
De novo genome assemblies of two cryptodiran turtles with ZZ/ZW and XX/XY sex chromosomes provide insights into patterns of genome reshuffling and uncover novel 3D genome folding in amniotes
- Basanta Bista1,7,
- Laura González-Rodelas2,3,7,
- Lucía Álvarez-González2,
- Zhi-qiang Wu1,4,
- Eugenia E. Montiel1,8,
- Ling Sze Lee1,9,
- Daleen B. Badenhorst1,
- Srihari Radhakrishnan1,
- Robert Literman1,
- Beatriz Navarro-Dominguez1,10,
- John B. Iverson5,
- Simon Orozco-Arias6,
- Josefa González6,
- Aurora Ruiz-Herrera2,3 and
- Nicole Valenzuela1
- 1Department of Ecology, Evolution, and Organismal Biology, Iowa State University, Ames, Iowa 50011, USA;
- 2Departament de Biologia Cel·lular, Fisiologia i Immunologia, Universitat Autònoma de Barcelona, 08193 Cerdanyola del Vallès, Spain;
- 3Genome Integrity and Instability Group, Institut de Biotecnologia i Biomedicina, Universitat Autònoma de Barcelona, 08193 Cerdanyola del Vallès, Spain;
- 4Guangdong Laboratory for Lingnan Modern Agriculture, Agricultural Genomics Institute at Shenzhen, Chinese Academy of Agricultural Sciences, Shenzhen 518124, China;
- 5Department of Biology, Earlham College, Richmond, Indiana 47374, USA;
- 6Institute of Evolutionary Biology, CSIC, UPF, 080003 Barcelona, Spain
-
↵7 These authors contributed equally to this work.
Abstract
Understanding the evolution of chromatin conformation among species is fundamental to elucidate the architecture and plasticity of genomes. Nonrandom interactions of linearly distant loci regulate gene function in species-specific patterns, affecting genome function, evolution, and, ultimately, speciation. Yet, data from nonmodel organisms are scarce. To capture the macroevolutionary diversity of vertebrate chromatin conformation, here we generate de novo genome assemblies for two cryptodiran (hidden-neck) turtles via Illumina sequencing, chromosome conformation capture, and RNA-seq: Apalone spinifera (ZZ/ZW, 2n = 66) and Staurotypus triporcatus (XX/XY, 2n = 54). We detected differences in the three-dimensional (3D) chromatin structure in turtles compared to other amniotes beyond the fusion/fission events detected in the linear genomes. Namely, whole-genome comparisons revealed distinct trends of chromosome rearrangements in turtles: (1) a low rate of genome reshuffling in Apalone (Trionychidae) whose karyotype is highly conserved when compared to chicken (likely ancestral for turtles), and (2) a moderate rate of fusions/fissions in Staurotypus (Kinosternidae) and Trachemys scripta (Emydidae). Furthermore, we identified a chromosome folding pattern that enables “centromere–telomere interactions” previously undetected in turtles. The combined turtle pattern of “centromere–telomere interactions” (discovered here) plus “centromere clustering” (previously reported in sauropsids) is novel for amniotes and it counters previous hypotheses about amniote 3D chromatin structure. We hypothesize that the divergent pattern found in turtles originated from an amniote ancestral state defined by a nuclear configuration with extensive associations among microchromosomes that were preserved upon the reshuffling of the linear genome.
Unraveling the dynamics of genome reshuffling that underlie karyotypic diversification (Bourque et al. 2004; Ruiz-Herrera et al. 2006, 2012; Faraut 2008; Longo et al. 2009; Berthelot et al. 2015; Damas et al. 2022), together with their causes and consequences, is essential to deciphering the evolution of genome function across the Tree of Life. This is facilitated in the “chromosomics” era (Deakin et al. 2019) by technical advances that move beyond the comparative analyses of linear DNA sequences alone. These include chromosome conformation capture (3C) techniques and derivatives (including Hi-C), which enhance our understanding of the three-dimensional (3D) organization of genomes (Lieberman-Aiden et al. 2009; Rao et al. 2014). As genome architecture and function are interdependent (Rowley and Corces 2018), 3D chromatin folding is expected to coevolve along with functional genomic changes. This view predicts that 3D chromatin remodeling should underlie phenotypic evolution and adaptation to changing environments that are associated with chromosome rearrangements (Kirkpatrick and Barton 2006; Hoffmann and Rieseberg 2008; Loxdale 2010; Álvarez-González et al. 2022a; Waters et al. 2023). While foundational work has deciphered fundamental principles of the 3D genome structure and function in model species (i.e., human and mouse) (Lieberman-Aiden et al. 2009; Rao et al. 2014), evolutionary aspects remain obscure.
Within nuclei, vertebrate genomes are compartmentalized in superimposed layers of organization, including chromosome territories (subnuclear regions), open (active) and closed (inactive) chromatin compartments named A and B, respectively, topologically associated domains (TADs), and DNA looping interactions, which are established and maintained by structural proteins (i.e., cohesins and CCCTC-binding factor—CTCF) (Dekker et al. 2013; Rao et al. 2017). Such 3D chromatin organization is tightly associated with transcriptional states and interdependent with gene expression (Rowley and Corces 2018; Schield et al. 2019), responds to environmental stimuli (Kumar et al. 2021; Waters et al. 2023), and is remodeled during embryonic development in ways that affect the determination of cell fate (Phillips-Cremins et al. 2013; Murphy et al. 2024).
Recent comparative studies identified distinct patterns of chromosome folding across vertebrate lineages, including chromosome territories (i.e., Type I pattern) and centromeric associations (i.e., Type II or Rabl-like pattern) (Hoencamp et al. 2021; Álvarez-González et al. 2022a). Moreover, chromatin folding may influence evolutionary patterns of genome reshuffling (Álvarez-González et al. 2022a), as chromosome folding is affected by inversions and fusions/fissions that can occur during germ cell formation (Vara et al. 2021; Álvarez-González et al. 2022b; Vara and Ruiz-Herrera 2022). Yet, how these levels of organization are linked to evolutionary innovations in nonmodel vertebrates remains underexplored.
Amniote vertebrates shared a last common ancestor ∼325 million years ago (Mya) (Shedlock and Edwards 2009) and are characterized by distinctive chromosome morphology and evolutionary adaptations. Sauropsids (lizards, snakes, turtles, crocodiles, and birds) vary in their chromosome number (2n = 22–138) (Ruiz-Herrera et al. 2012) and most possess generally well-conserved macro- and microchromosomes (Waters et al. 2021). Among nonavian sauropsids (reptiles), turtles are a charismatic group whose unique adaptations and potential as ecotoxicology sentinels render them emergent models for ecology, evolution, and biomedicine (Montiel et al. 2016; Sabath et al. 2016; Cortés-Gómez et al. 2018; Bista and Valenzuela 2020; Lee et al. 2020; Chaousis et al. 2021; Thépot 2021; Congdon et al. 2022; Mizoguchi et al. 2022; Zdyrski et al. 2024). Unlike mammals and crocodilians, that only possess large (macro) chromosomes, turtles (like birds) exhibit larger (macro) and tiny (micro) chromosomes, and vary widely in their diploid number between 2n = 28 and 2n = 68 (Montiel et al. 2016), likely as the result of fusions and fissions over >210 million years (Myr) of evolution (Montiel et al. 2016). Recent studies uncovered a distinct pattern of chromatin dynamics in micro- versus macrochromosomes shared by disparate vertebrates (Schield et al. 2019; Perry et al. 2021; Waters et al. 2021), including the Green Sea Turtle (Chelonia mydas) and the Slider Turtle (Trachemys scripta), two chelonians (extant turtles) possessing temperature-dependent sex determination (TSD), a mechanism mediated by chromatin remodeling (Ge et al. 2018; Weber et al. 2020). But it is unknown whether 3D chromatin structure differs in turtles with sex chromosome systems of genotypic sex determination (GSD) compared to those with TSD.
Understanding genome organization in turtles is relevant as sex chromosomes arose in turtles at least five times independently (and perhaps were lost twice) (for review, see Bista and Valenzuela 2020), and these transitions were accompanied by a 20× higher rate of evolution of diploid numbers (Valenzuela and Adams 2011) and accelerated molecular evolution (Literman et al. 2018). This high rate of turnover in diploid numbers in turtles implies profound reorganization of the genome that might have altered 3D chromosome folding and interchromosomal interactions during the diversification of the group. Thus, whether previously studied turtle genomes exhibit a conserved 3D genome structure due to retention of the ancestral TSD mechanism (Sabath et al. 2016; Kratochvíl et al. 2021) or whether such conservation is a fundamental aspect of turtle and vertebrate genome organization remains to be tested. Furthermore, the hierarchical 3D genome folding of chelonians and how it differs among turtle taxa with distinct diploid number are unknown.
Here, we report the generation of two de novo genome assemblies for cryptodiran (hidden-neck) turtles possessing contrasting GSD systems and chromosome numbers (Montiel et al. 2016), namely, the Spiny Softshell Turtle Apalone spinifera (ZZ/ZW, 2n = 66) (Badenhorst et al. 2013) and the Northern Giant Musk Turtle Staurotypus triporcatus (XX/XY, 2n = 54) (Sites et al. 1979). To illuminate amniote genome evolution, we not only test for changes to the linear organization of turtle genomes, but also for changes in the patterns of amniote chromosome folding, by comparing the 3D genome structure of these two genomes and selected vertebrates, including human (Homo sapiens), African Elephant (Loxodonta africana), Tasmanian Devil (Sarcophilus harrisii), Platypus (Ornithorhynchus anatinus), Emu (Dromaius novaehollandiae), and chicken (Gallus gallus).
Results
Chromosomal-level assemblies of two turtle genomes
We generated high-quality chromosome-level genome assemblies of Apalone and Staurotypus (species will be referred to by their genus name after first use hereafter) using a combination of high coverage Illumina short reads, Chicago, and Hi-C reads (Fig. 1; see Supplemental Table S1 for read numbers and coverage). Namely, an initial de novo genome assembly of >72× coverage per species from Illumina sequencing data was generated and used as input for the first HiRise genome scaffolding using Chicago library sequencing data (Supplemental Table S1). The resulting assembly was then used as input for the second HiRise genome scaffolding using Hi-C sequencing data (Fig. 1A; Supplemental Table S1).
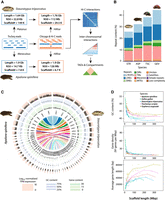
Genome assemblies. (A) Methodological workflow for the generation of turtle genome assemblies. (B) Repetitive DNA abundance in the turtle genomes assembled here for Staurotypus triporcatus (STR) and Apalone spinifera (ASP), plus those of Trachemys scripta elegans (TSC) (Simison et al. 2020) as corrected by Lee et al. (2020), and Gopherus evgoodei (GVE) (Rhie et al. 2021) that serve as reference for comparison. (C) Circos plots (of sequences >3 kbp) depicting genomic chromosomal syntenies and comparative genomic characteristics (Log10 of normalized Transcripts Per Million—TPM—gene expression, GC, and gene content) between ASP and STR. (D) GC content, gene density, and average gene length as a function of chromosome length in four turtle species and chicken, Gallus gallus (Warren et al. 2023).
We then generated final genome assemblies with a size of 1.76 Gbp for Staurotypus and 1.9 Gbp for Apalone, 50% of which was contained in five chromosomes (L50) in both cases. These genome assemblies have an N50 of 112 Mbp for Staurotypus and 126 Mbp for Apalone, and vertebrate_odb09 BUSCO scores indicate they are ∼96% and ∼88% complete, respectively (Table 1). Additionally, the 27 and 33 largest scaffolds in Staurotypus and Apalone, respectively, which correspond to the number of chromosome pairs in each species (i.e., their haploid number, n), contain 98% and 99% of the genome assembly. The species-specific RNA-seq reads from multiple tissues used for annotation mapped to the genome assemblies at a rate of 97.3% for Apalone (using available transcriptomic data from blood, liver, ovary, and testis), and 96.1% for Staurotypus (using available transcriptomic data from heart, ovary, and testis). Among the unique 18,340 genes annotated in the chromosome-level assembly of the reference T. scripta elegans genome (Simison et al. 2020) (denoted Trachemys hereafter), 16,621 and 15,945 unique genes (91% and 87%) were identified in the assemblies of Staurotypus and Apalone, respectively. We used the Trachemys genome (Simison et al. 2020) for comparison, as it is an annotated chromosome-level assembly that has been partially mapped physically and some chromosome assignments corrected accordingly (Lee et al. 2020), along with Gopherus evgoodei (Rhie et al. 2021).
Summary statistics of the de novo genome assemblies of Staurotypus triporcatus and Apalone spinifera
Repetitive DNA abundance (estimated from the assembled genomes), was higher in Apalone (32%) than Staurotypus (26%) (Fig. 1B) and higher yet in Trachemys (38%) and Gopherus (44%) whose assemblies were generated using longer read sequencing (Pacific Biosciences [PacBio] or Oxford Nanopore Technologies [ONT], respectively). Transposable elements (TEs) such as long terminal repeats (LTRs), long interspersed nuclear elements (LINEs), short interspersed nuclear elements (SINEs), and dictyostelium intermediate repeats (DIRs), increased in proportion to the size of the assemblies, whereas terminal inverted repeats (TIRs) and miniature inverted-repeat transposable elements (MITEs) varied independently. Satellite DNA and simple repeats represented <1% of the repetitive DNA in all taxa. This relative abundance of repetitive elements, which are difficult to assemble, may explain in part the smaller size of the assemblies obtained here (1.76 Gbp and 1.9 Gbp, respectively) compared to the estimated 2.6 Gbp genome size based on C-values from the Animal Genome Size Database calculated by flow cytometry for Apalone, and for Staurotypus when using as proxy its close relative Sternotherus odoratus (http://www.genomesize.com).
High degree of conservation of GC content in micro- and macrochromosomes
In general, we confirmed a well-established trend between macro- and microchromosomes in vertebrates (Andreozzi et al. 2001; Saccone et al. 2002; Federico et al. 2006) when analyzing the genomes of chicken (Gallus) and turtles (Apalone, Staurotypus, Trachemys, and Gopherus). Namely, GC content and gene density decreased with increasing chromosome size, whereas the average and standard deviation of gene size increased with chromosome length (Fig. 1D). Thus, compared to macrochromosomes, chicken and turtle microchromosomes are more GC-rich, and have fewer but more densely packed genes, which tend to be shorter (Fig. 1D) and more uniform in size (Supplemental Fig. S1A), and are more transcriptionally active (display higher Log10 Transcripts Per Million—TPM—values) (Fig 1C; Supplemental Fig. S1A).
Furthermore, GC content in the Apalone and Staurotypus genomes was higher toward the ends of chromosomes (Fig. 1C; Supplemental Fig. S2), as expected for telomeric regions (Paeschke et al. 2010). But GC content was also elevated in central chromosomal regions in Staurotypus (Fig. 1C), particularly those macrochromosomes that resulted from the fusion of two ancestral chromosomes, namely, STR-5, STR-7, STR-8, STR-9, and STR-10 (Fig. 2), as described below. Higher GC content was also detected in central chromosomal regions of STR-1, but not in STR-2, two chromosomes known to contain interstitial telomeric sequences detectable by molecular cytogenetics (Montiel et al. 2016). These observations also support the notion that the smaller than expected size of the genome assemblies of Apalone and Staurotypus is due in part to the underrepresentation of repetitive elements.
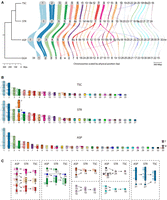
Genome conservation and chromosomal rearrangements. (A) Genespace plot depicting chromosomal syntenies (of sequences >1 Mb) and rearrangements (of >5 genes) between turtles and chicken. Chromosomes are scaled by physical size (bp). (TSC) Trachemys scripta elegans assembly from Simison et al. (2020) with corrected chromosomal nomenclature as per (Lee et al. 2020), (STR) Staurotypus triporcatus (this study), (ASP) Apalone spinifera (this study), (GGA) Gallus gallus (Warren et al. 2023). Suffixes “p” and “q” denote a fragment of the short- and long-arm of a chromosome, respectively, whereas “b” denotes a fragment of an acrocentric chromosome or of the same chromosome arm. (B) Ideograms depicting BAC positions and chromosomal homology across species (Badenhorst et al. 2015). (C) Major chromosomal fusions and fissions identified in this study between Apalone, Staurotypus, and Trachemys (chromosome nomenclature and homology as in B).
Whole-genome comparisons between chicken and turtles reveal several genome-reshuffling events
We next explored the dynamics of karyotype evolution and genome reshuffling in turtles through comparative genomics (Fig. 2A). This analysis included the two genome assemblies newly reported here, in addition to those of Red-eared Slider Turtle T. scripta elegans (Simison et al. 2020) (whose chromosome assignments were corrected via BAC-FISH [Lee et al. 2020] and denoted by the prefix TSC hereafter), using chicken (G. gallus, GGA) (Warren et al. 2023) as the reference genome. Accession numbers of these and all other turtle genomes used for comparative analyses in this study are listed in Supplemental Table S2. Scaffolds were physically assigned to chromosomes through the combination of in silico BAC mapping, in-metaphase BAC-FISH, and whole-chromosome painting (WCP) as described elsewhere (Fig. 2B; Supplemental Fig. S3; Badenhorst et al. 2015; Lee et al. 2020).
Considering chicken as the outgroup for turtles, we detected a high degree of syntenic collinearity between Apalone (ASP) and chicken chromosomes (Fig. 2A; Supplemental Table S3), mirroring previous large-scale observations using chicken WCP and BAC-FISH onto Apalone (O'Connor et al. 2018) and cDNA-FISH onto the Chinese softshell turtle Pelodiscus sinensis (Matsuda et al. 2005) (denoted Pelodiscus hereafter). This observation indicates that the fairly conserved karyotype seen in Apalone may be close to the ancestral state for turtles; i.e., the karyotype that this softshell turtle has retained since the divergence of its family Trionychidae from other hidden-neck turtles (Crawford et al. 2015). Indeed, a single identifiable large-scale fission event was observed for GGA-4 that led to the formation of two chromosomes, ASP-5 and ASP-17 (involving the “short”/“p” arm above the centromere ASP-17p, rather than the “longer”/“q” arm below the centromere) (Fig. 2A,B). Unlike the paucity of interchromosomal rearrangements between chicken and Apalone, around 70 intrachromosomal rearrangements (inversions and repositioning) of varying size (from 93 kbp to 2.3 Mbp) were observed in scaffolds >1 Mbp. This was the case of homologous chromosomes ASP-2/GGA-2, ASP-3/GGA-3, ASP-4/GGA-5, ASP-5/GGA-4, and ASP-6/GGA-Z. Altogether, our results demonstrate that a low degree of evolutionary change accrued during >260 Myr of independent evolution of trionychid softshell turtles and birds, paralleling the relatively lower rates of turtle molecular evolution compared to other sauropsids (Eo and DeWoody 2010; Shaffer et al. 2013; Takezaki 2018).
Importantly, our analysis also uncovered patterns of genome reshuffling and lineage-specific chromosome rearrangements within turtles. Namely, our comparison with chicken detected eight chromosomal fusions in the lineage leading to Staurotypus, of which one is unique to this taxon and seven are shared with Trachemys (i.e., are retained in Trachemys), plus two additional fusions that are unique to Trachemys (Fig. 2A,C). Most of these rearrangements involve micro- and midsize chromosomes, and some are more complex than others. For instance, we identified the fission of ASP-17 and ASP-12 with the concomitant fusion of their components into different Staurotypus chromosomes, i.e., the fusion of the short-arm of chromosome ASP-17 (ASP-17p) and the long-arm of ASP-12 (ASP-12q) into STR-9/TSC-9, and the fusion of the long-arm of ASP-17 (ASP-17q) and part of ASP-15 (labeled ASP-15b in Fig. 2A) into STR-8/TSC-8 (Fig. 2C). Additionally, part of ASP-8q split and fused with ASP-11 to form STR-7/TSC-7. In another case, ASP-4 and ASP-23 appear fused to form STR-5, though remain intact in Trachemys as TSC-4q and TSC-24, whereas ASP-19 remains separate as STR-18, but its homolog fused to become TSC-4p (Fig. 2C). Five rearrangements represent simpler fusions. For example, ASP-9 and ASP-20 fused into STR-14/TSC-13. Additionally, ASP-14 and ASP-16 fused to compose STR-10/TSC-10. Similarly, micros ASP-26 and ASP-27 are fused into STR-19/TSC-18. Likewise, ASP-32 and ASP-Z/W form a single micro in the other two turtles, STR-24/TSC-16. Lastly, ASP-31/STR-27 contributes to a portion of TSC-2 (Fig. 2A,C). We emphasize that the chromosome nomenclature used here for Trachemys corresponds to physical chromosomes following Lee et al. (2020) (i.e., labeled TSC) and not to the uncorrected assignment of the genome scaffolds to putative chromosomes labeled TSE (T. s. elegans) as initially reported (Simison et al. 2020).
Our comparative analysis suggests the presence of additional large-scale or whole chromosome species-specific reorientation (due to centromere repositioning) or inversion of chromosomal segments in turtles. This is the case of ASP-14, which appears reoriented in Apalone with respect to Gallus but not in Staurotypus or Trachemys, and the same is true for ASP-25. On the other hand, the portion of STR-10 homologous to ASP-16 is only inverted in Staurotypus. At this stage, our BAC-FISH and G-banding comparative data (Fig. 2B) do not allow us to rule out the possibility that the reorientations detected in ASP-14, ASP-16, and ASP-25 correspond to inversions or in silico artifacts. In contrast, chromosomes homologous to ASP-9, ASP-11, ASP-12, ASP-15, and ASP-27 appear reoriented in Staurotypus (i.e., STR-14, STR-7, STR-9, STR-13, and STR-27, respectively), a conformation that is retained in Trachemys. Cytogenetic data in our study provided support for these in silico patterns only when multiple BACs were hybridized successfully (Fig. 2B).
Overall, we detected two contrasting patterns of genome reshuffling in turtles, namely, high conservation in Apalone reflected in the high degree of syntenic collinearity to chicken chromosomes, whereas Staurotypus and Trachemys exhibited higher rates of chromosomal reorganizations.
Patterns of genome-wide chromosomal interactions across vertebrates
The generation of the de novo genome assemblies of Apalone (2n = 66) and Staurotypus (2n = 56) allowed us to further explore patterns of 3D genome folding across vertebrates. This was accomplished by evaluating the novel Hi-C data generated in the present study (Fig. 3A; Supplemental Table S4) in combination with publicly available Hi-C data for representative species of mammals and birds, including human (2n = 46) (Rao et al. 2014), African Elephant (2n = 56) (Álvarez-González et al. 2022a), Tasmanian Devil (2n = 14) (Álvarez-González et al. 2022a), Platypus (2n = 52) (Zhou et al. 2021), Emu (2n = 78) (Liu et al. 2021), and chicken (2n = 70) (Fishman et al. 2019). As in birds and mammals, the diagonal of the heatmaps of Apalone and Staurotypus revealed strong interactions between proximal loci within chromosomes (in cis) with the expected exponential decay in interaction signal as the distance between genomic regions increased (Fig. 3A). And we note that the diagonal thickness, which reflects the level of chromatin compaction along each chromosome (Belton et al. 2012), was greater for sauropsids and specially for birds, than for mammals (Fig. 3A).
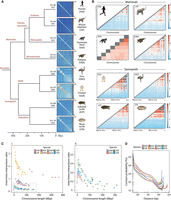
Patterns of chromosome folding across vertebrates. (A) Whole-genome Hi-C contact maps for human (Homo sapiens [HSA], Rao et al. 2014), African Elephant (Loxodonta africana [LAF], Álvarez-González et al. 2022a), Tasmanian Devil (Sarcophilus harrisii [SHA], Álvarez-González et al. 2022a), Platypus (Ornithorhynchus anatinus [ONA], Zhou et al. 2021), Emu (Dromaius novaehollandiae [DNO], Liu et al. 2021), chicken (Gallus gallus [GGA], Fishman et al. 2019), Spiny Softshell Turtle (Apalone spinifera [ASP], this study), and Northern Giant Musk Turtle (Staurotypus triporcatus [STR], this study]. (B) Heat maps representing interchromosomal mean interactions per chromosome for human, African Elephant, Tasmanian Devil, Platypus, chicken, Emu, Spiny Softshell Turtle, and Northern Giant Musk Turtle. (Ci) Inter-/intrachromosomal interactions as a function of chromosome length (in Mbp) in the target mammals and sauropsids compared in this study. In the case of turtles and birds, color intensity differentiates between macro (darker color) and micro (lighter color) chromosomes. (Cii) Enlarged view of the inter-/intrachromosomal interactions analyzed for birds and turtles (demarcated by the dashed line in Ci). (D) Chromosome-specific contact probability P(s) as a function of genomic distance in all species analyzed.
Our multispecies analysis of genome-wide interchromosomal interactions also revealed a general trend for smaller chromosomes (those containing <5% of the genome size) to interact more frequently among themselves (mean of interchromosomal interactions >1.5×) than larger chromosomes (defined as >5% genome size) in all species (Fig. 3B), with the exception of marsupials as previously reported (Álvarez-González et al. 2022a). The same was true for all sauropsids included in the analysis (chicken, Emu, and both turtles), which exhibited high interaction frequencies among microchromosomes (Chromosome pairs 12/13 and higher), mirroring previous studies (Schield et al. 2019; Perry et al. 2021; Waters et al. 2021). Additional higher-resolution Hi-C data are needed to test more directly the alternative 3D models of chromatin architecture suggested by our findings.
Importantly, comparison of Hi-C contact matrices revealed different patterns of chromosomal interactions between species as reflected by the inter-/intrachromosomal interaction ratios (Fig. 3C). Namely, compared to mammals, sauropsids (turtles and birds) presented lower interaction ratios per chromosome (<1) (Fig. 3Ci), with birds showing the lowest, and with further differences between macro- and microchromosomes (Fig. 3Cii). That is, microchromosomes in sauropsids showed an accentuated degree of inter-/intrachromosomal interaction ratios than macrochromosomes, as microchromosomes interact more frequently among themselves (Fig. 3B). Finally, such differences were also detected when analyzing the average distance-dependent interaction frequencies within chromosomes, represented by curves of contact probability (P(s)) as a function of genomic distance (Fig. 3D). Namely, the contact probability curves for sauropsids were elevated (reflecting more compacted chromatin) (Belton et al. 2012; Naumova et al. 2012) compared to mammals (except for Apalone at longer distances, for reasons that remain unknown). Altogether, these results suggest that sauropsids are characterized by a distinct chromosome compartmentalization pattern when compared to mammals, defined by higher values of intrachromosomal interactions and lower values of interchromosomal interactions. This can be interpreted as sauropsids having more compacted chromosomes than mammals (more intra- than interchromosomal interactions and higher contact probabilities curves across genomic distance), a pattern that is further accentuated in microchromosomes than macrochromosomes, mirroring previous studies (Perry et al. 2021; Waters et al. 2021; Li et al. 2022).
Divergent patterns of chromosome nuclear position
We next analyzed nuclear architecture across and within species, and uncovered different patterns of interchromosomal interactions, which provided a proxy of chromosomal relative position within the nucleus. In both turtle species, microchromosomes interacted more among themselves than with macrochromosomes (Figs. 3B, 4A). Consistent with the presence of chromosome territories, eutherian mammals exhibit high contact frequency between loci on the same chromosome (Fig. 4B). Moreover, we detected a novel pattern of centromere–telomere interactions in turtles, which was more prominent in macrochromosomes (Fig. 4). This was illustrated in both Circos plots of genome-wide interactions (Fig. 4A) and chromosome-specific heterologous interactions (Fig. 4B,C). Centromere–telomere interactions were not detected in chicken, whose telomere–telomere interactions were conspicuous instead.
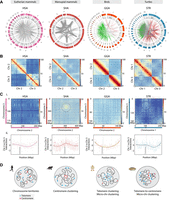
Divergent patterns of nuclear architecture. (A) Circos diagrams depicting interchromosomal interactions in human (Homo sapiens [HSA], Rao et al. 2014), Tasmanian Devil (Sarcophilus harrisii [SHA], Álvarez-González et al. 2022a), chicken (Gallus gallus [GGA], Fishman et al. 2019), and Northern Giant Musk Turtle (Staurotypus triporcatus [STR], this study). In each plot, the chromosomes are illustrated in the outer colored track, whereas the inner gray track illustrates the histogram of the absolute value of the interaction at each genomic position. The color of the links at the center of each Circos plot indicates different types of interactions: full set of interactions (gray links), interactions among macrochromosomes (red links), and interactions among microchromosome (green links). (B) Hi-C contact maps representing a pair of macrochromosomes for human (HSA, Rao et al. 2014), Tasmanian Devil (SHA, Álvarez-González et al. 2022a), chicken (GGA, Fishman et al. 2019), and Northern Giant Musk Turtle (STR, this study). Note the centromeric interactions in the Tasmanian Devil, the telomeric interactions in the chicken, and the telomeric–centromeric interactions in the turtle highlighted by dashed-line circles. (Ci) Heatmaps showing normalized interchromosomal interactions between heterologous chromosomes in human (Chromosomes 2 and 3), Tasmanian Devil (Chromosomes 2 and 3), chicken (Chromosomes 2 and 3), and Northern Giant Musk Turtle (Chromosomes 1 and 2). Telomeric, centromeric, and telomeric–centromeric interactions are highlighted by dashed-line circles. The position of centromeres is denoted by black lines for each chromosome. (Cii) Z-score interactions as a function of genomic distance for representative chromosomes of human, Tasmanian Devil, chicken, and musk turtle. Dashed horizontal lines denote the centromere location. (D) Representation of chromosome position inside nuclei of species showing chromosomal territories (CTs) without centromere clustering (human), centromere clustering (Tasmanian Devil), telomere clustering (chicken and musk turtle), and telomere–centromere associations (musk turtle). Centromeres are depicted in red and telomeres in blue.
Overall, we detected the presence of at least four different patterns of nuclear chromosomal architecture across species (Fig. 4D). Eutherian mammals (i.e., human) are characterized by the presence of chromosomal territories, whereas marsupials (i.e., Tasmanian Devil) present a divergent clustering of centromeres, as previously described (Álvarez-González et al. 2022a). Among sauropsids, we could distinguish two different patterns in birds versus turtles. Namely, while microchromosome clustering was common in both lineages, turtles exhibited telomere-to-centromere interactions, not present in birds (i.e., chicken) or other previously studied amniotes (Hoencamp et al. 2021). Consistent patterns of chromosomal interactions were detected in G. evgoodei (Rhie et al. 2021) when compared to Apalone and Staurotypus (Supplemental Fig. S4). Collectively, the difference identified among lineages in our results suggest that nuclear architecture is more diverse in vertebrates than initially thought.
Genomic compartmentalization features within vertebrates
We then scored both A/B compartments (“open”/“closed” chromatin, respectively) and TADs detected in Apalone and Staurotypus based on Hi-C data, and compared them to human, Emu and chicken (Fig. 5; Supplemental Fig. S5). Broadly, A compartments correspond to “open” chromatin and positive values for the first eigenvector of the contact matrix (see Methods), whereas “closed” chromatin B compartments have negative values. Here, the genome-wide distribution of A/B compartments was concordant across taxa, with ∼50% of the genome corresponding to A compartments (Fig. 5A; Supplemental Fig. S5A). Moreover, microchromosomes exhibited a greater proportion of A compartments and higher values of compartment strength (two-tailed t-test P < 0.001) (Fig. 5A; Supplemental Fig. S5B), indicating that microchromosomes have higher compartmentalization and are composed of more “open” chromatin (concordant with their relatively higher gene content) than macrochromosomes. These results mirror previous studies (Fishman et al. 2019; Perry et al. 2021), and are consistent with the higher values of inter-/intrachromosomal interaction ratios displayed by microchromosomes (Fig. 3C). We note that no differences between species were found when comparing TAD boundary strength genome-wide (Supplemental Fig. S5B).
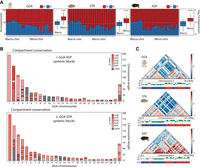
Genome compartmentalization in chicken and turtles. (A) Bar plots depicting the percentage of each chromosome assessed to be an A (red) or B (blue) compartment, distinguishing between macro and microchromosomes. A two-tailed t-test between macro and microchromosomes is also illustrated for each species; (****) P < 0.001 in all cases. (B) Chicken (Gallus gallus [GGA]) chromosome ideograms color-coded according to the compartment conservation of homologous syntenic blocks between GGA and Apalone spinifera (ASP) (upper panel), and between GGA and Staurotypus triporcatus (STR) (lower panel), expressed as P-value scores (statistically significant P-values are denoted in gray). Asterisks in Chromosome 2 indicate an exemplar syntenic region illustrated in detail in (C). (C) Enlargement of a structural conserved syntenic region between GGA, STR, and ASP Chromosome 2. Compartment conservation is represented by the compartments heatmap and the distribution of values of its first eigenvector, whereas TAD conservation is represented by the contact heatmaps and triangles delineated by black lines. Genome-wide TADs and A/B compartments are presented in Supplemental Figure S4.
Conservation of the higher-order chromatin organization in chicken and turtles
To test whether the higher-order structural organization of genome architecture was conserved in chicken and turtles, we considered the homologous syntenic blocks between Gallus and Apalone and between Gallus and Staurotypus (Fig. 2A), and compared their first eigenvector values, whereas TAD conservation was evaluated by comparing their insulator scores (Fig. 5B,C; Supplemental Fig. S6A,B). We found relatively modest differences at the A/B compartment level between chicken and turtles. Namely, 81.3% of the Apalone and 85.1% of the Staurotypus genomes have a conserved A/B compartment structure with the Gallus genome (Fig. 5B,C; Supplemental Fig. S6A). Moreover, no differences were observed in the same pairwise comparisons for TADs at 500 kbp resolution (>90% of which were conserved in syntenic blocks) (Fig. 5B,C; Supplemental Fig. S6A,B). Altogether, our results suggest that the higher-order chromatin organization is mainly conserved at lower levels of resolution (i.e., TADs) but not at intermediate and high levels of resolution (i.e., A/B compartments and chromosomal position within the nucleus), mirroring previous observations in mammals (Álvarez-González et al. 2022a).
Discussion
Understanding the function and evolution of vertebrate genomes requires deciphering patterns of chromosomal changes that accrue among lineages along with their 3D chromatin folding features. The initial view that the hierarchical 3D genome organization (i.e., TADs, A/B compartments, and chromosomal territories) is extensively conserved across mammals derives from studies conducted in Boreoeutherian species (human, mouse, dog, macaque, and gibbon) (Rao et al. 2014; Rudan et al. 2015; Dixon et al. 2016; Lazar et al. 2018). Recent studies in highly divergent mammalian orders (i.e., Afrotherians and marsupials) unveiled two important features of mammalian 3D genome organization and conservation: (1) the existence of divergent patterns of chromosome occupancy and (2) different degrees of structural conservation among species (Álvarez-González et al. 2022a). Yet, whether these novel patterns hold for nonmammalian vertebrates was untested until now. Our de novo genome assemblies of A. spinifera and S. triporcatus provide insight into patterns of genome reshuffling in sauropsids (avian and nonavian reptiles) and help unveil broader fundamental principles of 3D chromosome folding across amniotes (Rao et al. 2014; Fishman et al. 2019; Liu et al. 2021).
New genomic resources for hidden-neck turtles with genotypic sex determination
The first turtle genome assemblies were published over a decade ago for Chrysemys picta (Shaffer et al. 2013), C. mydas, and P. sinensis (Wang et al. 2013) (referred to by their genus names hereafter). Since then, twelve highly contiguous genome assemblies of chelonians have been reported, nine of them annotated (Mauremys mutica, Mauremys reevesii, Gopherus flavomarginatus, G. evgoodei, Malaclemys terrapin, T. scripta, Caretta caretta, C. mydas, and Dermochelys coriacea), and three unannotated (Rafetus swinhoei, Pelochelys cantorii, and Carettochelys insculpta) (Simison et al. 2020; Rhie et al. 2021; Liu et al. 2022a,b, 2023; Ren et al. 2022; Bentley et al. 2023; Chang et al. 2023; Li et al. 2024). These assemblies include large scaffolds (N50 ≈ 140 Mb), of which the five to six largest (L50) contain 50% of the assembly, whereas 90% of the assembly is contained in a more variable number of scaffolds (L90 = 16–24) (Supplemental Fig. S1B).
The two novel genome assemblies generated in this study add to this list, and constitute an important genomic resource for the scientific community as they represent two cryptodiran turtle lineages with independently evolved sex chromosomes of contrasting heterogamety, an XX/XY system (Staurotypus) (Sites et al. 1979), and a ZZ/ZW system (Apalone) of GSD (Badenhorst et al. 2013), and the first assembly for a Kinosternoid turtle and for a turtle with male heterogamety (Staurotypus). The Apalone assembly constitutes a significant improvement from the annotated Trionychid genome also assembled using Illumina sequencing a decade ago for Pelodiscus (Wang et al. 2013), which are the two softshell turtles most studied for eco-evo-devo-genomics in this family. Our de novo assemblies for Staurotypus and Apalone are comparable to the eleven highly contiguous assemblies listed above in N50 and L50, whereas their total size is smaller (Supplemental Fig. S1B), due in part to the underrepresentation of repetitive elements (Fig. 1B) and GC-rich regions which are notoriously difficult to assemble (Peona et al. 2021), especially with Illumina sequencing. In particular, repetitive DNA was less abundant in the assemblies of Staurotypus and Apalone compared to the more contiguous genomes of Trachemys or G. evgoodei obtained with PacBio or Nanopore longer read sequencing, respectively (Fig. 1B; Simison et al. 2020; Rhie et al. 2021). This is also evident in the larger size of the recently published and yet unannotated genome assemblies of the softshell turtles R. swinhoei (Ren et al. 2022) and P. cantorii (Liu et al. 2023), also produced by long-read sequencing (Supplemental Fig. S1B). Likewise, GC-rich microchromosomic regions were underrepresented in the Staurotypus and Apalone assemblies, which include portions of the sex chromosomes and the nucleolar organizing region (NOR) that is sex-linked in both of these GSD turtles (Montiel et al. 2016, 2022). The higher L90 observed in Apalone when compared to Staurotypus was expected given the high number of microchromosomes present in softshell turtles (Trionychidae) (Montiel et al. 2016).
Conserved ancestral synteny of the linear genome in softshell turtles
Our comparative genomic analysis strongly supports the existence of extensive conservation in synteny and collinearity between chicken and the Spiny Softshell Turtle (Apalone), both of which shared a distinct abundance of microchromosomes (Fig. 2A,B). This finding is consistent with earlier molecular cytogenetic comparisons of chicken to Apalone (O'Connor et al. 2018), or between chicken and the macrochromosomes of Pelodiscus (Matsuda et al. 2005) and Trachemys (Supplemental Table S2; Kasai et al. 2012). Combining the earlier data (O'Connor et al. 2018) with the synteny between the microchromosomes of chicken and Apalone reported in this study (Fig. 2A; Supplemental Table S3), we can conclude that Apalone, a representative of the hidden-neck turtle family Trionychidae which is sister to all other cryptodiran turtles (Crawford et al. 2015; Thomson et al. 2021), retained a karyotype likely close to the ancestral state for turtles. This conservation, however, does not extend to all chelonians, as multiple fusions occurred in more derived lineages that lead to the reduction in diploid number in Staurotypus (Kinosternidae) and Trachemys (Emydidae). Indeed, Chromosomes 7, 8, 9, 10, 14, 16, and 18 in Staurotypus and their homologs in Trachemys result from the combination of two distinct chromosomes found separate in Apalone (Fig. 2A,C). Furthermore, while there is extensive syntenic conservation between Trachemys and Staurotypus (Fig. 2A,B), additional chromosome fission/fusions events were also detected between them (Fig. 2C). Additionally, the larger scale fusion/fission events between Apalone and Staurotypus were outnumbered by intrachromosomal inversions between homologous chromosomes that took place during their ∼184 Myr of independent evolution (Thomson et al. 2021).
Lastly, our results indicate that the high conservation in synteny observed between the avian Z sex chromosome and human (Nanda et al. 1999) or squamate (Pokorná et al. 2011, 2012) chromosomes extend to chelonians as well (Fig. 2A). As such, GGA-Z is retained mostly intact in Apalone (ASP-6) and Trachemys (TSC-6), and as the pseudo-autosomal region in Staurotypus (STR-4XY) (Fig. 2; Kawagoshi et al. 2014; Montiel et al. 2016). We note that the X-limited portion of the sex chromosomes of Staurotypus which is evolutionarily derived (Sites et al. 1979), as well as a large portion of the ZW sex chromosomes of Apalone, were not represented in our assemblies. In both these cases, the missing genome portion contains the NOR which is sexually dimorphic in both species (Kawagoshi et al. 2014; Montiel et al. 2016), has been used as a sex diagnostic in Apalone (Literman et al. 2014), and is subject to contrasting modes of dosage compensation (Montiel et al. 2022) that differ from the dosage compensation affecting the rest of the Z in Apalone (Bista et al. 2021).
Nonrandom density of genes and GC content by chromosome size
We also observed that Apalone and Staurotypus microchromosomes are GC-rich and gene-rich, whereas macrochromosomes are heterogeneous in GC content (Fig. 1C,D), a pattern detected earlier in a subset of genes of Pelodiscus softshell turtles (Kuraku et al. 2006). Furthermore, peaks of GC content coincide with peaks of gene content along the chromosomes (Fig. 1C; Supplemental Fig. S2), which is expected given the positive correlation between these two variables. Further, Apalone and Staurotypus coincide in displaying an elevated GC content toward the end of the chromosomes likely from the telomeric regions that are GC-rich, as observed in chicken (Andreozzi et al. 2001). Yet these two turtle species differ in that elevated GC content is also observed in Staurotypus around the “scars” of ancestral chromosome fusions detected in STR-5, STR-7, STR-8, STR-9, and STR-10. The higher GC content in these regions may be due to higher gene content or to telomeric sequences leftover in the middle of these fused chromosomes in Staurotypus, although such interstitial telomeric DNA was detected only in STR-1 and STR-2 macrochromosomes via molecular cytogenetics (Montiel et al. 2016).
Earlier studies suggested that mammals, birds, amphibians, and reptiles shared a conserved pattern where GC-richer regions/chromosomes have a much less compact chromatin structure and are located toward the center of the interphase nucleus compared to GC-poorer regions (Saccone et al. 2002; Federico et al. 2006). It was further hypothesized that the overall higher GC content of endotherms compared to ectotherms evolved to stabilize open chromatin in the face of warmer mammalian body temperatures, in genomic regions not already stabilized by compact chromatin structure (Saccone et al. 2002). In turn, avian microchromosomes were proposed to be counterparts of these mammalian GC-rich chromosomal segments found within chromosomes in a relatively relaxed chromatin structure (Andreozzi et al. 2001). But our findings contradict these models. Indeed, the conserved karyotype between Apalone (ectotherm) and Gallus (endotherm), the homology of their microchromosomes and with the microchromosomes of Staurotypus, as well as the results from our 3D chromatin analyses presented below, all counter these hypotheses.
Turtle chromosome folding reveals novel 3D chromatin structural pattern in vertebrates
While some significant evolutionary events were uncovered in the linear genomes of turtles, including fusion/fission events described above (Fig. 2), the most profound changes revealed in our study are the changes to the 3D chromatin structure found in chelonians compared to other vertebrates.
Indeed, our data provide evidence for the existence of at least four different chromosome folding patterns within vertebrates. These included (1) chromosome territories, (2) centromere clustering, (3) telomere clustering in combination with clustering of microchromosomes, and (4) telomere-to-centromere clustering in combination with clustering of microchromosomes. Consistent with previous studies (Hoencamp et al. 2021; Álvarez-González et al. 2022a), eutherian chromosomes were organized into chromosomal territories, with marsupials depicting conspicuous centromere clustering (Fig. 4D). As for sauropsids, all species studied thus far (chicken, emu, tegu, rattlesnake, Python, and four turtles—Trachemys, C. mydas, Apalone, and Staurotypus) exhibited microchromosome association (this study and Waters et al. 2021), probably positioned toward the center of the nucleus (Fig. 4D), as previously suggested by cytogenetic studies (Cremer and Cremer 2001; Waters et al. 2021). This nuclear positioning would enable the greater interactions observed among microchromosomes (Fig. 3B), consistent also with their higher proportion of “open” chromatin (Fig. 5A) that is more transcriptionally active (Supplemental Fig. S1A), higher A compartment strength (Supplemental Fig. 4B), and associated denser gene content and higher GC content (Fig. 1D) when compared to macrochromosomes. We highlight that the combination of microchromosome interactions and telomere-to-centromere clustering observed in turtles is a novel pattern not previously reported for amniotes (Hoencamp et al. 2021). The overall low inter-/intrachromosomal interaction ratios observed in sauropsids are indicative of relatively more compacted chromatin when compared to all mammals (Fig. 3C) as reflected also in the higher distance-dependent interactions within chromosomes (Fig. 3D). Given the phylogenetic position of turtles and birds, who shared a common ancestor ∼260 Mya (Kumar et al. 2022) and split from synapsid reptiles that gave rise to mammals ∼325 Mya (Shedlock and Edwards 2009), and that microchromosomes are building blocks of bird, reptile, and mammal chromosomes (Waters et al. 2021), we propose that the nuclear configuration in the form of microchromosomal association is an ancestral state for amniotes. Whether this association is the ancestral state for all vertebrates remains an open question. If true, such an ancestral nuclear position would also suggest functional coherence, providing genome integrity since sauropsids are characterized by more stable diploid numbers compared to mammals (Ruiz-Herrera et al. 2012). In fact, the cryptodiran turtle species reported here also show relatively lower rates of genome rearrangements than mammals (Damas et al. 2022). Further studies are needed to test whether our findings extend to the Pleurodira turtle suborder.
In conclusion, our study provided an evolutionary view of the 3D genome folding patterns in turtles, contributing new insights into chromatin structure in nonmodel vertebrates. Through an integrative approach combining de novo assemblies and Hi-C data sets with the use of comparative genomics and cytogenetics, it was possible to infer lineage-specific reorganization events in turtles, capturing their divergence from Archosaurian (birds + crocodilian) reptiles, and inferring a putative ancestral chromosome folding state for amniotes. We can only speculate on the mechanisms responsible for this chromosome configuration in turtles. For instance, the contrasting patterns among vertebrate lineages may be mediated by the idiosyncratic evolution of TEs which are known to affect 3D chromatin structure and chromosomal interactions across mammals (Diehl et al. 2020; Choudhary et al. 2023; Lawson et al. 2023). Despite the challenges associated with using nonmodel species, future analysis with increasing species representation is warranted to test this hypothesis. Additionally, understanding the dynamics of chromatin conformation through 3D modeling will be crucial for decoding the structural plasticity of vertebrate genomes.
Methods
Sample collection
A female juvenile A. spinifera was sampled for this study and corresponds to the individual collected in Iowa as described previously (Gessler et al. 2023), whose genome assembly is available from the NCBI BioProject database (https://www.ncbi.nlm.nih.gov/bioproject/) under accession number PRJNA837702. Additionally, an adult male S. triporcatus from a captive private collection was sampled as well. In 2017, blood from both individuals was drawn using 1/10 volume of 2% sodium EDTA as an anticoagulant, flash frozen in liquid nitrogen, and sent to Dovetail Genomics (now Cantata Bio) for processing as described below. Another six Staurotypus hatchlings (three males and three females) were collected for a parallel study, and RNA-seq data obtained therein from heart, ovary, and testis were used for genome annotation as described below. Likewise, RNA-seq data obtained from male and female hatchlings and adults for a previous study of Apalone (Bista et al. 2021) were used to annotate its genome assembly. All animal procedures were approved by the IACUC of Iowa State University.
De novo assemblies of the Apalone and Staurotypus genomes
Initial de novo assemblies of the Apalone and Staurotypus genomes were generated from Illumina sequencing (110.8× and 72.1× coverage, respectively) (Supplemental Table S1), and improved by scaffolding with Chicago library sequencing and Dovetail Hi-C library sequencing data using HiRise v.2016 (Putnam et al. 2016), and manually curated using Hi-C and the Juicer/3D-DNA pipeline (Dudchenko et al. 2017), as described in detail in the Supplemental Methods.
Because the HiRise Apalone assembly thus obtained lacked the sex chromosome, a Z/W scaffold was identified by its homology with chicken GGA-15 (Kawagoshi et al. 2009; Badenhorst et al. 2013) from a publicly available fragmentary Apalone assembly (GCA_000385615.1) after scaffolding it using Ragtag2 v.2.1.0 followed by an Omni-C scaffolding as described by Alonge et al. (2022), lifted, and added to the de novo HiRise assembly for the comparative genomic analyses. BUSCO v.5.2.2 (Manni et al. 2021) was run on the genome assemblies with odb09 vertebrate, eukaryote, and tetrapod data sets to assess the completeness and quality of the genome assembly. Additional curation (beyond that performed with Hi-C and Juicer/3D-DNA) included the partial mapping of genome scaffolds to their physical chromosomal location via fluorescence in situ hybridization (FISH) onto metaphase chromosomes of Apalone and Staurotypus of BAC clones with known sequences from BAC library VMRC CHY3 of painted turtles (C. picta) (Supplemental Fig. S3; Shaffer et al. 2013), following our standard protocols (Badenhorst et al. 2015; Lee et al. 2020). This way, five errors in the genome assembly inferred by incongruence between BAC-FISH information or Hi-C data and scaffold sequences could be detected and manually corrected. Identifying specific microchromosomes cytogenetically is challenging by definition as their absolute assignment is somewhat arbitrary due to their equivalent small size, and thus, potentially subject to future modification.
Genome annotation and interspecies chromosome homology
The genome annotation for both species followed a combination approach including an evidence-based annotation that uses RNA-seq data and an ab initio annotation approach implemented once in Augustus/BRAKER v.3.0.3 (Stanke et al. 2006, 2008; Hoff et al. 2016, 2019; Brůna et al. 2021). The Mikado pipeline v.2.3 (Venturini et al. 2018) was used for the evidence-based approach which integrated multiple genome-guided transcriptome assemblies, namely: Trinity v.2.10.0 (Haas et al. 2013), Cufflinks v.2.2.1 (Trapnell et al. 2010), StringTie v2.1.4 (Pertea et al. 2015), and CLASS v2.2.3 (Song et al. 2016) along with robust splicing junction derived using Portcullis v.1.2.3 (Mapleson et al. 2018). The BRAKER pipeline used a combination of ab initio approach like GeneMark-ET and AUGUSTUS, and combined it with extrinsic evidence from RNA-seq and protein homology information. The final gene models reported by the two approaches were filtered based on homology with the protein sequences of the T. scripta elegans genome (Simison et al. 2020). The S. triporcatus annotation included 12 lllumina HiSeq RNA-seq libraries sampled from six hatchlings (three males and three females, heart and gonads). RNA-seq for A. spinifera included 24 libraries sampled from the liver, blood, and gonads of six hatchlings (three males, three female), and the liver and gonads of three adults (one males and two females), and corresponded to the transcriptomes reported earlier (Bista et al. 2021; Gessler et al. 2023).
The gene models from the annotations of the G. gallus (Warren et al. 2023) and T. scripta elegans (Simison et al. 2020) genomes, along with the assembled genomes, were used as the input for the synteny analysis. Circos plots (Krzywinski et al. 2009) were created using LAST v.1205 (Kielbasa et al. 2011) and homologous syntenic blocks were identified using GENESPACE v.1.2.1 (Lovell et al. 2022), an R package for synteny- and orthology-constrained comparative genomics along with the riparian plots. BAC-FISH data and WCP data from the literature or obtained here by flow-sorting Trachemys chromosomes also provided information about the homology of chromosomes between chicken and Apalone/Trachemys (Kasai et al. 2012; O'Connor et al. 2018), between Apalone/Staurotypus and Chrysemys (Badenhorst et al. 2015), and in turn, between Apalone/Staurotypus and Trachemys (Lee et al. 2020). Briefly, WCP data are obtained by separating individual metaphase chromosomes via flow cytometry and labeling them for subsequent FISH on metaphase chromosomes of the same or other species (Ferguson-Smith et al. 1998). We flow-sorted Trachemys chromosomes at the flow-cytometry facility of Iowa State University (Supplemental Fig. S3). We followed the chromosome nomenclature presented in Montiel et al. (2016) for all turtles, but note that because microchromosomes are by definition particularly difficult to distinguish from each other, the assignment of scaffolds to specific physical microchromosomes was somewhat arbitrary.
Estimation of broad repeat and transposable element content
To construct TE libraries for the two de novo assemblies of Apalone and Staurotypus, plus those of Trachemys and Gopherus used for comparison, RepeatModeler v.2.0.5 (RM2, Flynn et al. 2020) was executed using the TEtools Docker image available at GitHub (https://github.com/Dfam-consortium/TETools), with default parameters. Then, MCHelper v.1.6.5 (Orozco-Arias et al. 2023) was executed to automatically curate each of the four TE libraries. Briefly, MCHelper reduces redundancy, extends consensus sequences, filters false positives, does homology and structural checks, and classifies originally “unknown/unclassified” elements. To run the filtering step, we used the sauropsida_odb10 BUSCO data set. Additionally, we also annotated satellite sequences. For this, we first added the satellite sequences identified by RepeatModeler2 to the MCHelper library. Then, we ran RepeatMasker v.4.1.2-p1 (Smit et al. 2015) using the -lib parameter, and all other parameters by default. We used the OneCodeToFindThemAll script (Bailly-Bechet et al. 2014) to defragment the copies in each genome annotation. We calculated the TE genomic proportions directly from the outputs of the OneCodeToFindThemAll script, which summed up all the bases corresponding to each TE order divided by the total assembly size. Satellites, simple repeats, and low complexity repeat proportions were obtained from the RepeatMasker output, since the OneCodeToFindThemAll only accounts for TEs. No annotations were detected for PLEs, Helitron, and Crypton TE orders.
Hi-C data processing, binning, and normalization
Quality metrics of the Hi-C data used in this study per species is presented in Supplemental Table S5.
Hi-C data processing was conducted using the Juicer v.1.6.2 pipeline (Durand et al. 2016) with default parameters. At least 100 M reads per species were mapped against the curated genomes using BWA v.0.7.17 (Li 2013). Duplicated reads were removed with Juicer. Hi-C matrixes were generated in HIC format and transformed into COOL format using hicConvertFormat from HiCExplorer v.3.7 (Wolff et al. 2020). Cool matrices were created at 500 kbp resolution and corrected following the ICE method using “hicCorrectMatrix” from HiCExplorer v.3.7. Then, for comparison purposes, matrixes of both turtle species were normalized to 50M reads using hicNormalize HiCExplorer v.3.7. All Hi-C maps were plotted with hicPlotMatrix from HiCExplorer v.3.7 using a logarithmic scale.
Averaged contact probability P(s) curves
Using as an input the corrected 500 kbp matrices normalized to 50 M counts, the contact probability versus distance curves [P(s)] for each chromosome were calculated and averaged per species using “hicPlotDistvsCounts” from HiCExplorer v.3.7. The curves were plotted using Rstudio v.4.2.0 and maximum distance of 1 Gbp.
Interchromosome and intrachromosome interaction analysis
Using the program “hicConvertMatrix” from HiCExplorer v.3.7, intra- and interchromosomal interactions were obtained by converting the corrected and normalized 500 kbp matrices from h5 format to ginteractions format. The inter-/intrachromosome interaction ratio was then determined using Rstudio v.4.2.0 (R Core Team 2019) to calculate the mean number of inter- and intrachromosomal interactions per chromosome.
First eigenvector and insulator score calculation
Hi-C sequencing of cross-linked interacting DNA regions bound to proteins revealed the interaction frequency among genomic regions by their read abundance. The first eigenvector of this contact matrix reflects the overall tendency of each genomic region to interact with regions of similar or different activity: Positive values of the first eigenvector typically correspond to A compartments (active or open chromatin), and negative values to B compartments (inactive or closed chromatin), while higher absolute values denote greater compartment definition (compartment strength). On the other hand, insulation scores (points of no contact, i.e., score ≈ 0) were calculated from the read depth of this contact matrix to interpret how well-defined TADs are. This score measured the insulation strength of each boundary by assessing the ratio of interactions within the TAD compared to interactions across the TAD boundary (Lajoie et al. 2015). Both the first eigenvector and insulator score values were obtained using the Hi-C analysis tools package, FAN-C v.0.9.1 (Kruse et al. 2020). The first eigenvector was calculated using the “fanc compartments” tool, on the normalized 500 kbp matrices with default settings. The compartment matrices were produced using “fancplot.” The tool “fanc insulation” was used to calculate the insulator score on normalized 500 kbp matrices and the following commonly used window sizes (Dixon et al. 2012; Phillips-Cremins et al. 2013; Rao et al. 2014; Schmitt et al. 2016; Álvarez-González et al. 2022a): 1 Mbp, 1.5 Mpb, 2 Mbp, and 3 Mbp.
TAD calling
TADs were called using the tool find_tads from TADbit v.1.0 (Serra et al. 2017) with default parameters and normalized 500 kbp matrixes. Only TAD boundaries with a TADbit score >6 were considered robust (Serra et al. 2017) and included in downstream analyses.
Conservation of the higher-order chromatin organization between species
To evaluate the higher-order structural conservation between pairwise syntenic blocks, we conducted pairwise comparisons of the first eigenvector and insulator score values. Our comparative analysis included syntenic blocks larger than 2 Mbp, each spanning at least four 500 kbp Hi-C bins, as the average TAD size was 1 Mbp in the species analyzed. We then performed pairwise comparisons by testing the similarity of the average first eigenvector and insulator score values between pairs of homologous syntenic blocks, which were extracted from the genome-wide data using BEDTools intersect (Quinlan and Hall 2010). The significance of these pairwise differences was assessed using a two-tailed t-test at an alpha = 0.01 (a conservative 99% confidence level), such that homologous syntenic blocks with a P-value > 0.01 were considered conserved in their chromatin structure.
Data access
All sequencing data generated in this study have been submitted to the NCBI BioProject database (https://www.ncbi.nlm.nih.gov/bioproject/) under accession number PRJNA1021228.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
This work was funded in part by National Science Foundation grants IOS 1555999 and IOS 2127995 to N.V., by the Ministry of Economy, Industry and Competitiveness (CGL2017-83802-P to A.R.-H.), the Spanish Ministry of Science and Innovation (PID2020-112557GB-I00 funded by AEI/10.13039/501100011033 to A.R.-H.), the Agència de Gestió d'Ajuts Universitaris i de Recerca, AGAUR (2021SGR00122 to A.R.-H.) and the Catalan Institution for Research and Advanced Studies (ICREA). L.A.-G. was supported by an FPI predoctoral fellowship from the Ministry of Economy and Competitiveness (PRE-2018-083257). J.G. was supported by grant PID2020-115874GB-I00 funded by MICIU/AEI/10.13039/501100011033 and by grant 2021 SGR 00417 funded by Departament de Recerca i Universitats, Generalitat de Catalunya.
Author contributions: N.V., B.B., and A.R.-H.: conceptualization; N.V., A.R.-H., and J.G.: supervision; B.B., L.G.-R., L.A.-G., Z.Q.W., E.E.M., L.S.L., D.B.B., S.R., R.L., B.N.-D., and S.O.-A: investigation; J.B.I.: resources; J.G., A.R.-H., and N.V.: funding; B.B., L.G.-R., L.A.-G., A.R.-H., and N.V.: writing of the original draft; all authors: writing review and editing.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.279443.124.
-
Freely available online through the Genome Research Open Access option.
- Received April 5, 2024.
- Accepted September 20, 2024.
This article, published in Genome Research, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








