, as a candidate marker of disease relapse. These candidate prognostic biomarkers of relapse will need to be validated in the future in independent cohorts.
A unique strength of long-read sequencing is the ability to determine preferential use of specific UTRs by specific SVs, which can suggest tissue- or disease-specific cotranscriptional processing mechanisms. Indeed, we demonstrated this for recently identified ccRCC-associated SVs (Chang et al. 2022), including MVK and HPCAL1. Focusing these analyses on immune checkpoints led to the discovery of a novel sPD-L1 transcript with a longer 3′ UTR than the currently annotated sPD-L1. This means that the novel sPD-L1 is likely to be controlled by additional posttranscriptional mechanisms, including microRNA-mediated silencing or regulation by RNA-binding proteins. Of note, regulation through the 3′ UTR is a major determinant of mPD-L1 expression (Sun et al. 2018; Yamaguchi et al. 2022). Indeed, the novel sPD-L1 transcript demonstrates lower stability than the other PD-L1 transcripts under homeostatic or inflammatory conditions in vitro. We found that there is a trend for downregulation for tumor mPD-L1 but no differences in sPD-L1 in patients that experience recurrence. This is consistent with the observed loss of CD8+ T cells from these tumors and the enhanced responsiveness of mPD-L1 to IFNG and TNF observed in vitro. Clinically, this is important as PD-1/PD-L1-targeted checkpoint inhibitors are currently being explored in the adjuvant setting (Gorin et al. 2022; Motzer et al. 2023; Choueiri et al. 2024) and expression of sPD-L1 has been linked with ccRCC prognosis and immunotherapy treatment outcome (Larrinaga et al. 2021; Mahoney et al. 2022). Future studies will have to explore the relative contributions of the different PD-L1 transcripts, including the novel one reported here, to tumor immune evasion and response to immunotherapy.
Overall, we demonstrate the feasibility of both DRS and PCS in archival clinical samples with significant overlap between the two methods with regard to detectable transcripts, differential gene expression analysis, pathway enrichment analysis, and novel transcript and gene discovery. We also identify a common limitation in that when using historical samples that have been stored for long periods of times (years) both methods might result in relatively shorter read length. Higher depth can be achieved for PCS, which might be beneficial for initial comparative analyses. On the other hand, demonstrating the feasibility of DRS using archival clinical samples opens the way for future studies exploring questions that can only be addressed by DRS (e.g. RNA posttranscriptional modifications) avoiding biases associated with reverse transcription and PCR amplification. We provide evidence for the existence of thousands of novel transcript isoforms and hundreds of novel genes detected by both DRS and PCS in primary ccRCC tumors but also in vitro in ccRCC cell lines. We describe the loss of TOX and LINC04216 and upregulation of RECART as novel candidate predictors of relapse. We discover and validate through orthogonal methods a novel sPD-L1 isoform with differential stability. These findings demonstrate that the application of long-read RNA sequencing, even in long-term stored tissue samples, has the potential to lead to a radical revision of our understanding of cancer transcriptomes.
Methods
Study participants and ethics
In this observational study, we used 32 ccRCC tumor nephrectomy samples (16 nonrecurrent and 16 recurrent cases) collected between 2000 and 2012 and stored in the Leeds multidisciplinary research tissue bank. Twelve samples were used as a discovery cohort for DRS and PCS sequencing (six nonrecurrent and six recurrent) and 20 (10 in each group) were used as an independent validation cohort. For the recurrence group, the median time to relapse was 23 months (5–176). For the control group, median follow-up without relapse was 11 years (7–18). Groups were matched for demographic, pathological, and clinical characteristics including TNM and Leibovich score (Supplemental Table S1, age is shown in 5-year intervals). The sample/kidney IDs were not known to anyone outside the research group. Mutation status of each sample was determined as described (Scelo et al. 2014; Vasudev et al. 2023). This study was approved by regional ethics committee approval: Yorkshire and the Humber—Leeds East Research Ethics Committee, reference 15/YH/0080. The research conforms with the principles of the Declaration of Helsinki. All patients gave written informed consent for their participation in this study.
Tissue sample preparation
Following surgical removal, tissue samples were washed in phosphate-buffered saline (PBS), blotted on a tissue before being enveloped in aluminum foil and snap frozen in liquid nitrogen. Once thawed, samples were immediately used for RNA extraction without further freeze-thawed cycles. All cases underwent pathology review of a parallel formalin-fixed paraffin-embedded (FFPE) block to confirm ccRCC histology and tumor cell viability, as part of a separate study (Scelo et al. 2014).
Cell culture and cytokine treatment
RCC4 cells were maintained at 37°C in a humidified atmosphere of 5% CO2 and grown in complete Dulbecco's modified Eagle's medium (DMEM, Gibco 21969-05), supplemented with 10% fetal bovine serum (FCS) (Gibco A5256701), 1% 200 mM l-glutamine (Gibco 25030), and 1% penicillin/streptomycin (Gibco 15140). For RCC4 DRS experiment, 1 × 106 RCC4 cells were seeded in 15 mL of complete DMEM in T75 flasks. Twenty-four hours after seeding, media were changed into complete DMEM, with or without the addition of IFNG (1000 U/mL, PeproTech 300-02) and TNF (25 ng/mL, Peprotech 300-01). Cells were harvested 24 h later for RNA extraction. Three flasks of T75s were used for each replicate for the sequencing experiment.
RNA extraction
Total RNA was extracted from nephrectomy specimens or cultured cells using QIAzol (Qiagen 79306) and RNeasy kits (Qiagen 74004) with on-column DNase I digestion step, according to the manufacturer's instructions. Nephrectomy specimens were homogenized in QIAzol using a TissueLyser LT (Qiagen 85600) with stainless steel beads (Qiagen 69997). RIN was determined using the 2100 Bioanalyzer with RNA Nano kit (Agilent 5067) and quantified using Qubit RNA HS assay kit (Invitrogen Q32852). Total RNA from RCC4 for DRS was enriched for poly(A)+ RNA molecules using the Dynabeads Oligo(dT)25 mRNA isolation kit (Invitrogen 61002).
Library preparation and RNA sequencing
Sequencing libraries used for PCR-cDNA-seq and Direct RNA-seq were generated using the SQK-PCS111 and SQK-RNA002 kit (Oxford Nanopore Technologies, ONT), respectively. For the nephrectomy specimens, 200 ng and 2 μg of extract total RNA were used as input for each sequencing library for PCR-cDNA-seq and Direct RNA-seq, respectively. Five hundred nanograms of poly(A)+ RNA was used for each sequencing library for Direct RNA-seq of RCC4 cells. For PCR-cDNA-seq, cDNA libraries were prepared with the SQK-PCS111 kit according to the manufacturer's instructions with 14 cycles of PCR cycles. For Direct RNA-seq, libraries were prepared with the SQK-RNA002 kit according to the manufacturer's instructions including the optional reverse transcriptase step. Sequencing libraries for each experiment were prepared together to mitigate batch effects. All sequencing libraries were sequenced on ONT PromethION sequencer with R9.4.1 PromethION flow cells (ONT) for 72 h. Basecalling and FASTQ file generation were performed with Guppy (v5.1.12, ONT).
Quality control and reads alignment
Sequencing reads generated from Direct RNA-seq, and PCR-cDNA-seq with a minimum read quality score (Q score) of 7 were used for mapping and downstream analysis. FASTQ files generated from sequencing runs were concatenated using catfishq (v1.4.0, https://github.com/philres/catfishq). PCR-cDNA-seq reads were orientated by Pychopper v2 (v2.5.0, https://github.com/epi2me-labs/pychopper), filtered for the presence of 5′ and 3′ sequencing adaptors and trimmed by cutadapt (v4.1) (Martin 2011). Direct RNA-seq and processed PCR-cDNA-seq reads were aligned to either human genome, transcriptome (GRCh38, Ensembl release 105) or StringTie2 assembly using minimap2 (v2.24), with recommended parameters (Genome alignment: -ax splice –uf –k14; Transcriptome alignment: -ax map-ont –p 0 -N 10) (Li 2018). Aligned reads were sorted, merged, and indexed to BAM files with SAMtools (v1.13) (Li et al. 2009). For subsampling, reference genome-aligned PCS reads were randomly selected using the SAMtools view command with “-s 0.05”. The workflows for reads alignment are available at Supplemental Code and https://github.com/joshuacylee/DRS and https://github.com/joshuacylee/PCR-cDNAseq. Mapping data quality and statistics of sequencing data were analyzed by NanoPlot and BamSlam (De Coster et al. 2018) (https://github.com/josiegleeson/BamSlam). Illumina sequencing reads were processed with FastQC, trimmed using cutadapt (version 1.18) to remove sequence adaptors, followed by reference genome alignment with HISAT2 (Kim et al. 2019).
Differential gene expression
Gene-level expression quantification was performed using featureCounts with long-read counting mode (-L) (subread v2.0.0) (Liao et al. 2014). Transcript isoform quantification was performed using Salmon (v1.7.0) with Oxford Nanopore long-reads mode (‐‐ont) (Patro et al. 2017). Normalization and identification of DEGs (Padj ≤ 0.1 and |log2FoldChange| ≥ 2) were performed using the R package (R Core Team 2022) DESeq2 (v1.40.2) (Love et al. 2014). PCA plots were generated by DESeq2 and volcano plots were generated with the R package EnhancedVolcano (v1.18.0). Workflow for differential gene expression identification is available at Supplemental Code and (https://github.com/joshuacylee/DESeq2).
Gene set enrichment analysis and tumor-infiltrating immune cell analysis
Gene set enrichment analysis was performed using clusterProfiler (v4.4.4) (Wu et al. 2021).
GO BP and molecular function databases were used for functional enrichment analysis. Parameters used for GO enrichment were as follows: Permutations (nPerm):10,000, minimum gene set size (minGSSize): 5, Maximum gene set size (maxGSSize): 500, Minimum P-value (PvalueCutoff) = 0.05, Organism (Orgdb) = org.Hs.eg.db, PAdjustMethod = Benjamini–Hochberg (BH). Tumor purity and tumor-infiltrating immune cell population abundance were estimated using two gene signature-based algorithms: ESTIMATE (v1.0.13) and xCell (v1.1.0) (Yoshihara et al. 2013; Aran et al. 2015). Tumor-infiltrating immune cell type deconvolution was performed using CIBERSORTx and EPIC (Newman et al. 2019; Racle and Gfeller 2020).
Differential transcript usage
DTU analysis of DRS and PCS data was performed with RNAseqDTU (version 3.14) workflow, which employs both DRIMSeq and DEXSeq, followed by stageR statistical postprocessing. Isoform quatification was scaled and normalized (dtuScaledTPM) before analysis. Analysis was performed on transcripts which had minimum expression levels of 5 (normalized TPM) across all 12 tumor samples, with 5% of total gene expression in at least half of the samples in at least half of the samples. Genes with Padj values below 0.1 were considered significant.
Transcriptome assembly and novel gene/isoform discovery
Using reference genome-aligned PCR-cDNA-seq BAM files, transcript assembly was performed with StringTie2 (v2.2.1) and FLAIR (Kovaka et al. 2019). StringTie2 assembly was performed with long-reads processing mode (-L), guided by reference gene annotation (Ensembl release 105). StringTie2 transcriptome assemblies from all sequenced nephrectomy specimens were then merged using the –merge option to generate transcript annotation file (GTF file). StringTie2 annotation used for novel gene mapping was performed by merging all nephrectomy assemblies with reference gene annotation (Ensembl release 105). FLAIR assembly was generated using the “flair correct” and “flair collapse” commands, with the long-read optimized option selected (‐‐trust_ends). Generated transcript annotation files from StringTie2 and FLAIR were compared to Ensembl reference gene annotation (with –r option) where each assembled transcript was classified with a classcode using GffCompare (v0.12.6). In accordance with Gleeson et al. (2022), transcripts were categorized into three main categories: “Known” (“=”: Complete intron chain match, “c”: Partial intron chain match), “Novel” (“j”: Multiexon with at least one matched junction”, “k”: Containing reference, “m”: Retained intron(s)—all covered, “n”: Retained intron(s)—not all covered, “i”: Contained within intron, “o”: Overlapped exon, “x”: Overlapped antisense, “y”: Containing reference within intron, “u”: None of above/Unknown), and “Potential Artefacts” (“p”: No overlap, “e”: Single exon partially covering an intron, “s”: Intron matched on opposite strand, “r”: Repeat).
StringTie2 assembled transcripts were also characterized by SQANTI3 (v5.1.2), which classifies genes as “annotated” or “novel,” and isoforms as FSM, ISM, NIC, NNC, antisense, genic intron, genic genomic, and intergenic (Pardo-Palacios et al. 2024). FSM represents isoforms with the exact same splice junctions and number of exons with the reference annotation. ISM represents isoforms with fewer exons from the 5′ end but with the remaining internal splice junction sites matching with the reference annotation. NIC isoforms contain novel combinations of known splice junctions/exons compared with the reference annotation. NNC represents isoforms with at least one novel, unannotated splice site. In the SQANTI3 model, FSM and ISM represent the “Known” transcripts, whereas NIC, NNC, antisense, genic intron, genic genomic, and intergenic isoforms represent the “Novel” transcripts. SQANTI3 also predicts coding potential of transcripts using the GeneMarkS-T model (Tang et al. 2015). IGV tracks and reference genome mapped reads aligned to the region of novel genes and isoforms were visualized using IGV viewer (Robinson et al. 2011). Evidence of novel transcript expression was derived from analysis of the GTEx project RNA-seq data of normal tissues (The GTEx Consortium 2013) and LocExpress RNA-seq of cancer cell lines (Hou et al. 2016).
cDNA synthesis and qRT-PCR
RNA molecules were reverse transcribed to cDNA molecules using Oligo(dT) primer (Novagen 69896) and SuperScript II reverse transcriptase (Invitrogen 18064022). qPCR assays were performed using Fast SYBR Green master mix (Applied Biosystems 4385612) and prevalidated primers (Eurofins) on a StepOnePlus Real-Time PCR system (Applied Biosystems) for 40 amplification cycles. Relative transcript levels were determined using the ΔΔCt (cycle threshold) method with GAPDH and ACTB used as loading controls. Details on the primers used can be found in Supplemental Table S20.
RNA stability assay
In total, 4 × 104 RCC4 cells were seeded in 12-well plates. Twenty-four hours after seeding, media were changed into complete DMEM, with or without the addition of IFNG (1000 U/mL) and TNF (25 ng/mL). Twenty-four hours later, Actinomycin D (2 μg/mL, Thermo Fisher Scientific, 11805017) was added and cells were harvested after 0, 2, 4, and 8 h of incubation for RNA extraction and qPCR. Three wells were used as technical replicates for each biological replicate (n = 3) for each time point.
Statistical analysis
Statistical analysis was performed using GraphPad Prism 9. Two-tailed Mann–Whitney U tests were used to compare nonparametric analysis of gene or transcript isoform expression levels, tumor purity estimations, immune scores, and relative immune cell populations between experimental groups, with P ≤ 0.05 considered statistically significant. For comparison of more than two groups, Kruskal–Wallis test was used with P ≤ 0.05 considered significant. For correlative analysis, R2 (coefficient of determination) was used to calculate the goodness of fit between data sets, and P-values were generated from F-test, with P ≤ 0.05 considered statistically significant. Differential gene expression analysis by DESeq2 implements the Wald test, followed by false discovery rate correction by the BH method. Genes with Padj < 0.05 and |log2FoldChange| ≥ 2 are considered to be significantly differentially expressed. All P-values of nonsignificant results are indicated in graphs.
Data access
All raw and processed sequencing data generated in this study have been submitted to the NCBI Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/) under accession numbers: GSE242204 (PCS), GSE241932 (DRS), GSE242084 (RCC4 DRS), GSE246408 (RCC4 short-read Illumina sequencing). The workflows for read alignment are available at GitHub (https://github.com/joshuacylee/DirectRNAseq and https://github.com/joshuacylee/PCR-cDNAseq) and as Supplemental Code. Workflow for differential gene expression identification is available at GitHub (https://github.com/joshuacylee/DESeq2) and as Supplemental Code.
Competing interest statement
E.A.S. and D.J.T. are employees of and stock option holders in Oxford Nanopore Technologies. As of November 2023, J.L. is also an employee of Oxford Nanopore technologies. N.S.V. has received grants, speaker honoraria, and/or advisory fees from Bristol Myers Squibb, Ipsen, EUSA pharma, Eisai, and Pfizer, all outside the submitted work. The remaining authors declare no competing interests.
Acknowledgments
This work was funded by the Biotechnology and Biological Sciences Research Council (BBSRC) White Rose doctoral training partnership (BB/J014443/1) through an Industrial Cooperative Awards in Science and Engineering (iCASE) studentship supported by Oxford Nanopore Technologies. Additional support was provided by the Hull York Medical School and Oxford Nanopore Technologies. We are indebted to the study participants and their families for contributing to medical research. We thank Aino Järvelin for the support with bioinformatics analyses of long-read sequencing data. We thank staff at the Genomics Lab in the University of York Bioscience Technology Facility for technical assistance with short-read Illumina sequencing.
Authors contributions: J.L. contributed to experimental design, data generation, data analysis, figure design, and manuscript writing; E.A.S. contributed to experimental design and data generation; C.E.B. contributed to data generation; J.B. and R.E.B. managed clinical sample and data collection and maintenance; D.J.T. assisted with study conception and design; N.S.V. contributed to clinical sample and data collection, study design, and data interpretation; D.L. assisted with study conception, study design, data interpretation, study supervision, and manuscript writing. All authors read and approved the final manuscript.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.278801.123.
-
Freely available online through the Genome Research Open Access option.
- Received December 19, 2023.
- Accepted September 10, 2024.
This article, published in Genome Research, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








