
AGAP duplicons associate with structural diversity at Chromosome 10q11.22
- Stefania Fornezza1,4,
- Vincenza Simona Delvecchio1,4,
- William T. Harvey2,
- Philip C. Dishuck2,
- Evan E. Eichler2,3 and
- Giuliana Giannuzzi1
- 1Department of Biosciences, University of Milan, 20133 Milan, Italy;
- 2Department of Genome Sciences, University of Washington School of Medicine, Seattle, Washington 98195, USA;
- 3Howard Hughes Medical Institute, University of Washington, Seattle, Washington 98195, USA
-
↵4 These authors contributed equally to this work.
Abstract
The 10q11.22 chromosomal region is a duplication-rich interval of the human genome and one of the last to be fully assembled. It carries copy number–variable genes associated with intellectual disability, bipolar disorder, and obesity. In this study, we characterized the structural diversity at this locus by analyzing 64 haploid assemblies produced by the Human Pangenome Reference Consortium. We identified 11 alternative haplotypes that differ in the copy number and/or orientation of large genomic segments, ranging from hundreds of kilobase pairs (kbp) to over one megabase pair (Mbp). We uncovered a 2.4 Mbp size difference between the shortest and longest haplotypes. Breakpoint analysis revealed that genomic instability results from nonallelic homologous recombination between segmental duplication (SD) pairs with varying similarity (94.4%–99.6%). Nonetheless, these pairs generally recombine at positions where their identity is higher (>99.6%). Recurrent inversions occur with different breakpoints within the same inverted SD pair. Inversion polymorphisms shuffle the entire SD arrangement, creating new predispositions to copy-number variations. The SD architecture is associated with a catarrhine-specific subgroup of the AGAP gene family, which likely triggered the accumulation of SDs at this locus over the past 25 million years of human evolution. Our results reveal extensive structural diversity and genomic instability at the 10q11.22 locus, and expand the general understanding of the mutational mechanisms behind SD-mediated rearrangements.
The human genome and the genomes of African apes show an enrichment of large, recent, and interspersed segmental duplications (SDs) (Marques-Bonet et al. 2009). These duplications are not evenly distributed along chromosomes but are clustered at some loci, especially pericentromeric and subtelomeric regions (Bailey et al. 2001; Giannuzzi et al. 2014). SD clusters exhibit a mosaic and complex arrangement due to the duplication of multiple segments from various regions at different times. Within these clusters, a high-copy ancestral “core duplicon” can be identified (Jiang et al. 2007). Genomic intervals with SDs are generally structurally polymorphic because of copy-number variation and inversion of some segments. They also show extensive reorganization when the human sequence is compared to the one at orthologous locations in nonhuman primate genomes (Bailey and Eichler 2006; Antonacci et al. 2014; Nuttle et al. 2016). In fact, due to the high identity between copies, SDs are substrates for nonallelic homologous recombination (NAHR) that generates structural changes of the SDs themselves as well as of the flanking unique regions (Carvalho and Lupski 2016).
Given their impact on genetic diversity, these genomic intervals are expected to significantly influence phenotypic variation and susceptibility to common diseases. Although their role in predisposing to pathogenic recurrent deletions and duplications is well established (Bailey et al. 2002; Stankiewicz and Lupski 2002; Migliavacca et al. 2015), their contribution to trait variation remains largely unexplored due to limited knowledge of their diverse structures and challenges associated with genotyping. The recent development of long-read sequencing platforms and new assembly algorithms has enhanced our ability to obtain their sequences and holds promise for overcoming these limitations (Vollger et al. 2019).
The 10q11.22 chromosomal region is one of these complex intervals of the human genome (Chaisson et al. 2015). It has an euchromatic gap in the GRCh38 human reference that derives from the incomplete assembly of a pair of SDs. The gap, which includes GPRIN2B, has been resolved by using long reads (Vollger et al. 2019) and a contiguous sequence of the region is available from the T2T (Telomere-to-Telomere) CHM13 human genome sequence (Nurk et al. 2022). The architecture of this locus is the result of several structural changes that occurred in the human lineage after divergence with the chimpanzee (Dennis et al. 2017).
Previous works showed that genes mapped on this interval, like GPRIN2 and NPY4R, are copy-number polymorphic (Handsaker et al. 2015; Vollger et al. 2019) and additional GPRIN2 copies derive from tandem duplications (Liao et al. 2023). 10q11.22 copynumber variation has been associated with obesity (Sha et al. 2009; Jarick et al. 2011; Aerts et al. 2016; Shebanits et al. 2018) and bipolar disorder (Priebe et al. 2012; Chen et al. 2016), whereas the association with intellectual disability (Cooper et al. 2011; Stankiewicz et al. 2012) was not confirmed in a subsequent study that used a larger cohort (Coe et al. 2014).
In this study, we sought to characterize structural variation at the Chromosome 10q11.22 region and gain insights into the molecular mechanisms of its genomic instability.
Results
Duplicon and gene annotation at Chromosome 10q11.22 in the T2T-CHM13 assembly
We assessed SDs, protein-coding genes, and pseudogenes annotated at Chr 10: 46,500,000–49,100,000 in the T2T-CHM13 genome assembly, as this is the only human reference having a contiguous and gapless sequence of the interval. Overall, 60% of the sequence is composed of SDs (Fig. 1A). We underline the presence of three SD pairs exhibiting inverted orientation and high sequence similarity (>99%): a 460 kbp pair containing the NPY4R/R2, GPRIN2A/2B, SYT15/15B protein-coding genes and overlapping the NPY4R-A and NPY4R-B segments; a 122 kbp pair containing the PTPN20 gene (designated as PTPN20-A and PTPN20-B segments); a 68 kbp pair containing the ANXA8 and ANXA8L1 genes (designated as ANXA8-A and ANXA8-B segments) (Fig. 1A).
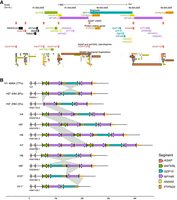
Structural variation at the 10q11.22 chromosomal region. (A) Annotation of segments, genes, and SDs. View in the UCSC Genome Browser on human T2T-CHM13 v2.0 of the region Chr 10: 46,500,000–49,100,000. The following segments are drawn: ANTXRL (green), NPY4R-A/B (violet), ANXA8-A/B (yellow), PTPN20-A/B (orange), and GDF10 (blue). ANXA8-A overlaps NPY4R-A whereas ANXA8-B is just outside the NPY4R-B segment. The location of AGAP copies (protein-coding genes and pseudogenes) is shown with red boxes. Protein-coding genes and the ANTXRL pseudogene are indicated, with the text colored according to the segment to which they belong. At the bottom, SD annotation with colors reflecting the level of similarity: light to dark gray for 90%–98%; yellow for 98%–99%; orange for similarity >99% (Numanagic et al. 2018; Vollger et al. 2022). Black rectangles mark the three pairs of inverted SDs that overlap the NPY4R, ANXA8, and PTPN20 segments, respectively. (B) Schematic of 11 alternative haplotypes identified in 64 haploid genomes. Numbers next to H1, H2, and H3 haplotypes represent their frequency. All other haplotypes were identified in a single genome. H1 corresponds to the T2T-CHM13 haplotype and is the most common. Asterisks denote haplotypes validated by the GAVISUNK tool. Gray links between haplotypes highlight the underlying structural variation.
Classification of diverse 10q11.22 structural haplotypes
We leveraged available de novo assembled and phased genomes to characterize and catalog large-scale (>50 kbp) structural variation at Chromosome 10q11.22. We first assessed which combination of sequencing technology and algorithm better performed in the phased assembly of this locus. We analyzed available sequences of the HG002 and HG00733 genomes that were obtained by using the High Fidelity (HiFi) Pacific Biosciences (PacBio) and/or Nanopore long-read sequencing technologies and the Hifiasm, HiCanu, and/or Shasta tools (Shafin et al. 2020; Cheng et al. 2021). This comparison showed that the PacBio HiFi technology combined with the Hifiasm algorithm produced a contiguous sequence in one out of four haplotypes and had a better performance compared to the other tools that never succeeded in the assembly.
Next, we analyzed our region of interest in diploid and phased assemblies for 47 samples (94 haplotypes) produced by the Human Pangenome Reference Consortium (Liao et al. 2023). These assemblies were produced with the Trio-Hifiasm assembly tool that uses PacBio HiFi long-read sequences and parental Illumina short-read sequences. The locus is assembled in a single contig in 64/94 (68%) genomes. Of these 64 sequences, 49 (77%) have the same structure as T2T-CHM13, which turns out to be the most common haplotype (H1), whereas 15 sequences have a different structure (Fig. 1B). Specifically, eight sequences (8/64 = 12%; haplotypes H2, H9, H10, and H11) display an inversion of the region between the NPY4R duplicons, hereby referred to as the GDF10 segment, which includes the PTPN20-A duplicon (Fig. 1A,B). This inversion polymorphism was previously described and shown to be a case of mutational toggling (Porubsky et al. 2020, 2022). Five haplotypes exhibit expansions (H4, H6, and H7) or contractions (H10 and H11) in either the proximal or distal copy of the NPY4R segment. We noted that whereas the ANXA8-A segment overlaps with the beginning of the NPY4R-A segment, ANXA8-B is located outside the NPY4R-B segment and overlaps the end of the GDF10 segment susceptible to inversion (Fig. 1A). The H5 variant shows a tandem duplication of the ANTXRL and NPY4R segments. We observed another structure (H3) in two assemblies that has an ∼170 kbp deletion overlapping the ANTXRL segment (Fig. 1A,B).
Overall, we identified 11 different structures of the 10q11.22 region, with the T2T-CHM13 configuration being the most prevalent. H2, H3, H5, H9, H10, and H11 haplotypes were also validated by GAVISUNK (genome assembly validation via inter-SUNK distances in nanopore reads) (Dishuck et al. 2023) using Oxford Nanopore Technologies (ONT) reads from the same samples. H8 was not assessed, whereas H4, H6, and H7 structures could not be validated due to insufficient ONT coverage across the region.
Copy number estimates of 10q11.22 genes
We sought to validate the identified diverse structures using another orthogonal approach. We assessed Illumina read-based copy number estimates of 10q11.22 genes that map to copy-number variant segments, that is, ANTXRL, NPY4R, GPRIN2, SYT15, NPY4R2, SYT15B, PTPN20, ANXA8, and ANXA8L1, as well as of ArfGAP with GTPase domain, ankyrin repeat and PH domain (AGAP) genes (Fig. 1A; Table 1). We applied both the whole-genome shotgun sequence detection (WSSD) and singly unique nucleotide k-mers (SUNK) methods in 3436 humans from 1000 Genomes Project (1KGP) and Human Genome Diversity Project (HGDP) panels, and 57 nonhuman primates. The WSSD method estimates the total copy number for sequences ≥95% identity and over 1 kbp, whereas the SUNK method estimates paralog-specific copy number.
AGAP genes annotated in the human genome (Ensembl release 110)
NPY4R, GPRIN2, SYT15, NPY4R2, and SYT15B WSSD copynumber plots looked highly similar, with copy numbers ranging from 2 to 8. Most humans carry four diploid copies, whereas all nonhuman primates have two diploid copies (the GPRIN2 plot is depicted in Fig. 2A as representative). Consistently, human values have high pairwise correlations (0.59 ≤ ρ ≤ 1, P < 2.2 × 10−16) (Fig. 2B). These data are in line with our results showing that (i) H1 with two copies of the NPY4R duplicon is the most common structure and (ii) GPRIN2A, NPY4R, and SYT15 as well as GPRIN2B, NPY4R2, and SYT15B are located within the same duplicon and are jointly duplicated or deleted.

Copy number of 10q11.22 genes. (A) WSSD or SUNK estimates of GPRIN2, ANXA8, ANTXRL, PTPN20, and AGAP4 copy number in 1KGP, HGDP, and nonhuman primate panels. The color of the line to the left of each plot corresponds to the color code of the segment where the gene is located (see Fig. 1A). (B) Spearman's correlations between copy number estimates of NPY4R/R2, GPRIN2, SYT15/15B, AGAP4, ANXA8L1, ANXA8, ANTXRL, and PTPN20. All values are significant. The color of rectangles around gene symbols corresponds to the color code of the segment where the genes are located (see Fig. 1A).
ANXA8 and ANXA8L1 WSSD copy numbers were significantly correlated with copy numbers of NPY4R duplicon genes (0.43 ≤ ρ ≤ 0.48, P < 2.2 × 10−16) (Fig. 2B). This is in line with the location of ANXA8L1 in the proximal NPY4R-A segment. ANXA8 is duplicated also in African apes (Fig. 2A).
We next weighed the ANTXRL copy number and used SUNK-based estimates, as WSSD ones seemed to be influenced by the presence of the ANTXRLP1 pseudogene, whose genomic sequence has a 92% similarity with that of ANTXRL (Fig. 1A). ANTXRL SUNK estimates showed that most humans (3209/3436, 93%) and all nonhuman primates have two diploid copies. This gene is copynumber polymorphic, with copy number ranging from 1 to 6 and ∼6% of humans (197/3436) carrying one additional copy (CN = 3) (Fig. 2A). ANTXRL copy number is moderately correlated with the copy number of genes located within the NPY4R duplicon (0.24 ≤ ρ ≤ 0.26, P < 2.2 × 10−16) (Fig. 2B); 205/214 (96%) of humans carrying more than two ANTXRL copies also carry more than four GPRIN2 copies. This is in line with the structure of H5, H6, and H7 haplotypes presenting a joint duplication of ANTXRL and NPY4R segments. The lower correlations of ANTXRL with genes mapped to the NPY4R segment compared to the correlations of genes within the NPY4R segment are consistent with our haplotype scheme, in which the NPY4R segment varies in its copy number either alone or in concert with the ANTXRL segment.
PTPN20 copy number is also variable and moderately correlated with the copy number of genes in the NPY4R segment (0.20 ≤ ρ ≤ 0.27, P < 2.2 × 10−16) (Fig. 2A,B). This is consistent with the H11 structure that carries a deletion of the NPY4R-B segment as well as one less copy of the PTPN20 segment.
In 3421/3436 humans, AGAP1, AGAP2, and AGAP3, which map outside Chromosome 10, are in single copy. Conversely, the other 10 AGAP genes, which map on Chromosome 10, have a WSSD diploid copy number ranging from 15 to 32, with the mean value equal to 19 or 20 (representative AGAP4 copynumber plot and correlations are shown in Fig. 2). In fact, as these 10 AGAP copies share an identity in their genomic sequence ranging from 97.5% to 99.6% (Supplemental Table S1) and the WSSD method considers a 95% identity threshold, the estimates for each gene reflect the diploid copy number of all 10 copies together. In line with this, WSSD estimates of Chromosome 10 AGAP genes show high pairwise correlations (0.65 ≤ ρ ≤ 0.94, P < 2.2 × 10−16). The copy number of Chromosome 10 AGAP genes also correlates with the copy number of the other 10q11.22 genes assessed (Fig. 2B). This is in line with the concomitant copy-number variation of AGAP7P, AGAP14P, AGAP13P, AGAP9, and/or AGAP12P with the other 10q11.22 genes (Fig. 1B).
Next, we checked copy number estimates in 15 samples that are homozygous for the H1 haplotype. NPY4R, GPRIN2, SYT15, NPY4R2, and SYT15B WSSD estimates, ANTXRL and AGAP10P SUNK estimates, and PTPN20 WSSD and SUNK estimates are fully consistent with expected values. ANXA8 and ANXA8L1 WSSD estimates correspond to expected values except for one sample. AGAP WSSD estimates range from 17 to 20 (predicted value of 20, considering 10 AGAP copies on each Chromosome 10). SUNK estimates for the NPY4R/R2, SYT15/15B, ANXA8/ANXA8L1, and AGAP paralogs do not always coincide with the expectation of two copies (Supplemental Table S2).
We evaluated copy number estimates in samples carrying alternative haplotypes (from H2 to H11) (Table 2; Supplemental Table S2). ANTXRL SUNK estimates align with its duplication in H5, H6, and H7 haplotypes. Similarly, NPY4R, GPRIN2, NPY4R2, SYT15B, and ANXA8L1 WSSD estimates correspond to the expected values and are consistent with the duplication of the NPY4R segment in H4, H5, and H6 haplotypes, as well as the deletion in H10. AGAP WSSD estimates usually do not match expectations; however, the general trend aligns with a higher or lower copy number compared to H1–H1 genomes. NPY4R, NPY4R2, SYT15, and SYT15B SUNK estimates do not generally align with expectations. Overall, copy-number data support the polymorphic structural configurations that we identified at Chromosome 10q11.22 region.
Copy number estimates in samples with alternative haplotypes
Lastly, we considered copy-number data of samples with unassembled haplotypes to evaluate whether fragmented sequences correspond to more complex and longer structures with further expansions of these segments. In particular, we retrieved data of samples with one H1 haplotype and the other haplotype unassembled (n = 10) and of samples with both haplotypes unassembled (n = 9). Copy-number data were available for 5 out of 10 and 4 out of 9 samples, respectively, corresponding to an H1–H1 genotype. This suggests that the assembled haplotypes provide an unbiased sample of the large-scale structural variation occurring at Chromosome 10q11.22. It also implies that difficulties in assembling this interval in some samples are not caused by the presence of a high-copy-number of these large segments.
Refinement of structural variation breakpoints
To gain insights into the mechanism originating 10q11.22 structural diversity, we sought to (1) identify the location of the variant breakpoints; (2) narrow down as much as possible the interval; (3) assess sequence features at the refined locations. We considered the alternative haplotypes that were validated by GAVISUNK and assumed that the H1 haplotype, the most common one, is the parental structure from which new haplotypes were formed.
As we noted that all variant breakpoints map to SD clusters, we posited that the new haplotypes were generated by SD-mediated NAHR. As recombination activity is higher in females than in males at this locus, we infer that most 10q11.22 NAHR events might occur during maternal meiosis (Supplemental Fig. S1). Precisely, interchromosomal or interchromatidal NAHR between SDs with direct orientation can generate alleles with the duplication or deletion of the intervening sequence together with the gain or loss of one SD copy (Fig. 3A). Additionally, the new SD copy or the remaining one are “chimeras,” with the first portion derived from the downstream SD copy and the second part derived from the upstream SD copy in the duplication allele, and vice versa in the deletion allele (Fig. 3A). Similarly, intrachromatidal NAHR between SDs with inverted orientation can generate alleles with inversion of the segment in-between. The inversion also reshuffles the flanking SDs and mutates them to two hybrid copies (Fig. 3A). The junction between the two parts derived from the original SD sequences corresponds to the exact NAHR breakpoint.
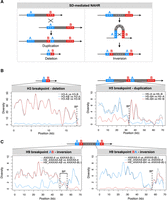
Characterization of rearrangement breakpoints in H3, H5, and H9 haplotypes. (A, left) Schematic of interchromosomal or interchromatid NAHR between directly oriented SDs resulting in reciprocal duplication and deletion. The misalignment of two directly oriented SDs—block A (blue) and block B (red)—produces two chromatids: one carrying a duplication and the other a deletion of the sequence between them (gray), as well as of the SDs themselves. In the chromatid with the duplication, the new SD copy is a “chimera,” with the first portion derived from the downstream SD (B, red) and the second portion from the upstream SD (A, blue). Conversely, in the chromatid with the deletion, the remaining SD copy is a “chimera,” with the first portion derived from the upstream SD (A, blue) and the second portion derived from the downstream SD (B, red). The switch from A to B (or vice versa) corresponds to the exact NAHR site. (Right) Schematic of intrachromatid NAHR between SDs with inverted orientation generating an inversion. The misalignment of two SDs with inverted orientation—block A (blue) and block B (red)—generates an inversion of the sequence between the SDs (gray), as well as two hybrid “AB” SDs, with each copy having an A-derived and a B-derived portion. The switch site between the A and B blocks depends on the position of the NAHR breakpoint. (B) Diversity plots of H3 and H5 breakpoint regions compared with the putative SDs mediating the deletion/duplication. The plots show the sliding window pairwise diversity between the H3/H5 breakpoint regions (H3-AB, H5-BA) and the original “A” (blue line) or “B” (red line) SDs. The gray line refers to the comparison between the parental SDs (H3-A vs. H3-B; H5-A vs. H5-B), as a reference. The dashed line indicates the location of the breakpoint, which is the point where the derived hybrid SD copy switches similarity from one copy (H3-A or H5-B) to the other (H3-B or H5-A) of the original SDs. (C) Diversity plots of H9 breakpoint regions compared with the putative SDs mediating the inversion. Diversity plots of the H9 ANXA8-AB duplicon (left) or H9 ANXA8-BA duplicon, reverse strand (right) compared with the H1 ANXA8-A (blue line) or ANXA8-B (red line) duplicons. The gray line refers to the comparison between H1 ANXA8-A and ANXA8-B duplicons, as a reference. Dotted lines designate the breakpoint region.
We first identified the SD pair that putatively underlies the NAHR event at the origin of the variant. Next, we conducted a sliding window diversity analysis, comparing the SD hybrid sequence/s (one in case of deletion or duplication and two in case of inversion) with each parental SD sequence from the T2T genome assembly (“A” and “B” duplicons). Additionally, we assessed the diversity between the two parental SD sequences as a reference. We visually examined the resulting patterns to identify the position where the derived hybrid SD sequence transitions from being a better match to one SD of the pair to being a better match to the other SD of the same pair. Finally, the breakpoint corresponds to the entire interval at the switch that cannot be confidently assigned to either parental SD.
SDs generating the deletion in the H3 allele belong to clusters containing AGAP7 and AGAP14 copies, whereas those that underlie the joint duplication of ANTXRL and NPY4R segments in the H5 allele encompass, respectively, AGAP7 and AGAP13 copies (Fig. 1A,B). We refined the breakpoints of H3 deletion and H5 duplication to, respectively, 369 and 867 bp intervals with perfect identity and no genes annotated (Table 3; Fig. 3B; Supplemental Figs. S2, S3; Supplemental Table S3). Whereas the first interval is characterized by a high GC content (66%), the second one corresponds to a long interspersed nuclear element (LINE) (Table 3; Supplemental Figs. S2, S3).
Features of rearrangements and breakpoints
We next analyzed the five different haploid genomes corresponding to the H2 haplotype with inversion of the GDF10 segment. This inversion is mediated by inverted copies of NPY4R duplicons (Fig. 1A). We compared the sequence of the parental duplicons with that of the derived hybrid duplicons at both sides of the inversion from H2 genomes (Supplemental Fig. S4A). As the breakpoint region was consistent between the two approaches (analysis of “AB” and “BA [reverse complement]” duplicons), we considered the intersection between the two breakpoint intervals as the final breakpoint (Table 3; Supplemental Table S3). We found that the five H2 inverted chromosomes derive from five distinct inversion events with different breakpoints (Supplemental Fig. S4). We observed that H2.3-BA, H2.4-BA, and H2.5-AB plots show unforeseen intervals where the duplicon switches again its higher similarity from the “A” copy (blue line) to the “B” copy (red line) (Supplemental Fig. S4A).
Our analysis of the H9 haplotype with inversion of a region that includes both the GDF10 segment and the NPY4R-A duplicon showed that this rearrangement was mediated by inverted copies of ANXA8 duplicons and the breakpoints occur at ANXA8 loci (Table 3; Fig. 3C; Supplemental Fig. S5; Supplemental Table S3). Breakpoint analysis of H11 and H10 haplotypes, respectively, with the deletion of the distal and proximal copy of the NPY4R duplicon, showed that they likely derived, respectively, from the H2 and H9 inverted haplotypes. In fact, the inversion in H2 changes the reciprocal orientation of PTPN20 duplicons from indirect to direct and these can thus mediate the H11 deletion (Fig. 4A). Similarly, the inversion in H9 changes the reciprocal orientation of NPY4R duplicons from indirect to direct and locates them in a tandem configuration. This new configuration is susceptible to the deletion of one copy of NPY4R duplicon as observed in H10 haplotype (Fig. 4B). We restricted H11 breakpoint to a 1641 bp interval that corresponds to intron 26 of FRMPD2/2B genes (Table 3; Fig. 4A; Supplemental Fig. S6; Supplemental Table S3) and H10 breakpoint to an ∼20 kbp interval overlapping FAM245B (Table 3; Fig. 4B; Supplemental Fig. S7; Supplemental Table S3).
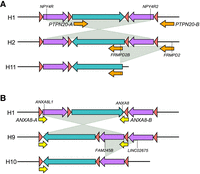
Inversion haplotypes generate new predispositions to copy-number variations. (A) Schematic of H2 and H11 haplotypes. The H2 haplotype differs from H1 by an inversion mediated by NPY4R inverted duplicons (purple arrows). Following this inversion, the PTPN20 duplicons (orange arrows) become in direct orientation and mediate the deletion identified in H11. Genes mapped at the breakpoints are specified. (B) Schematic of H9 and H10 haplotypes. The H9 haplotype differs from H1 by an inversion mediated by ANXA8 inverted duplicons (yellow arrows). Following this inversion, the NPY4R duplicons (purple arrows) become in tandem configuration and direct orientation and mediate the deletion of one copy in H10 haplotype. Genes mapped at the breakpoints are specified.
Finally, we evaluated features that have been previously associated with breakpoint sequences of NAHR events, that is, long stretches of homology between nonallelic sequences, high GC content, and the presence of PRDM9-binding motifs. We assessed both the length and homology of SDs mediating the rearrangements, as well as those of the refined breakpoint intervals. In the latter, we also checked GC content and the presence of PRDM9 motifs (Table 3). Whereas the length and homology of the entire SD pairs are variable, with the H3 rearrangement mediated by a pair with low similarity, diversity plots indicate that NAHR crossing overs tend to occur at positions with higher identity between parental SDs (99.64%–100%), as shown by reference lines (Table 3; Fig. 3B,C; Supplemental Figs. S2, S3, S5–S7). We also observed a GC content higher than the human genome average of 40.9% (Piovesan et al. 2019) at seven out of 10 breakpoints (Table 3), whereas we identified no enrichment in PRDM9 motifs when we compared breakpoint intervals versus the rest of the SD sequences (Fisher's exact test, P = 1).
Phylogenetic analysis of AGAP duplicons
Breakpoint sequence analysis revealed that alternative haplotypes derive from NAHR between SD pairs located in clusters that share the occurrence of a functional or pseudogenized AGAP copy (Fig. 1A). Among the 13 AGAP copies annotated in the human reference genome, AGAP1, AGAP2, and AGAP3 have 1:1 orthologs in vertebrate genomes, including marmoset and mouse lemur (Table 1, GENCODE v44 and Ensembl release 110). The remaining 10 copies map on Chromosome 10, with the majority (7/10) at the q-arm pericentromeric region, precisely 10q11.22 (Table 1). They consist in four protein-coding genes, five pseudogenes, and one noncoding RNA gene. These 10 copies correspond to an ∼22 kbp genomic segment, except AGAP11 that is shorter and corresponds to an ∼9 kbp segment. Except AGAP9 that has a 1:1 ortholog in Gorilla gorilla, these 10 copies do not have 1:1 orthologs in other genomes. Conversely, they have 1:many or many:many orthologs in species belonging to the Catarrhini parvorder of Primates that includes Old World monkeys and apes.
We analyzed the organization, synteny relationships, and phylogeny of human AGAP copies located on Chromosome 10 in comparison with other great apes. Through BLAT search using human AGAP4 genomic sequence as query, we identified the location of Chromosome 10 AGAP genes in the chimpanzee (panTro6), gorilla (gorGor6), and orangutan (ponAbe3) genomes. In chimpanzee, we identified AGAP sequences at five locations on Chromosome 10, as well as complete or partial alignments in four unlocalized contigs (chrUn). In gorilla, three AGAP sequences are located on Chromosome 10, with two additional partial matches in unlocalized contigs that probably correspond to one copy. In orangutan, we identified partial alignments at five locations in a 5 Mbp gap-rich 10q11 pericentromeric interval as well as additional shorter matches in unlocalized contigs. These data show that a cluster of AGAP copies is present at the pericentromeric region of the long arm of Chromosome 10 in human, chimpanzee, and orangutan and it likely reflects the ancestral ape organization of this catarrhine-specific subgroup of the AGAP family (Fig. 5A). Although genome references report a higher number of copies in humans and chimpanzees compared to other apes, AGAP copy number estimates suggest an expansion in gorillas similar to that in chimpanzees (Fig. 2A).
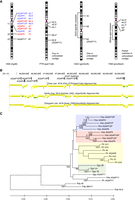
Genomic organization and phylogeny of Chromosome 10 AGAP copies in great apes. (A) Chromosome location (in Mbp) of AGAP copies in human, chimpanzee, gorilla, and orangutan reference genomes according to the most recent releases. In the common ancestor of African apes, a pericentric inversion moved AGAP11 outside the pericentromeric region. Copies with an asterisk (*) are shorter than the others and are thus incomplete. Human AGAP copies are colored according to the clade in the phylogenetic tree shown in C. (B) Pairwise genomic sequence alignments of the q-arm pericentromeric region of human Chromosome 10 with great ape genomes (UCSC track of primate net alignments). The location of AGAP duplicons is shown. (C) Phylogeny of Chromosome 10 AGAP copies in great apes. The tree was inferred by using the Maximum Likelihood method and Kimura 2-parameter model. The tree is drawn to scale, with branch lengths measured in the number of substitutions per site. All positions containing gaps and missing data were eliminated (complete deletion option). There were a total of 4961 positions in the final data set. Bootstrap values >75 are shown next to significant nodes. Timing estimates in millions of years of split events between human AGAP copies are shown in italics.
Pairwise genomic sequence alignments of human versus other primates (UCSC track of net alignments) revealed numerous breaks of synteny between genomes and complex rearrangements at this locus (Fig. 5B). This feature, together with the presence of several AGAP sequences in unlocalized contigs of ape genome assemblies, hampered the identification of 1:1 orthology relationships between the majority of Chromosome 10 AGAP copies. An evolutionary pericentric inversion in the African ape ancestor moved one AGAP copy (AGAP11) more distally and outside the pericentromeric cluster. This is the only copy for which the orthology relationship between human, chimpanzee, and gorilla genes is straightforward.
Next, we built a phylogeny based on a 5 kbp segment that is shared among all human AGAP copies. We retrieved AGAP genomic sequences for human (n = 10), chimpanzee (n = 8), gorilla (n = 4), and orangutan (n = 2). As the presence of gaps in the orangutan genome assembly might limit the recovery of sequences corresponding to our query, we searched for fully sequenced Pongo abelii BAC clones containing AGAP sequences. We identified two clones (CH276-56H17 and CH276-327M15) that embedded one missing sequence. We noted that these clones correspond to the proximal African ape inversion breakpoint and AGAP and FAM25 sequences are located within the 26 kbp breakpoint interval. The phylogeny reveals a monophyletic clade that includes human, chimp, and gorilla AGAP11 copies. Conversely, all other AGAP genes cluster by species, probably because of gene conversion among closely located AGAP members (Fig. 5C). Human pericentromeric AGAP copies cluster in two main clades both supported by high bootstrap values. One clade includes AGAP4, AGAP6, AGAP7P, AGAP10P, and AGAP13P, whereas the other comprises AGAP5, AGAP9, AGAP12P, and AGAP14P. AGAP copies at 10q11.22 SD clusters belong to both groups. By applying the molecular clock approach and calibrating the tree based on a human/chimpanzee divergence time of 6.4 million years ago, we estimated that split events between human pericentromeric AGAP copies happened between 5.7 and 1.4 million years ago (Fig. 5C).
Discussion
In this work, we took advantage of recent advancements in research resources, that is, the release of the first gapless human genome (Nurk et al. 2022) and the de novo assembly of diverse human genomes (Chaisson et al. 2019; Liao et al. 2023), to study the genomic organization and structural variation of human Chromosome 10q11.22. Our sequence analysis revealed extensive structural diversity, including large-scale (>50 kbp) deletions, duplications, and inversions that originate from SD-mediated NAHR. We identified 11 alternative structures, each differing in size from the most common haplotype (H1) by as much as 20 kbp up to 1.7 Mbp. We believe that this catalog does not encompass the full spectrum of structural variation at this locus. For instance, eight structures were unique to a single genome, indicating that analyzing additional genomes might reveal novel arrangements. We note that according to our WSSD and SUNK copy number estimates, some individuals carry more than four PTPN20 copies, yet all haplotypes that we describe have one or two copies. It is possible that some individuals carry the reciprocal product (duplication) of the NAHR generating the H11 deletion haplotype and therefore carry five PTPN20 diploid copies. This region was unassembled in about one-third of the genomes in the initial set (30 out of 94). As copy-number data for these samples, when available, align with an H1–H1 genotype, the lack of successful assembly could be due to the presence of highly repetitive DNA, such as long stretches of simple repeats, or structural mosaicism within the same cell line. However, neither possibility would impact the conclusions of the present study.
SUNK estimates of paralogous genes within copy-number variant segments do not always align with expectations. We believe that some discrepancies result from inversion toggling mediated by NPY4R inverted duplicons (Porubsky et al. 2020, 2022), which mixes paralogous copies of genes located on these segments. Additionally, gene conversion events might also contribute to the observed differences. Human AGAP WSSD estimates typically range around 19 or 20 in H1–H1 samples, with 20 being the expected value. This discrepancy is likely due to limitations in WSSD genotyping, particularly for high-copy-number genes.
Breakpoint analysis in five genomes with the same inversion showed they derive from different NAHR events mediated by the same SD pair but with breakpoints at diverse positions. We noticed intervals within some of the derived SD hybrid sequences where the similarity to the parental SD copies is not as anticipated. These patterns might derive from either subsequent gene conversion with the SD copy on the homologous chromosome or additional inversion events (inversion toggling) and subsequent recombination in the region between the inverted SDs. Indeed, there is evidence of mutational toggling for this inversion (Porubsky et al. 2020, 2022) and of recombination in the GDF10 segment (Supplemental Fig. S1).
Previous studies have demonstrated that several factors influence the likelihood of NAHR, including the length, homology, and distance between nonallelic duplicate sequences, as well as the GC content, presence of tracts of perfect sequence identity, PRDM9-binding sites, G-quadruplex forming sequences, and local recombination activity (Lupski 1998; Lindsay et al. 2006; Liu et al. 2011; Dittwald et al. 2013; Hillmer et al. 2016; Summerer et al. 2018). In this study, we analyzed the occurrence of some of these features in 10 finely mapped breakpoint sequences and found that the presence of tracts of perfect or almost perfect sequence identity is a selective requirement of NAHR crossing-over sites, regardless of the overall similarity of the SD pair. The presence of PRDM9-binding sites, G-quadruplex forming sequences, and local recombination activity could not be assessed due to missing data on SD regions in genome-wide studies that evaluated these features (Altemose et al. 2017; Hänsel-Hertsch et al. 2018).
The 10q11.22 SD architecture and rearrangement breakpoints are associated with a catarrhine-specific subgroup of the AGAP gene family that likely triggered the accumulation of SDs at this locus in the last 25 million years of human evolution. The AGAP family includes AGAP1, AGAP2, and AGAP3 that have 1:1 orthologs in vertebrate genomes, as well as AGAP4 to AGAP14 that map on Chromosome 10 and are more recent genes. These additional copies began to emerge in the common ancestor of apes and Old World monkeys by duplication at the pericentromeric region of ancestral Chromosome 10. Evolutionary genomic rearrangements, assembly gaps in ape genome references, and interlocus gene conversion hamper the identification of ancestral Chromosome 10 copies, 1:1 orthology relationships between ape genomes, and lineage-specific more recent copies. The incompleteness of AGAP sequences in ape reference genomes is reflected by numerous matches in unlocalized contigs and by the discrepancy between the number of copies annotated in the genomes and the number of WSSD-predicted copies, especially in gorillas.
AGAP proteins, previously referred to as members of the gamma subgroup of the centaurin superfamily of small GTPases (centaurin gamma), are characterized by a pleckstrin homology (PH) domain, an Arf GTPase-activating (ArfGAP) domain, and ankyrin repeats (Kahn et al. 2008). AGAP1 and AGAP2 have been the most studied members and have roles in endosomal trafficking and cytoskeleton dynamics (Nie et al. 2002, 2005). In particular, AGAP1 was shown to control cytoskeleton remodeling involved in cell movement (Luo et al. 2016), whereas AGAP2 was shown to regulate retrograde transport between early endosomes and the trans-Golgi network (Shiba et al. 2010) and to modulate the disassembly of focal adhesions during cell migration (Zhu et al. 2009). AGAP proteins have specialized functions in neurons. AGAP1 was shown to participate in dendritic spine morphology (Arnold et al. 2016) and endocytic recycling of muscarinic receptors (Bendor et al. 2010). AGAP2 has several roles in the central nervous system, including neurite outgrowth (Dwane et al. 2014), myelination (Chan et al. 2014), regulation of glutamate receptors at synapses (Chan et al. 2011a), neuronal survival (Tang et al. 2008), and memory formation (Chan et al. 2011b). AGAP3 was proposed to have a role in contextual novelty-induced memory consolidation by regulating glutamate receptor trafficking in the synapse (Oku and Huganir 2013; Højgaard et al. 2023). To our knowledge, functional studies on the primate-specific AGAP genes have not been reported in the literature. It is possible that primate-specific functional AGAP copies have similar roles at neuronal synapses. This suggests that AGAP expansion might have enhanced synaptic plasticity and advanced memory functions in primates and humans.
The promotion of further duplicative transposition events and the creation of complex SD clusters around AGAP duplicons recall the behavior of other elements in human and primate genomes, like LRRC37 copies on Chromosome 17 (Giannuzzi et al. 2013b), GOLGA copies on Chromosome 15 (Giannuzzi et al. 2013a; Antonacci et al. 2014; Maggiolini et al. 2019), and NPIP copies on Chromosome 16 (Nuttle et al. 2016; Cantsilieris et al. 2020). LRRC37, GOLGA, and NPIP are all classified as core duplicons (Jiang et al. 2007).
The extensive diversity at Chromosome 10q11.22 recalls that of other regions, such as 3q29 (Yilmaz et al. 2023), 22q11 (Demaerel et al. 2019), 17q21.31 (Steinberg et al. 2012), and 16p11.2 (Nuttle et al. 2016; Loviglio et al. 2017; Giannuzzi et al. 2019, 2022), where copy-number variant segments and inversion polymorphisms give rise to multiple structural haplotypes differing in up to a few millions of base pairs. Additionally, this locus provides another example of how large inversion polymorphisms may alter the whole SD arrangement, generating new predispositions to copy-number variations. In fact, a similar pattern occurs at the 17q21.31 Koolen de Vries syndrome region (Koolen et al. 2006) and 7q11 Williams–Beuren syndrome region (Osborne et al. 2001).
Our detailed map of polymorphic structural variants will aid in identifying potential pathogenic structures at this locus. The pathogenicity and association of 10q11 deletions and duplications involving GPRIN2 and other genes with neurodevelopmental and neuropsychiatric diseases remain unclear, possibly due to the structural complexity and diversity of the locus, as well as the potential low penetrance and variable expressivity of the variants (Cooper et al. 2011; Stankiewicz et al. 2012; Coe et al. 2014).
Methods
Structural variation analysis
We downloaded diploid assemblies of 47 samples (94 haplotypes) from GitHub (https://github.com/human-pangenomics/HPP_Year1_Assemblies) (Liao et al. 2023). We obtained Chr 10: 44,500,000–50,500,000 sequence from the T2T-CHM13 genome (v2.0) and aligned it to each genome using minimap2 v2.24 (Li 2018). We kept for further analysis the haplotypes where the 10q11.22 region was fully assembled (i.e., present in a single contig). We compared the sequences with the reference T2T-CHM13 sequence using the re-DOT-able tool (https://www.bioinformatics.babraham.ac.uk/projects/redotable/). We verified the assemblies using GAVISUNK (Dishuck et al. 2023). Figure 1B was created in R (R Core Team 2023) using the ggplot2 v3.4.4 (Wickham 2009) and gggenomes (Hackl et al. 2023) packages.
Copy numbers were estimated using the WSSD (Bailey et al. 2002) and SUNK (Sudmant et al. 2010) methods. The WSSD method estimates the total copy number for sequence ≥95% sequence identity in nonoverlapping 1 kbp genomic windows based on the depth of coverage of whole-genome Illumina short reads aligned to the reference genome. The SUNK-based method estimates the paralog-specific copy number. Correlations were calculated using Spearman's method and the correlation matrix between copy number estimates was obtained using the scales v1.2.1 (Wickham and Seidel 2022) and ggcorrplot v0.1.4.1 (Kassambara 2023) R packages.
Breakpoint analysis
We identified approximate breakpoint regions by aligning each haplotype sequence to the H1 sequence using BLAST (Basic Local Alignment Search Tool) (Altschul et al. 1990). We mapped the approximate breakpoint region in the T2T-CHM13 v2.0 genome by BLAT (Kent 2002) and pinpointed the SD pair that putatively mediated the NAHR event originating the new haplotype. We multialigned the original SD sequences from the T2T genome and the sequence of the “chimeric” SD sequence/s at the breakpoint from the variant haplotype using MAFFT (Katoh et al. 2009). We calculated pairwise nucleotide diversity in 500 bp sliding windows with a 100 bp increment by using the PopGenome R package (Pfeifer et al. 2014) and narrowed down the location of the breakpoint by visual inspection of the resulting plots and the multialignment. We refined the breakpoint at the position where the breakpoint sequence becomes more similar from one duplication to the other of the pair.
We assessed the annotation of genes (CAT + Liftoff Gene Annotations UCSC track), repetitive elements content (RepeatMasker Repetitive Elements UCSC track), and GC content (GC Percent in 5-Base Windows UCSC track) of breakpoint sequences. We analyzed the presence of PRDM9 motif (5′ CCNCCNTNNCCNC 3′) by using the FIMO MEME-suite (Grant et al. 2011).
Evolutionary analyses
We multialigned human AGAP genomic sequences using MAFFT and calculated pairwise divergences. We located AGAP sequences in ape genomes through BLAT search and visual inspection of the resulting alignments. To reconstruct AGAP phylogeny, we used a 5 kbp sequence nearly corresponding to exon 6—intron 6—exon 7—intron 7—exon 8 of AGAP4 genomic sequence (gene structure based on MANE transcript annotation). We retrieved human and ape AGAP sequences through BLAT search in human (hg38), chimpanzee (panTro6), gorilla (gorGor6), and orangutan (ponAbe3) genomes. Sequences were multialigned by ClustalW (Thompson et al. 1994). The phylogeny was inferred by using the Maximum Likelihood method and Kimura 2-parameter model (Kimura 1980). All positions containing gaps and missing data were eliminated (complete deletion option). The statistical significance of nodes was evaluated by using the bootstrap test with 100 replicates (Felsenstein 1985). To date the expansion of AGAP copies in humans, we built a phylogenetic tree of human AGAP pericentromeric copies (i.e., excluding AGAP11) with one chimpanzee (Ptr_42) and one orangutan (Pab_45) sequence. After testing the molecular clock hypothesis using the Maximum Likelihood method, we inferred the evolutionary timing of human duplications. We used the orangutan sequence as the outgroup and calibrated the tree by assigning a fixed time of human–chimpanzee split at 6.4 million years ago. Evolutionary analyses were conducted in MEGAX (Kumar et al. 2018; Stecher et al. 2020).
Competing interest statement
E.E.E. is a scientific advisory board (SAB) member of Variant Bio, Inc.
Acknowledgments
This study received funding from the European Union—Next Generation EU—National Recovery and Resilience Plan (NRRP), Mission 4, Component 2, Investment 1.1, call PRIN 2022 D.D. 104 02.02.2022 to G.G. (2022RB88C7). This work was supported, in part, by US National Institutes of Health (NIH) grant HG002385 to E.E.E. We thank Paolo Carnevali and Sara Simmonds for providing access to data. We thank Paolo Gandellini for the discussions and Tonia Brown for assistance in manuscript editing. E.E.E. is an investigator of the Howard Hughes Medical Institute.
Author contributions: G.G. conceived and supervised the study; S.F., V.S.D., and G.G. analyzed pangenome data; P.C.D. performed the GAVISUNK analysis; S.F. performed the breakpoint analysis; W.T.H. and E.E.E. provided copy number estimates; S.F. and G.G. analyzed copy number estimates. V.S.D. and G.G. performed an evolutionary analysis of AGAP genes. G.G. wrote the manuscript. All authors read and approved the manuscript.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.279454.124.
- Received April 11, 2024.
- Accepted September 10, 2024.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see https://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








