
Evidence for compensatory evolution within pleiotropic regulatory elements
- Zane Kliesmete1,
- Peter Orchard1,2,
- Victor Yan Kin Lee1,3,
- Johanna Geuder1,
- Simon M. Krauß1,4,
- Mari Ohnuki1,5,
- Jessica Jocher1,
- Beate Vieth1,
- Wolfgang Enard1 and
- Ines Hellmann1
- 1Anthropology and Human Genomics, Faculty of Biology, Ludwig-Maximilians Universität München, 82152 Munich, Germany;
- 2Department of Computational Medicine and Bioinformatics, University of Michigan, Ann Arbor, Michigan 48109-2218, USA;
- 3Section for Molecular Ecology and Evolution, Globe Institute, University of Copenhagen, 1350 Copenhagen, Denmark;
- 4Department of Hematology, Cell Therapy, Hemostaseology and Infectious Diseases, University Leipzig Medical Center, 04103 Leipzig, Germany;
- 5Faculty of Medicine, Institute for the Advanced Study of Human Biology (ASHBi), Kyoto University, Kyoto 606-8501, Japan
Abstract
Pleiotropy, measured as expression breadth across tissues, is one of the best predictors for protein sequence and expression conservation. In this study, we investigated its effect on the evolution of cis-regulatory elements (CREs). To this end, we carefully reanalyzed the Epigenomics Roadmap data for nine fetal tissues, assigning a measure of pleiotropic degree to nearly half a million CREs. To assess the functional conservation of CREs, we generated ATAC-seq and RNA-seq data from humans and macaques. We found that more pleiotropic CREs exhibit greater conservation in accessibility, and the mRNA expression levels of the associated genes are more conserved. This trend of higher conservation for higher degrees of pleiotropy persists when analyzing the transcription factor binding repertoire. In contrast, simple DNA sequence conservation of orthologous sites between species tends to be even lower for pleiotropic CREs than for species-specific CREs. Combining various lines of evidence, we propose that the lack of sequence conservation in functionally conserved pleiotropic CREs is owing to within-element compensatory evolution. In summary, our findings suggest that pleiotropy is also a good predictor for the functional conservation of CREs, even though this is not reflected in the sequence conservation of pleiotropic CREs.
One of the initial perplexing revelations of the human genome project was the seemingly limited number of genes, which did not align with the increase in complexity compared with organisms such as yeast, worms, and flies. It became evident that this complexity must stem from gene regulation, with the probability that most genes play roles in multiple contexts throughout development and in various tissues. Considering the varying contexts of utilization in terms of location as well as timing, it follows that mutations within the same gene can exert influence on multiple traits. This phenomenon is widely recognized as pleiotropy. In a molecular context, pleiotropy is frequently measured as the number of tissues in which a gene is expressed, a metric called expression breadth (Hastings 1996; Duret and Mouchiroud 2000).
The advent of microarrays and subsequent RNA-seq technology allowed for an impartial, genome-wide evaluation of expression breadth. As data accumulated, it became evident that expression breadth is in fact a very good predictor of the conservation of protein sequences. In particular, the ratio of the nonsynonymous over synonymous substitution rate shows that pleiotropic genes tend to be more conserved than tissue-specific genes (Hastings 1996; Duret and Mouchiroud 2000; Zhang and Li 2004). Moreover, the amount of sequence constraint associated with expression in a given tissue can vary considerably: Genes expressed in the brain tend to be more conserved than genes specific to other tissues, such as the liver (Kuma et al. 1995; Khaitovich et al. 2005; Wang et al. 2007). A similar pattern emerges in terms of expression level conservation: Brain-expressed as well as pleiotropic genes tend to have more similar expression levels across species than other genes (Khaitovich et al. 2005; Brawand et al. 2011; Wang et al. 2020).
Naively, one would expect that a higher level of expression conservation would be achieved via a higher level of activity and sequence conservation of the associated cis-regulatory elements (CREs). However, most enhancers are tissue specific (Gasperini et al. 2020) and show low activity conservation across species, although the target gene expression appears conserved (Paris et al. 2013; Villar et al. 2014; Berthelot et al. 2018). In contrast to the majority of tissue-specific CREs, the activity of more pleiotropic CREs indeed appears to be more conserved across species (Roller et al. 2021).
In line with this pattern, previously identified highly active pleiotropic enhancers in humans were found to have higher sequence conservation than tissue-specific enhancers across a large phylogeny (Andersson et al. 2014; Singh and Yi 2021). Of note, this analysis included only a couple of hundred enhancers, using a rather stringent definition of pleiotropy. Similar trends were found analyzing human population data, that is, a much shorter evolutionary timescale (Huang et al. 2017).
Promoters are much more likely to be functionally conserved than enhancers (Berthelot et al. 2018). In addition, promoters are more pleiotropic than enhancers, which is probably because core promoters are more restricted in their spatial genomic location than enhancers, which can be located megabases away from the targeted transcription start sites (TSSs) (Villar et al. 2014; Andersson and Sandelin 2020). Promoters are further distinguished by their shape: Broad promoters are large, thought to harbor multiple TSSs, and tend to be more pleiotropic. In contrast, narrow promoters are small, probably have only one TSS, and are more likely to be tissue specific (Andersson and Sandelin 2020). Furthermore, evidence suggests that expression from broad promoters is less noisy and more robust toward mutations (Carninci et al. 2006; Schor et al. 2017; Sigalova et al. 2020; Floc'hlay et al. 2021), and in humans, these broad promoters also show strong enrichment for CpG islands (Morgan and Marioni 2018). At least in flies, this results in the counter-intuitive observation that although broad promoters are more robust and thus also more likely to be functionally conserved across species, overall, they exhibit lower sequence conservation between species than do narrow promoters (Schor et al. 2017). In summary, the relationship between pleiotropy and sequence conservation for CREs appears to be much more complicated than that between pleiotropy and protein-coding sequence conservation.
Here, we investigate the impact of pleiotropy on sequence and functional conservation in primates. To gauge pleiotropy, we thoroughly reanalyzed DNase hypersensitivity data from nine primary fetal tissues (Bernstein et al. 2010), integrating across seven or more biological replicates per tissue to also identify tissue-specific CREs robustly. To assess functional conservation of the identified CREs, we obtained RNA-seq and ATAC-seq data from two human and two cynomolgus macaque neural progenitor cell lines (NPCs). Furthermore, we obtained four different measures of sequence conservation: (1) a population genomic measure, (2) a conservation measure for the human lineage since the most recent common ancestor of humans and chimpanzees (Gronau et al. 2013), (3) a conservation score calculated for the primate phylogeny (Pollard et al. 2010), and (4) a scaled measure of transcription factor binding site (TFBS) conservation.
Results
To investigate different aspects associated with varying degrees of regulatory pleiotropy, we identified putative CREs as DNase hypersensitive sites (DHSs) in the Roadmap Epigenomics data, which provide comparable experiments for a wide selection of tissues (Bernstein et al. 2010). To ensure reproducibility, we included only tissues for which at least seven biological replicates of DNase-seq data were available, resulting in a set of nine tissues: adrenal gland, brain, heart, kidney, large intestine, lung, muscle, stomach, and thymus (Fig. 1A,B). Hence, in this study, we focus on the regulatory evolution across somatic tissues. We called DHS for each tissue separately using a peak caller that utilizes replicate information to gauge certainty (Ibrahim et al. 2015), resulting in more than 1.1 million DHSs ranging from about 80,000 sites detected in the large intestine to about 175,000 sites detected in the stomach (Fig. 1C). Of note, the number of detected DHSs does not correlate strongly with the number of replicates across tissues (Pearson's ρ = 0.23, P-value = 0.55).
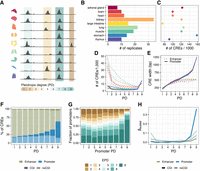
Study overview. (A) Open chromatin and expression data from the Roadmap Epigenomics Project (Bernstein et al. 2010) were used to infer the effect of pleiotropy on sequence and TFBS evolution, as well as associated gene expression in primates. Overlapping DHS peaks between tissues were merged to determine the degree of tissue specificity per CRE. (B) DHS data from nine human fetal tissues. The number of biological replicates per tissue varies between seven and 34. (C) The number of CREs per tissue varies 2.3-fold. There is no association between the number of replicates and the number of accessible regions per tissue. (D) Most enhancers (dotted line) are tissue specific, whereas promoters (solid line) are mostly pleiotropic. (E) CRE length increases with the number of tissues, particularly at the promoters. This increase was also observed at the peak level prior to merging (Supplemental Fig. S1A). (D,E) The colors represent the tissues as introduced in A,B. (F) The majority of PD9 CREs are CpG island promoters (solid blue), whereas tissue-specific elements are rarely CpG islands and mainly enhancers (transparent green). (G) Pleiotropic promoters are more commonly associated with pleiotropic gene expression patterns. The promoter pleiotropic degree (PD) indicates the highest PD of the associated promoters per gene. The y-axis shows the proportions of those x-categories (promoter PD) with associated gene expression pleiotropy ranging from one to nine. (EPD) Expression pleiotropic degree. (H) Scaled coefficients of a linear mixed model to predict gene expression levels using distance scaled CRE counts of different types.
In analogy to how expression breadth has been used as a proxy for pleiotropy of genes, we merge overlapping DHS from different tissues and define the pleiotropic degree (PD) as the number of tissues in which we found a DHS if this DHS was significantly differentially accessible (DA) compared with tissues without a DHS at this location, resulting in about 443,000 union CREs stratified by PD (Supplemental Methods). We distinguish promoters and enhancers based on genomic distance, in which we designate CREs within 2 kb of an active, annotated TSS (GENCODE v.32) as promoters and all other CREs within 1 Mb as enhancers (McLean et al. 2010; Fishilevich et al. 2017). Consistent with expectations, the majority of enhancers are tissue specific (PD1) (Gasperini et al. 2020), whereas promoters are more likely to be pleiotropic (PD9), and CREs with an intermediate PD (1 < PD < 9) are generally rare (Fig. 1D). With a median size of 1.2 kb, PD9 promoters are the largest CREs (Fig. 1E; Supplemental Fig. S1A). Also, the overlap among DHS is highest in PD9 promoters (Supplemental Fig. S1B), suggesting that their larger size is owing to a higher information content rather than being an artifact of concatenation. A large proportion of the pleiotropic promoters are CpG islands (76.7%), and the proportion of CpG island promoters increases with increasing PD (Fig. 1F). The same is true for enhancers, although enhancers are only very rarely CpG islands (3.2%).
Next, we wanted to investigate whether the PD of CREs has an impact on the expression of the associated genes. To this end, we integrated DHS with gene expression estimates from matching samples from the Epigenomics Roadmap Project (Fig. 1G; Supplemental Fig. S2). As expected, we find a good correspondence between promoter PD and gene expression PD (χ2-test P-value < 2.2−16). Specifically, PD9 promoters are associated with genes that are expressed in all nine tissues (Pearson residual = 34.1, rank 1), and we also find tissue-specific promoters to be associated with tissue-specific genes (Pearson residual = 14.7, rank 2) (Fig. 1G; Supplemental Fig. S1C). We further modeled the associated gene expression using the presence of different CpG and non-CpG island CREs of varying PDs as predictors (Fig. 1H; Supplemental Fig. S1D). The total amount of variation in expression levels that can be explained by the model is 24% (CI: 23.8%–24.3%), whereas the same model with a shuffled PD label can only explain 19% (CI: 18.8%–19.8%; see Methods). Inspecting the scaled coefficients of the mixed-effect model suggests that PD9 promoters have the largest activating effect on expression, followed by PD9 and PD1 enhancers.
Characterization of TFBS repertoire across PDs
Under the premise that CREs regulate gene expression by binding transcription factors, we continued by characterizing TFBS associated with CREs of varying PDs. To this end, we collected nonredundant position-weight matrices (PWMs) of 643 binding motifs (Fornes et al. 2020) belonging to 561 TFs that we found to be expressed in at least one of the investigated tissues (Fig. 2A). Almost half of all expressed TFs (237 out of 561, 42%) were present in all tissues, namely, pleiotropic, whereas 94 (17%) showed tissue-specific expression. In agreement with others (Vaquerizas et al. 2009), we found that the brain has the highest proportion of tissue-specific TFs (8.5%).
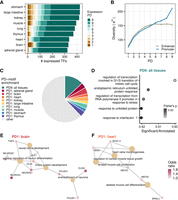
TFBS repertoire diversity and enrichment across tissue-specific and pleiotropic CREs. (A) An overview of the PD of TF expression (EPD) across tissues. (B) TFBS repertoire diversity increases with PD, particularly across promoters. Depicted are mean values ± SEM. (C) Overview of the overrepresented motifs in PD9 and PD1 CRE sequences. Depicted are binding site preferences for 643 motifs, the respective TFs of which are detected in the expression data. If a motif showed the highest binding proportion to PD9 CREs consistently within each tissue-CRE, it was assigned as overrepresented in PD9. If a motif showed the highest binding proportion to a PD1 of a particular tissue, but not that of others, it was assigned as PD1 enriched for that tissue. (D) Top five categories of gene set enrichment analysis of PD9-enriched motifs using all motifs as background (Gene Ontology, biological process, Fisher's exact P-value < 0.05). (E,F) Top four categories of gene set enrichment analysis of tissue-specific PD1 enriched motifs using all motifs as background (Gene Ontology, Biological process, Fisher's exact P-value < 0.05). (E) Brain-specific PD1 overrepresented motifs. (F) Heart-specific PD1 overrepresented motifs. (E,F) Odds ratio is based on the proportion of tissue-specific PD1 CREs with the motif over the global average proportion for that motif.
Next, we evaluated the overall binding potential of a TF to a CRE using Cluster-Buster (see Methods) (Frith et al. 2003). We found that TFBS diversity increases with pleiotropy for both enhancers and promoters (Fig. 2B). When normalizing TFBS diversity by the CRE width and yielding a measure for TFBS density, we observe decreasing density with increasing PD (Supplemental Fig. S6A; for GC content normalization, see Supplemental Fig. S6B). This suggests that the complex binding repertoire in PD9 CREs is mainly achieved through their increasing width. Next, we investigated whether tissue-specific and pleiotropic CREs have binding sites for the same TFs or whether preferences exist. For most TFs (62%), we do not find an enrichment of predicted TFBS in any PD category (Fig. 2C). Of the remaining motifs, 159 (24.9%) are overrepresented in CREs that are specific for one of the tissues, and 84 (13.1%) motifs are enriched in the PD9 CREs. Gene-set enrichment analysis shows that overrepresented TF motifs in brain-specific CREs are associated with neural differentiation (Fig. 2E). Most prominently, this is driven by OLIG2, which is essential for oligodendrocyte development (Zhou and Anderson 2002; Jakovcevski et al. 2009; Yu et al. 2013), as well as by NEUROD1, NEUROD2, and NEUROG1, which are important for neuron development (Olson et al. 2001; Sun et al. 2001; Messmer et al. 2012; Pataskar et al. 2016). Other tissues also show an enrichment for TFBS of tissue-specific TFs: For example, TFBS that are overrepresented in heart-specific CREs include motifs of MEF2C, TBX20, and NKX2-5 (Fig. 2F), which are essential for cardiac muscle development (He et al. 2011; Schlesinger et al. 2011; Grunert et al. 2016).
In contrast, PD9 CREs contain more predicted binding sites for TFs associated with basic cellular processes, such as transcription regulation in connection with cell cycle and stress response (Fig. 2D). These motifs are more GC-rich and tend to have a higher information content than PD1-enriched motifs or motifs without any preference (Supplemental Fig. S6C,D). In addition, these elements are enriched for the so-called “stripe” TFs (odds ratio = 10.53, Fisher's exact P-value = 3−12) (Zhao et al. 2022), which include SP, KLF, and ZBTB family members.
The impact of pleiotropy on the evolutionary conservation of regulatory activity
To investigate the relationship between CRE PD and their evolutionary conservation, we generated RNA-seq and ATAC-seq data from iPSC-derived NPCs from humans and cynomolgus macaques (Supplemental Fig. S3A,B; Supplemental Methods). We intersected the detected genes and accessible peaks with the processed Epigenomics Roadmap data to assign a PD to genes and peaks (Fig. 3A). Of about 66,000 and about 69,000 called peaks in human and macaque NPCs, 76.8% and 64.3% show an overlap with our tissue-based CRE annotations, respectively (for the cross-species liftOver strategy, see Methods) (Supplemental Fig. S3C). In line with the expectation that pleiotropic CREs are more conserved, the amount of CRE overlap with NPC ATAC-seq peaks increases with increasing PD and is generally higher for promoters than for enhancers (Fig. 3B). Moreover, the activity of PD9 CREs is also more conserved between humans and macaques: Of all overlapping PD9 CREs, 88% was detected to be active in NPCs from both species, whereas this was the case for 15% of the PD1 CREs. Of note, the observed association between PD and activity conservation in human and macaque NPCs is not only owing to the increased regulatory activity that might generate a higher probability for PD9 elements being detected as peaks (Fig. 3B). Instead, even without stratifying by whether a peak was called, we observe a decrease in differential activity with increasing PD, measured by absolute log2-fold changes using DESeq2 (Fig. 3E; Love et al. 2014). A very similar conservation pattern emerges when we leverage the data from Roller et al. (2021) that encompasses CRE activity measures for 10 mammalian species in four tissues. To begin with, the assigned PD in our study that was solely based on human tissues is similar to the CRE PD in other primates, that is, rhesus macaque and marmoset, and is also consistent with the average level of pleiotropy across all 10 mammalian species (Supplemental Fig. S4A–D). Moreover, also the CRE activity conservation across the 10 species confirms that a higher PD is associated with evolutionarily conserved activity (Fig. 3C,D).
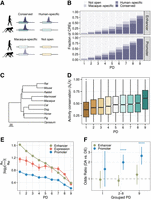
PD and evolutionary conservation of expression and regulatory activity. (A,B) The fraction of enhancers and promoters of different PDs as defined using data from nine tissues from the Epigenomics Roadmap Project, which overlapped with ATAC-seq peaks called in neural progenitor cell lines (NPCs) from cynomolgus macaques and humans. The colors indicate whether a human DHS-derived CRE overlapped with an NPC ATAC-seq peak from humans, cynomolgus macaques, both, or none. (C) The phylogeny from Bininda-Emonds et al. (2007) of species for which Roller et al. (2021) provided activity estimates for four tissues based on histone marks. Scale, million years ago. (D) Activity conservation for orthologous CREs based on Roller et al. (2021) (n = 47,377). We calculate the ratio subtree length λS for species in which the CRE is active over the total tree length λT. (E) Mean absolute log2-fold changes of gene expression and activities between humans and cynomolgus macaques. Error bars, 95% bootstrap CIs. PD9 genes (CREs) show more conserved expression (activity) than do more tissue-specific genes (CREs). (F) Enrichment (odds ratio > 1) of differentially accessible CREs with differentially expressed genes between humans and cynomolgus macaques using matched regulatory landscapes (n = 6882). Error bars, 95% CIs of the odds ratio. Asterisks indicate BH-adjusted Fisher's test P-value: (*) <0.05, (**) <0.01, and (***) <0.001.
To investigate the effect of differentially active CREs on the expression of associated genes, we test whether genes with a DA CRE (BH-adjusted P-value < 0.1) are also more likely to be DE (BH-adjusted P-value < 0.1; |log2-fold change| > 1) in our NPCs. When comparing the effect of CREs from different PD categories, PD9 promoters have a larger impact on the DE status of the associated gene than do PD1 promoters, whereas the difference between enhancers from different PD categories is nonsignificant (Fig. 3F). In summary, in agreement with Roller et al. 2021, we also find that the activity of pleiotropic CREs is evolutionarily more conserved between species than the activity of tissue-specific CREs. Moreover, if the activity of a pleiotropic promoter changes, such changes are more likely to have downstream effects, that is, to impact the expression of associated genes.
Cross-species sequence conservation is lowest in pleiotropic CREs
Up to this point, pleiotropy shows the expected effect on gene regulation and CRE activity conservation. Here, we investigate how this functional conservation is reflected in the underlying DNA sequence. We focus on three measures of sequence conservation: (1) recent selection quantified as the number of weakly deleterious sites in humans (E.W.) (Fig. 4A,B; Gronau et al. 2013), (2) selection on the lineage since the most recent common ancestor of humans and chimpanzees, quantified as the fraction of sites under (strong) negative selection ρ (Fig. 4C,D; Gronau et al. 2013), and (3) the average phyloP and phastCons scores across a primate phylogeny (Supplemental Fig. S5A,B; Supplemental Methods; Pollard et al. 2010). The main difference among the three measures is the evolutionary time across which sequence conservation is averaged. Because PD was assessed in human samples, the E.W. measure provides the closest match to our measure of pleiotropy. For ρ, phyloP, and phastCons, we average the strength of selection over longer evolutionary times. It should be noted that variants emerging within a population may undergo recombination, whereas mutations occurring after speciation remain on separate haplotypes. In line with our expectations, we indeed find that the number of E.W.s increases with the PD for both promoters and enhancers (Fig. 4A). This observation aligns with the conservation of CRE accessibility: Across all PD categories, we observe a higher prevalence of weakly deleterious sites in CREs that are open in both species (Fig. 4B). In contrast, when using ρ as a measure of conservation, we only find a higher sequence conservation for tissue-specific CREs (PD1–PD3) with conserved accessibility, whereas it appears that accessibility conservation is not reflected in the sequence conservation of pleiotropic CREs (Fig. 4D). Overall, ρ suggests that PD9 CREs have the lowest fraction of negatively selected sites compared with other PD categories (Fig. 4C). These patterns persist also when inferring E.W. and ρ for the CGI and non-CGI fractions of the PD categories separately (Supplemental Fig. S5C,D). This result also remains when we use the average phyloP or average phastCons score across a 10-species primate phylogeny as a measure of conservation, which confirms PD9 CREs as the PD category with the lowest cross-species sequence conservation (Supplemental Fig. S5A,B). In summary, even though the number of weakly deleterious sites within a CRE increases with pleiotropy in humans, this is not reflected in sequence conservation across species.
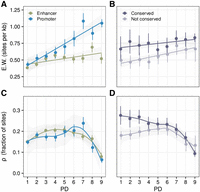
CRE sequence conservation patterns across varying degrees of pleiotropy. (A,B) Number of weakly deleterious sites in humans (E.W.) inferred from human polymorphisms. It increases with increasing PD. (A) Separated by enhancers/promoters. (B) Separated by human–macaque accessibility conservation in NPCs. (C,D) Fraction of sites under (strong) negative selection on the lineage since the MRCA of humans and chimpanzees is the highest at the intermediately specific CREs and lowest in the pleiotropic CRE sequences. (C) Separated by enhancers/promoters. (D) Separated by human–macaque accessibility conservation in NPCs. (A–D) Depicted are mean values ± SEM.
Tissue-specific effects on CRE sequence conservation
So far, we have not considered the possibility that different tissues might impose different amounts of constraint to the DNA sequence of the CRE. By separating CREs by the tissues in which they are utilized, we find that brain CREs are clearly under more constraint than those of other tissues. Nevertheless, also for the brain, the number of weakly deleterious sites increases with PD, showing that, although to smaller amounts, activity in other tissues still adds to the overall constraint (Fig. 5A). Again, this is not true when considering substitutions on the human lineage as used in the measure ρ (Fig. 5B). Here, brain-specific CREs show the most constraint, much more than pleiotropic PD9 CREs, which by definition are utilized also in the brain. To exclude the possibility that the brain effect on the PD9 elements is diluted by the merging of DHS across tissues, we contrast the ρ of the brain peak sequence with adjacent sequences that are part of the same merged CRE but are open only in other tissues (Fig. 5C). For PD9 CREs, brain peak sequences show lower sequence conservation on the human lineage than the adjacent sequence utilized only by other tissues, whereas for less pleiotropic CREs, the part that is used in the brain is under much more constraint (Fig. 5D). In summary, even though we find tissue-specific effects, this cannot explain the overall pattern of the relatively low sequence conservation of pleiotropic CREs. It remains that for pleiotropic CREs, there is no simple relationship between sequence and functional conservation between species.
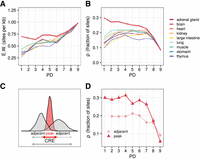
CRE sequence conservation patterns per tissue. (A) Weak negative selection inferred from human polymorphisms, separated by the tissue that utilizes the CREs. (B) Fraction of sites under (strong) negative selection, separated by the tissue that utilizes the CREs. (C) Brain CRE sequences were separated into peak and adjacent sequences. (D) The part of the sequence that is used by the brain shows a considerably higher fraction of sites under negative selection than the respective adjacent sequences. (A,B,D) Depicted are mean values ± SEM.
Pleiotropic CRE TF repertoire is conserved, not the binding sites
To explain the apparent mismatch between functional and sequence conservation in PD9 CREs across primates, we continued to
analyze levels that are intermediate between sequence conservation (farther from function) and accessibility conservation
(closer to function), which are CpG content, TFBS repertoire, and position conservation between human CREs and their orthologous
sequences in cynomolgus macaques. To begin with, we find that the conservation of CpG and GC content increases with PD and
is highest for pleiotropic promoters (Supplemental Fig. S5E,F). This coincides with the increase of the proportion of CpG island CREs (Fig. 1F) and suggests that the CpG island property at high PD CREs is conserved across species, thereby showing patterns closer
to the functional side. Next, for each CRE, we calculated the binding potential for all expressed TF motifs and approximated
TFBS repertoire conservation using the average pairwise Canberra distance between species (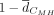 ). When we contrast CREs with conserved and nonconserved openness between humans and macaques, we find that functionally conserved
CREs also show higher repertoire conservation across all PD categories (Fig. 6B).
). When we contrast CREs with conserved and nonconserved openness between humans and macaques, we find that functionally conserved
CREs also show higher repertoire conservation across all PD categories (Fig. 6B).
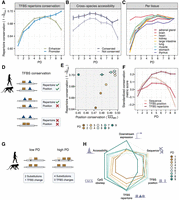
TFBS repertoire and position conservation between orthologous human and macaque CREs. (A–C) TFBS repertoire conservation across PDs. Depicted are mean values ± SEM. (A) TFBS repertoire conservation increases with higher PD among promoters and decreases slightly at high PD enhancers. (B) CREs that overlap NPC peaks with conserved openness show higher TFBS repertoire conservation than species-specific NPC peaks.
(C) TFBS repertoire conservation differs across tissues, in which the brain shows the highest conservation at lower PDs. (D) Simplified schematic of the scenarios of repertoire and position conservation. (E) TFBS position conservation versus repertoire conservation across PD categories. Depicted are mean values ± SEM. (F) Standardized scores (z-scores) of sequence (primate phyloP), TFBS repertoire, and binding site conservation between human and cynomolgus macaque.
(G) A schematic depicting how lower sequence conservation might lead to higher TFBS repertoire conservation through compensatory
mechanisms. (H) A summary of the scaled average conservation metric scores across PDs. Sequence, primate phyloP scores; TFBS position, 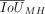 ; TFBS repertoire,
; TFBS repertoire, 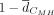 ; CpG observed/expected,
; CpG observed/expected, 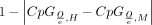 ; accessibility, 1 − |LFC| of NPC-DA; and downstream expression, 1 − |LFC| of NPC-DE.
; accessibility, 1 − |LFC| of NPC-DA; and downstream expression, 1 − |LFC| of NPC-DE.
TFBS repertoire conservation generally increases with pleiotropy in all tissues (Fig. 6C). There is a simple relationship for promoters for which repertoire conservation is highest for PD9 and lowest for PD1 CREs (Fig. 6A). Also for enhancers, albeit less pronounced, the average repertoire conservation in PD9 CREs (0.66, SEM 0.166) is considerably higher than in PD1 CREs (0.62, SEM 0.241), which is in contrast to what we observed for sequence conservation, again showing higher similarity to the patterns of functional conservation (Fig. 3E). To validate our findings about TFBS repertoire conservation, we integrated our PD scores with ChIP-seq data on four liver TFs in five mammalian species (Supplemental Fig. S7A,B; Supplemental Methods; Ballester et al. 2014). Consistent with our estimates, experimentally inferred TF binding is also more conserved if it resides within pleiotropic CREs compared with binding in liver-specific CREs (Supplemental Fig. S7C,D).
To understand how the high repertoire conservation is achieved for PD9 CREs in spite of having the lowest sequence conservation, we analyzed the positional conservation of TFBS as a third intermediate metric using Jaccard similarity index (IoUMH) (Fig. 6D). We find that the average repertoire conservation appears to be unrelated to the positional conservation in high PD categories (Fig. 6E). The pattern of positional conservation across PD categories resembles the pattern that we observe for sequence conservation (Fig. 6F). In summary, although CRE sequence and TFBS positions are the least conserved in PD9 elements, CpG content and TFBS repertoire are in agreement with the more functional metrics—accessibility and expression conservation—in that they show the highest conservation in PD9 elements. These patterns are consistent with a mechanism of compensatory evolution (Fig. 6G).
The PD9 promoter of ATXN3 gene as an example
To illustrate the within-CRE compensatory evolution of TFBS within a PD9 promoter, we took a closer look at the promoter of the ubiquitously expressed protein-coding gene ATXN3. It is an important factor for the regulation of the degradation of damaged proteins (Schmitt et al. 2007; Gao et al. 2015; Feng et al. 2018). This gene plays a role in the brain, as its malfunction can lead to neurodegenerative diseases such as spinocerebellar ataxia (Evers et al. 2014). The ATXN3 promoter has relatively low sequence conservation (34th percentile) and low TFBS binding site conservation (49th percentile) but has high TFBS repertoire (77th percentile), accessibility, and expression conservation (Fig. 7A–E).
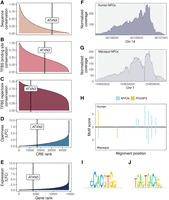
Ranks of ATXN3 PD9 promoter compared with other CREs in terms of the following: (A) sequence conservation (mean phastCons), (B) TFBS binding site conservation, (C) TFBS repertoire conservation, (D) CRE openness conservation between human and cynomolgus macaque in NPCs, and (E) ATXN3 gene expression conservation between human and cynomolgus macaque in NPCs. (F) ATXN3 PD9 promoter ATAC-seq read coverage in human NPCs. (G) ATXN3 PD9 promoter ATAC-seq read coverage in macaque NPCs. (F,G) ATXN3 PD9 promoter is accessible in NPCs from both species. Gray background area indicates the positions of the called ATAC-seq peak in the respective species. (H) ATXN3 promoter shows diverged TFBS positions between species among two TFs (MYCN, POU3F2) with validated binding based on ChIP-seq neural cell data that are involved in neurogenesis. Motif binding sites and scores are depicted in the aligned sequence space: human at the top and cynomolgus macaque at the bottom. (I,J) PWM logos from JASPAR2020 vertebrate core set of the investigated TF motifs with ChIP-seq data available: MYCN (I) and POU3F2 (J).
To investigate a few likely relevant TFs closer, we overlapped our TFBS data with published ChIP-seq data from human neural cells available in the GTRD database (Yevshin et al. 2019) and visualized the binding sites of the two TFs (MYCN, POU3F2) annotated to be involved in neurogenesis (Gene Ontology biological process term GO:0022008) (Fig. 7H). Both of their motifs are moderately complex as shown by their information content (MYCN: IC = 11.8, POU3F2: IC = 13.7) (Fig. 7I,J). Both promoter orthologs show high binding potential for both TFs. Humans have six and macaques have five MYCN binding sites, and both have one POU3F2 binding site, which is also reflected in similar ATAC-seq peak-shapes (Fig. 7F,G). However, only three of the 10 binding sites are positionally conserved between the species. This serves as an example of how the large disagreement between sequence, TF binding site conservation, and TFBS repertoire might co-occur.
Discussion
Pleiotropy has been shown to be the best predictor of both protein coding sequence conservation (Hastings 1996; Duret and Mouchiroud 2000; Zhang and Li 2004) and gene expression levels (Khaitovich et al. 2005; Brawand et al. 2011; Wang et al. 2020). Here, we investigate the effect of pleiotropy on the evolution of CREs. In agreement with the findings by Roller et al. (2021), we find that measures close to CRE function, such as accessibility and TFBS repertoire conservation, indeed show the expected higher conservation for more pleiotropic CREs. Similarly, a measure of conservation based on human diversity data also shows a trend for higher conservation in more pleiotropic CREs. However, we found that this is not reflected in the sequence and TFBS positional conservation between species (Fig. 6H). These observations imply that a simple model of purifying selection alone is insufficient to explain the effect of pleiotropy on CRE evolution and suggest a role for compensatory evolution.
Zooming into tissue effects, in line with previous investigations on brain evolution (Kuma et al. 1995; Wang et al. 2007; Brawand et al. 2011; Roller et al. 2021), we find that brain-specific CREs show by far the highest sequence conservation irrespective of the measure. Therefore, we would expect that pleiotropic CREs, which are by definition also open in the brain, show the highest sequence conservation. However, looking at between-species sequence conservation, the subsequences of PD9 CREs that are open in the brain are even less conserved than the adjacent sequences (Fig. 5D). This confirms the notion that the structure and evolution of PD9 CREs are inherently different, in that it allows for functional conservation without much sequence conservation.
Indeed, several basic structural properties of PD9 elements distinguish them from less pleiotropic CREs. They tend to be larger and have more CpGs and a higher GC content. Moreover, PD9 elements show an overrepresentation of GC-rich motifs that is associated with TFs that tend to be involved in more basic cellular processes. Among those, we also find enrichment for binding sites of a recently described group of highly cooperative TFs that prolong CRE openness (Zhao et al. 2022). It should also be noted that the majority of PD9 CREs are promoters, and PD9 promoters share many properties with broad promoters that were defined via the size of CAGE peaks (Andersson et al. 2014). Even though this classification is based on a completely different concept, broad promoters were also shown to be more pleiotropic, active, and CpG-rich. Indeed, as observed for PD9 CREs, broad promoters also have an increased substitution rate. Moreover, broad promoters have been shown to be more robust than narrow promoters in that they show less expression noise across haplotypes in Drosophila (Schor et al. 2017; Floc'hlay et al. 2021). Similarly, CpG island promoters have been found to induce more stable expression in humans (Morgan and Marioni 2018). Mechanistically, this picture fits with the notion that GC-rich regions facilitate combinatorial binding (Zhao et al. 2022), which has been shown to lead to evolutionarily more stable TF binding across closely as well as distantly related mammalian species (Stefflova et al. 2013; Ballester et al. 2014). In the same vein, Hagai et al. (2018) found that the regulatory response of genes associated with CpG islands to an immune stimulus is more conserved than that of genes associated with a TATA-box. In summary, there is ample evidence that large CpG island promoters are functionally robust while having high substitution rates.
We also find high substitution rates in PD9 enhancers, which share many features with PD9 promoters. Moreover, promoter and enhancer functionality frequently switches over evolutionary time (Roller et al. 2021). Hence, we suggest that the main differences in the evolutionary patterns observed for promoters and enhancers are closely linked to their degree of pleiotropy. Most enhancers show strong tissue preferences, placing them in our PD1 category. Consistent with multiple other studies (Schmidt et al. 2010; Berthelot et al. 2018; Danko et al. 2018; Roller et al. 2021), we find that only a relatively small fraction of enhancers is conserved between species in terms of accessibility and that this fraction is strongly enriched for pleiotropic CREs irrespective of their classification as enhancers or promoters. In fact, CRE conservation across the genome is so low (Doniger and Fay 2007; Horton et al. 2023) that a simple evolutionary model cannot explain it. Instead, the presence of proto-enhancers is required (Tuğrul et al. 2015; Emera et al. 2016).
In addition, the observed high CRE turnover rates appear to be inconsistent with the relatively low rates of change in gene expression levels. This discrepancy has prompted the proposal of compensatory evolution as a prevalent mechanism for CREs (Schmidt et al. 2010; Berthelot et al. 2018). The phenomenon of CREs at nonorthologous genomic positions in different species exhibiting the same function and being able to compensate for one another has been documented in detail for several cases (Ludwig et al. 2000; Domené et al. 2013; Arnold et al. 2014; Emera et al. 2016). It is related to the observation that a lot of function is encoded redundantly within a gene's regulatory landscape by so-called shadow enhancers (Hong et al. 2008; Wunderlich et al. 2016; Osterwalder et al. 2018). Osterwalder et al. (2018) showed that the deletion of one strong enhancer did not have an effect on the phenotype as long as the shadow enhancer was still active. This clearly demonstrates the presence of epistasis. If multiple similarly fit haplotypes coexist and different ones can get fixed in different species, this would facilitate compensatory evolution across CREs.
Other properties of CREs suggest that there is also a lot of epistasis within the same CRE. The billboard model (Kulkarni and Arnosti 2003; Arnosti and Kulkarni 2005) and the TF-collective model (Junion et al. 2012) of enhancer activity suggest that two CRE haplotypes with shifted but similar TFBSs should be functionally equivalent. It follows that the mutations that create these two haplotypes will also have nonadditive effects on fitness. Moreover, some studies showed that binding to a high-affinity site is facilitated by many neighboring low-affinity binding sites (Crocker et al. 2016), thus providing the raw material for high TFBS turnover rates (Tuğrul et al. 2015).
Combining all the evidence, we suggest that within-element compensation of TFBS is a common mode of evolution for pleiotropic CREs. This mode of evolution would explain the apparent disparity between the cross-species and within-species sequence conservation (Fig. 4). Moreover, it would also explain the disparity between the low sequence and the high functional conservation between species as observed in our ATAC-seq and RNA-seq data: If different, functionally equivalent haplotypes got fixed in different species, this should lead to high sequence divergence, whereas the open chromatin state and downstream gene expression remain conserved (Fig. 6H). Furthermore, we show that this is achieved through binding repertoire, not binding site conservation. In summary, we think that compensatory evolution is a prevalent mode for evolution of regulatory elements. Although for tissue-specific CREs compensation occurs between CREs over larger genomic distances, for pleiotropic CREs, compensation mainly occurs within the same element, leading to high sequence divergence.
Methods
CRE effect on gene expression across tissues
GENCODE v.32 (Harrow et al. 2012) was used to identify TSSs (5′ of transcripts) of the expressed genes (RNA-seq RPKM > 1 in 50% of the samples) in each tissue. CREs within 2 kb of a TSS are designated as promoters and are associated with all TSSs within that distance. CREs within 1 Mb of a TSS are deemed enhancers and are associated with the two closest TSSs in each direction, unless the distance to one TSS is >10× smaller than to the other TSS; in that case, only the closest TSS is assigned. In total, 397,228 CREs (89.6%) were assigned.
Log mean expression is estimated using a linear mixed-effect model with tissues as a random effect and the distance-to-TSS-weighted
(d) numbers of CpG Island and non-CpG Island promoters and enhancers as fixed effects: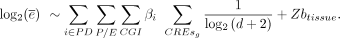 To assess the effect of PD, we shuffled the PD labels across all CREs.
To assess the effect of PD, we shuffled the PD labels across all CREs.
Cross-species gene expression and accessibility analysis
Previously generated iPSCs from humans (Homo sapiens) and cynomolgus macaques (Macaca fascicularis) (Geuder et al. 2021) were differentiated to NPCs via dual-SMAD inhibition (Chambers et al. 2009; Ohnuki et al. 2014).
To generate RNA-seq data, we used three clones of three human individuals and four clones of two cynomolgus macaque individuals. After 5 days of differentiation, the cells showed characteristics of neural progenitors and were harvested at days 5, 7, and 9. cDNA libraries were generated using prime-seq (Janjic et al. 2022) and processed with zUMIs (Parekh et al. 2018). Human samples were mapped to GRCh38, GENCODE v.32. Cynomolgus samples were mapped to macFas6 (Jayakumar et al. 2021), and for gene annotation, we transferred human GENCODE v.32 to macFas6 using Liftoff (Shumate and Salzberg 2021). Only genes from GRCh38 detected also in macFas6 were further included. Genes with UMI counts in at least 28.57% (six of 21) samples were kept, resulting in a 14,608 gene set.
To generate ATAC-seq data, iPSCs of two clones from two human individuals and two clones from two cynomolgus macaque individuals were differentiated as described above. Libraries were generated using the Omni-ATAC protocol (Corces et al. 2017) with minor modifications. Reads were mapped to GRCh38 and macFas6 using BWA-MEM2 (Vasimuddin et al. 2019). Peak calling was done using Genrich (https://github.com/jsh58/Genrich). Reciprocal liftOver (RLO) was used to convert CRE coordinates from GRCh37 to GRCh38 and from GRCh38 to macFas6. For example, coordinates from GRCh38 were converted to macFas6 coordinates and defragmented by merging all hits within 40 bp. The resulting macFas6 region was then reciprocally converted to GRCh38 coordinates, and we kept only CREs for which those coordinates overlapped with the original ones. We identified RLO matches for 99.7% CREs in GRCh38 and 88% in macFas6 and removed CREs with a RLO match width beyond [1.2 × GRCh37; 0.8 × GRCh37] and 31 CREs that contained N's. This resulted in an orthologous coordinate set containing 385,111 CREs.
Macaque ATAC-seq reads in macFas6 and human reads in GRCh38 were counted within the RLO PD-CRE coordinates. Only CREs that overlapped with an ATAC-seq peak by 10% relative to the width of both the PD-CRE and the ATAC-seq peaks in at least one species were kept for differential accessibility (DA) analysis (n = 65,753). Differential gene expression (DGE) and DA analyses were performed separately using DESeq2 (Love et al. 2014), using species as the predictor (BH-adjusted P-value < 0.1). For DGE, we also require |log2-FC| > 1.
To analyze the relationship between CRE DA and gene DE, for each gene's landscape, we quantified the number of DA and non-DA CREs in each of the six categories: assignment (promoter/enhancer) × PD1/PD2–PD8/PD9. To investigate what effect DA of a certain CRE group has on the odds of being associated with a DE gene, we chose matching landscapes that differed only by the DA status of one CRE in the respective category and used a BH-corrected Fisher's exact test to detect association with DE genes. This resulted in comparing 6882 gene landscapes.
For analysis in which we used the state of the ATAC-seq peak (open/closed) as an indicator for peak conservation, we required that conserved peaks overlap by 10% of their width between human LO peaks and macaque peaks in macFas6, keeping only one-to-one, zero-to-one, one-to-zero, and zero-to-zero overlaps.
Estimation of CRE phylogenetic conservation and PD across mammals
We used cross-species histone modification ChIP-seq data from Roller et al. (2021). They contain samples from four tissues of 10 mammalian species. Because they do not contain human data, we used the macaque genome space as an anchor. Coordinates of our human PD annotations were RLO to rheMac10, yielding 426,102 RLO CREs. All macaque CRE coordinates (174,860) were collected across the nine pairwise species files. Overlapping macaque CRE coordinates carrying different functional annotations in different tissues were merged, resulting in 141,649 merged CREs. Orthologous human and macaque CREs from the two data sets were identified by requiring that the overlapping part is >10% relative to the width of both species CREs. This resulted in 47,377 (66%) one-to-one orthologs and 580 (1%) one-human-to-many-macaque and 24,160 (33.4%) many-human-to-one-macaque CREs. PD of the orthologous CRE in the other species was inferred using macaque ortholog annotation tables from Roller et al. (2021). The phylogenetic tree from Bininda-Emonds et al. (2007) was trimmed to the 10 species. Activity conservation for each CRE was calculated using relative branch length of the phylogenetic tree for that CRE versus the full 10-species tree.
Quantification of transcription factor binding
Two sets of TF PWMs of the expressed TFs in (1) nine human tissues (643 motifs from 561 TFs) and (2) our human and cynomolgus macaque NPCs (521 motifs from 446 TFs) were extracted from the JASPAR core vertebrate set (Fornes et al. 2020). These PWMs were provided to Cluster-Buster (Frith et al. 2003). In each species for each TFBS cluster of a CRE, we ranked TF motifs based on their strongest binding site. For all subsequent analyses, for each CRE, we only considered TF binding motifs that were among the 10% strongest in at least one cluster in at least one species.
Calculation of TFBS diversity
For each CRE in each species, we measured TFBS diversity by Shannon entropy (H) (Shannon 1948) in which we considered a CRE as a collection of i = 1, 2, .., n motifs of varying frequency (p):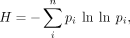 where
where 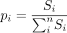 , and Si is the cumulative motif score per motif i per CRE. H is further converted to the true diversity eH (Hill 1973; Jost 2006).
, and Si is the cumulative motif score per motif i per CRE. H is further converted to the true diversity eH (Hill 1973; Jost 2006).
CRE PD ranking per motif to detect overrepresented motifs
Per tissue
We considered only expressed TF binding to CREs that are open in that tissue. For each PD category and motif, the relative
binding frequency was obtained as the fraction of CREs that have binding sites for that motif: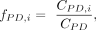 where PD indicates a PD category, i indicates a motif, CPD,i is the count of CREs with motif i binding site(s) present, and CPD is the total CRE count. Having obtained relative frequencies per PD, we ranked PD categories for each motif. Fold changes
of the binding fraction of rank-one PD relative to the average fraction for each motif i were
where PD indicates a PD category, i indicates a motif, CPD,i is the count of CREs with motif i binding site(s) present, and CPD is the total CRE count. Having obtained relative frequencies per PD, we ranked PD categories for each motif. Fold changes
of the binding fraction of rank-one PD relative to the average fraction for each motif i were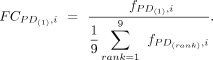
Across tissues
We focused on motifs that had the highest binding fractions (rank-one) to either PD9 or PD1. To obtain the PD9-enriched motifs, we identified TF motifs for which PD9 CREs had rank-one in all tissues. As the PD1-tissue-specific motifs, we considered the ones that have PD1 with rank-one only in that particular tissue but not in the others.
We selected universal “human stripe factors” from Zhao et al. (2022), using a detection rate cutoff of 0.9 across samples.
Calculation of TFBS repertoire conservation
The average Canberra distance for each CRE across the i = 1, 2, .., n motif cumulative scores (S) was calculated as follows: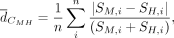 where M indicates the orthologous CRE in macaque and H in human. As a control, we shuffled CRE identifiers of the macaque profiles within the respective PD class and calculated
the average random TFBS profile similarity between species (Supplemental Fig. S6E,F).
where M indicates the orthologous CRE in macaque and H in human. As a control, we shuffled CRE identifiers of the macaque profiles within the respective PD class and calculated
the average random TFBS profile similarity between species (Supplemental Fig. S6E,F).
TFBS position overlap between human and macaque orthologous CREs
Orthologous human and macaque CRE sequences were aligned with MAFFT (Katoh and Standley 2013). Using the alignment of a CRE, the positions of TFBS with motif binding score of three or more in either species were projected
onto the common alignment space. Binding site agreement per motif i was calculated as the intersection of binding positions in base pairs between species over the union (Jaccard index) and
summarized by taking the mean across all i = 1, 2, .., n motifs that bind to the CRE: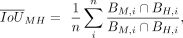 where B is a set of positions in the alignment that overlap with a binding site of motif i in the respective species macaque M or human H.
where B is a set of positions in the alignment that overlap with a binding site of motif i in the respective species macaque M or human H.
Quantification and statistical analyses
Data visualizations and statistical analysis were performed using R (version 4.2.3) (R Core Team 2023); session info is on GitHub. Details of the statistical tests performed in this study can be found in the main text and Supplemental Materials. Schematics were made using BioRender (https://www.biorender.com).
Data access
RNA-seq and ATAC-seq data generated in this study have been submitted to ArrayExpress (https://www.ebi.ac.uk/biostudies/arrayexpress) under accession numbers E-MTAB-13494 and E-MTAB-13373. A compendium containing processing scripts, tables, and detailed instructions to reproduce the analysis is available as Supplemental Code and on GitHub (https://github.com/Hellmann-Lab/Evidence-for-compensatory-evolution-within-pleiotropic-regulatory-elements). Data files and tables are available as Supplemental Material and on Zenodo (https://doi.org/10.5281/zenodo.11244831).
Competing interest statement
The authors declare no competing interests.
Acknowledgments
We thank Lucas Esteban Wange, Johannes Bagnoli, and Aleks Janjic for helping with bulk RNA-seq preparation. We also thank Paulina Spurk for contributing to the initial ATAC-seq analysis, Diego Emiliano Ruiz Navarro for contributing to the initial RNA-seq analysis, Swati Parekh for assistance with Perl scripts, and Eva Briem for helping with a schematic. We also thank Boyan Bonev for generating the liftOver files from GRCh38 to macFas6 and Andrea Betancourt for helpful discussions. We furthermore thank the reviewers whose insightful comments helped to improve the manuscript. This work was supported by Deutsche Forschungsgemeinschaft (DFG) project HE 7669/2-1 (project number 458247426) and DFG project HE 7669/1-2 (project number 407541155).
Author contributions: I.H. proposed the project and conceived the approaches of this study. W.E. provided the resources for data generation and helpful discussions. P.O. processed the human tissue accessibility data. B.V. provided expertise during initial steps. V.Y.K.L. contributed to TFBS evolutionary analyses. J.G. and M.O. generated the primate cell lines and the expression data. S.M.K. and J.G. generated the primate accessibility data. I.H. supervised the work and provided guidance in data analysis. Z.K. collected, integrated, and analyzed all data. Z.K. and I.H. wrote the manuscript. All authors read, corrected, and approved the final manuscript.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.279001.124.
-
Freely available online through the Genome Research Open Access option.
- Received January 17, 2024.
- Accepted August 19, 2024.
This article, published in Genome Research, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








