
Telomerase-independent survival leads to a mosaic of complex subtelomere rearrangements in Chlamydomonas reinhardtii
- Frédéric Chaux1,4,
- Nicolas Agier1,3,
- Clotilde Garrido1,3,
- Gilles Fischer1,
- Stephan Eberhard2 and
- Zhou Xu1
- 1Sorbonne Université, CNRS, UMR7238, Institut de Biologie Paris-Seine, Laboratory of Computational and Quantitative Biology, 75005 Paris, France;
- 2Sorbonne Université, CNRS, UMR7141, Institut de Biologie Physico-Chimique, Laboratory of Chloroplast Biology and Light-Sensing in Microalgae, 75005 Paris, France
-
↵3 These authors contributed equally to this work.
Abstract
Telomeres and subtelomeres, the genomic regions located at chromosome extremities, are essential for genome stability in eukaryotes. In the absence of the canonical maintenance mechanism provided by telomerase, telomere shortening induces genome instability. The landscape of the ensuing genome rearrangements is not accessible by short-read sequencing. Here, we leverage Oxford Nanopore Technologies long-read sequencing to survey the extensive repertoire of genome rearrangements in telomerase mutants of the model green microalga Chlamydomonas reinhardtii. In telomerase-mutant strains grown for hundreds of generations, most chromosome extremities were capped by short telomere sequences that were either recruited de novo from other loci or maintained in a telomerase-independent manner. Other extremities did not end with telomeres but only with repeated subtelomeric sequences. The subtelomeric elements, including rDNA, were massively rearranged and involved in breakage–fusion–bridge cycles, translocations, recombinations, and chromosome circularization. These events were established progressively over time and displayed heterogeneity at the subpopulation level. New telomere-capped extremities composed of sequences originating from more internal genomic regions were associated with high DNA methylation, suggesting that de novo heterochromatin formation contributes to the restoration of chromosome end stability in C. reinhardtii. The diversity of alternative strategies present in the same organism to maintain chromosome integrity and the variety of rearrangements found in telomerase mutants are remarkable, and illustrate genome plasticity at short timescales.
Protection of chromosome extremities is essential for genome integrity. For most eukaryotes, it is achieved by repeated DNA sequences called telomeres and by telomere-bound factors, which collectively prevent chromosome ends from being processed as DNA damage (Jain and Cooper 2010; de Lange 2018). Telomeres shorten with each round of replication owing to the end replication problem and are, in general, maintained by telomerase, a dedicated reverse transcriptase able to elongate telomeres de novo. In its absence, some telomeres eventually reach a critical length that triggers replicative senescence, an arrested state induced by the DNA damage checkpoint. Replicative senescence was shown in some species to increase genome instability owing to repair attempts and bypass of the checkpoint arrest through the adaptation to DNA damage process (Blasco et al. 1997; Lee et al. 1998; Chin et al. 1999; Artandi et al. 2000; Hackett et al. 2001; Hackett and Greider 2003; Maciejowski et al. 2015; Coutelier et al. 2018; Henninger and Teixeira 2020). In senescent cells that eventually escape cell cycle arrest, such as some precursor cancer cells, telomeres become dysfunctional and induce further genomic instabilities, a phenomenon termed telomere crisis (Artandi and Depinho 2009; Maciejowski and de Lange 2017).
The absence of telomerase therefore generates genome instabilities that stem from telomeres and take many shapes: point mutations, deletions/insertions, translocations, aneuploidy, duplications, and even more dramatic rearrangements, such as chromothripsis during telomere crisis (Maciejowski et al. 2015). The precise molecular mechanisms underlying these alterations are not all well understood but often involve classical and alternative nonhomologous end-joining (c- and a-NHEJ), homology-directed repair (HDR), including homologous recombination and break-induced replication (BIR), together with missegregation of chromosomes, breakage–fusion–bridge (BFB) cycles, and other dynamic phenomena that act in cascades over multiple cell divisions (McClintock 1941; Blasco et al. 1997; Hackett et al. 2001; Hackett and Greider 2003; Capper et al. 2007; Davoli et al. 2010; Jones et al. 2014; Maciejowski et al. 2015; Maciejowski and de Lange 2017).
Subtelomeres are the genomic regions adjacent to telomeres and often contain families of paralogous genes or pseudogenes, ribosomal DNA (rDNA) arrays, transposable elements, and other repeated sequences (Corcoran et al. 1988; Louis 1995; Kim et al. 1998; Fabre et al. 2005; Richard et al. 2013; Yue et al. 2017; Chaux-Jukic et al. 2021). Subtelomeres are often involved in telomere-associated rearrangements owing to their repetitive nature promoting HDR, replication fork stalling and template switching (FoSTeS), and BIR (Corcoran et al. 1988; Louis and Haber 1990; Linardopoulou et al. 2005; Kuo et al. 2006; Rudd et al. 2007; Maestroni et al. 2017; Takikawa et al. 2017; Chen et al. 2018; Kim et al. 2019). Consistently, subtelomeres evolve rapidly even in closely related species and within species (Anderson et al. 2008; Yue et al. 2017; Otto et al. 2018; Kim et al. 2019; Young et al. 2020). In some species, in the absence of telomerase, telomeres can be stabilized using the alternative lengthening of telomeres (ALT) pathway, which depends on homologous recombination and uses repeated sequences found in telomeres and subtelomeres as substrates (Lundblad and Blackburn 1993; Nakamura et al. 1998; Zellinger et al. 2007; Cesare and Reddel 2010).
Genome alterations, especially structural variations (SVs), initiated by telomere shortening and dysfunction, despite being widely studied in different models including cancer cells, have been difficult to map exhaustively owing to the complex nature of the rearrangements and the frequent involvement of repeated sequences such as the ones found in subtelomeres (Maciejowski and de Lange 2017; Ho et al. 2020). Only recently were long-read sequencing technologies used to enable the resolution of complex rearrangements at chromosome extremities, in response to telomere shortening and dysfunction in Caenorhabditis elegans and Saccharomyces cerevisiae (Kim et al. 2021; Kockler et al. 2021; Sholes et al. 2021).
We recently provided a comprehensive map of all 34 subtelomeres of the unicellular green alga Chlamydomonas reinhardtii (17 chromosomes as haploid, 111 Mb) (Chaux-Jukic et al. 2021). All contain arrays of repeated elements, the most common being the Sultan element, present in 31 out of the 34 chromosome extremities in a haploid strain, arranged in tandem repeats of up to 46 elements (Supplemental Fig. S1A). The basic Sultan element has a length of ∼850 bp and forms class A subtelomeres. The Sultan element of class B subtelomeres contains additional insertions. Next to most Sultan arrays (29 out of 31), a sequence of ∼500 bp called Spacer is unique to each subtelomere and may serve as promoter for downstream noncoding RNA genes. The three remaining subtelomeres are entirely composed of rDNA, for a total of approximately 350 copies corresponding to ∼3 Mb. Two other repeated elements, called Suber and Subtile, were found next to Sultan elements at three subtelomeres, called class C. The Suber element, initially named pTANC (Hails et al. 1993), contains the most abundant interstitial telomere sequence (ITS) of the genome. We previously found experimental evidence of telomere-associated genome rearrangements potentially involving subtelomeres in telomerase mutants of C. reinhardtii, correlated with long-term survival (Eberhard et al. 2019). Indeed, although some telomerase-negative mutant subclones underwent senescence-induced cell death, many managed to survive telomerase absence and must have therefore found a solution to maintain and protect telomeres. In this work, using long-read Oxford Nanopore Technologies (“Nanopore”) sequencing able to traverse large repeated regions, we investigated genome instability in telomerase mutants in C. reinhardtii with the aim of providing an exhaustive view of the landscape of genome rearrangements. The rearranged genomic regions, most importantly telomeres and subtelomeres, were then scrutinized to provide insights into the mechanisms of chromosome-end plasticity and stability.
Results
Long-read Nanopore sequencing of prolonged cultures of telomerase mutants
To investigate the genome rearrangements induced by the long-term absence of telomerase, we used two different telomerase-mutant strains, tel-m1 and tel-m2 (Fig. 1A). The two mutant strains were obtained from the “Chlamydomonas Library Project” (CLiP) library of random insertion mutants (Li et al. 2016) and contained the paromomycin resistance gene inserted in the RNA-binding domain (tert1‐1 allele, corresponding to strain tel-m1) and catalytic domain (tert1‐2 allele, corresponding to strain tel‐m2) of TERT1 (also called CrTERT), the gene encoding the catalytic subunit of telomerase. For each mutant strain, the single insertion of the paromomycin resistance gene in TERT1, leading to an “ever-shorter telomere” phenotype, was confirmed previously (Eberhard et al. 2019). Upon receiving them from the Jonikas laboratory, we cultivated the mutant strains alongside the corresponding wild-type strain CC-4533 for an estimated 450 generations and collected the samples called “tel-m1-1,” “tel‐m2‐1,” and “WT CC-4533,” respectively (Fig. 1A). As noted previously, we did not observe any obvious growth defect in these telomerase-mutant strains grown in standard culture conditions. For tel-m1, we also collected an earlier sample named “tel-m1-0,” which corresponded to the earliest time point we could obtain upon reception of the strain in our laboratory. The strains had potentially experienced several additional prior passages, needed for the procedure of transformation, propagation, and freezing as described by Li et al. (2016).
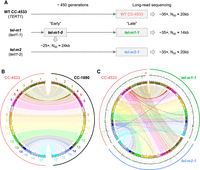
Independent genome assemblies of CC-4533 and telomerase-mutant strains tel-m1-1 and tel-m2-1 reveal few rearrangements at the genome assembly level. (A) Experimental setup and main sequencing output. (B,C) Circos plot (Krzywinski et al. 2009) of chromosome scaffolds from the long-term culture of CC-4533 compared with CC-1690 (B) and with tel-m1-1 and tel-m2-1 (C), shown as colored boxes, with black marks for centromere clusters (composed of the Zepp-L1 repeat). Links connect homologous blocks, colored according to CC-4533 chromosomes. Outer plot displays sequencing depth, averaged over 100-kb regions, with 0×, 35×, and 70× grid lines shown in black, green, and red, respectively. Depth over 50× is highlighted in red, and genomic position is indicated in megabases.
We optimized a CTAB/phenol/chloroform purification protocol followed by a size-selection step to extract and obtain long DNA molecules (Chaux-Jukic et al. 2022). We then sequenced the genomic DNA of these four samples by long-read Nanopore sequencing and obtained four sets of high-quality reads of N50 > 14 kb and depth > 25× (Fig. 1A; Supplemental Table 1). We previously proved that long reads allowed the accurate reconstruction of the highly repeated subtelomere structures in C. reinhardtii (Chaux-Jukic et al. 2021), which is instrumental for the analysis of rearrangements potentially implicating telomeres, subtelomeres, and other repeated elements.
The long-read data sets were processed by three assemblers, allowing us to generate de novo genome assemblies of CC-4533 and each telomerase mutant (Fig. 1B,C; Supplemental Table 2). We reached large chromosome-scale scaffolds similar to the most contiguous reference genome of CC-1690 (Fig. 1B; O'Donnell et al. 2020). Sequencing depth (Fig. 1C, outer circles; Supplemental Fig. S1B) indicated a partial duplication of Chromosome 1 in CC-4533 not present in a previous sequencing (Gallaher et al. 2015), suggesting that a duplicated mini-Chromosome 1 emerged during the long-term culture in our laboratory, a phenomenon that is quite common because duplications were relatively frequent (although shorter) in mutation accumulation lines (López-Cortegano et al. 2023) and large duplications (up to ∼400 kb) were observed in other laboratory wild-type strains (Flowers et al. 2015). The copy number variation (CNV) on Chromosomes 15 and 16 was because of frequently misassembled regions known to contain repeated sequences (Craig et al. 2023). No other chromosome-scale difference was found between CC-4533 and CC-1690 (Fig. 1B). The Sultan elements being present in 31 out of 34 subtelomeres and well assembled, we compared the number of Sultan elements at each chromosome extremity between CC-4533 and CC-1690 to evaluate their divergence at subtelomeres. We found that the number of Sultans for each extremity in these two strains was very close with few exceptions (Supplemental Fig. S1C). The subtelomeres of the wild-type CC-4533 strain have thus not been substantially altered during growth in this experiment and even since its selection as a laboratory strain decades ago, consistent with our previous finding that the subtelomeres have been very stable in laboratory strains (Chaux-Jukic et al. 2021).
Compared with CC-4533, tel-m1-1 displayed a number of large-scale SVs and CNVs, corresponding to translocations and duplications of chromosome extremities (Fig. 1C; Supplemental Fig. S2A). The other mutant, tel-m2-1, showed fewer large-scale rearrangements in the assembly, but CNVs were detected at several locations, suggesting genome alterations (Supplemental Fig. S2B). We then used the SV caller MUM&Co (O'Donnell and Fischer 2020) to detect insertions, deletions, duplications, inversions, and translocations compared with CC-1690, and we found similar numbers for CC-4533 and the telomerase mutants assemblies, with about half of SVs shared by all three strains (n = 370 out of a total of 746) (Supplemental Fig. S2C,D). The assemblies of the telomerase mutants displayed a moderate number of unique SVs (n = 52 and 49 for tel-m1-1 and tel-m2-1, respectively), involving sequences of median size < 600 bp.
However, we found that the quality of read mapping to subtelomeres was lower than that corresponding to the core genome both in CC-4533 and in the telomerase mutants (Supplemental Fig. S2E), presumably because the repeated elements found at subtelomeres present a challenge for genome assembly even using long-read sequencing data (Filloramo et al. 2021). Consistently, the assembled genomes reached only 25, two, and 19 telomeres in CC-4533, tel-m1-1, and tel-m2-1, respectively. We thus suspected that the assembly-level analysis would miss genome rearrangements affecting chromosome extremities. Additionally, assemblers were not designed to handle genome heterogeneity at the subpopulation level, which is likely to be the case in telomerase mutants. We therefore directly looked for genome alterations at the level of the sequencing reads instead of the assemblies in the rest of this work.
Telomere shortening and loss in telomerase mutants
We were first interested in investigating changes related to telomere sequences. To detect all telomere sequences at the level of individual reads, we developed a new method called TeloReader that scores 8-mers with respect to their level of identity to any of the canonical 8-mers (TTTTAGGG/CCCTAAAA and circular permutations) in sliding windows and uses thresholds for the average score to find the boundaries (see Methods). TeloReader allowed us to detect all telomere sequences of at least 16 bp, whether they were at a chromosome extremity (terminal) or not (interstitial, i.e., ITS), in the read data sets.
We measured the length of all terminal telomere sequences in the wild-type and mutant reads and found shorter telomeres in the mutants (mean ± SD for CC-4533: 293 ± 126 bp, tel-m1-0: 167 ± 109 bp, tel-m1-1: 162 ± 120 bp, tel-m2-1: 181 ± 88 bp) (Fig. 2A). Telomeres were already short in the tel-m1-0 sample, and their length did not further decrease in the tel-m1-1 sample, suggesting the stabilization of telomere length by telomerase-independent mechanisms or de novo telomere formation after complete loss.
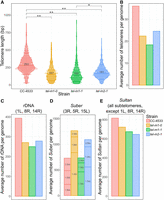
Telomere length distribution and CNVs of subtelomeric elements. (A) Telomere length distribution of terminal telomere sequences detected by TeloReader from the reads data set in CC-4533 and three telomerase mutant samples. Statistically significant difference: (*) P-value = 6.8 × 10−9 and (**) P-value < 2.2 × 10−16, using a nonparametric Kruskal–Wallis test. (B) Number of telomeres in reads normalized by the sequencing depth. (C–E) CNVs for repeated elements normally found at subtelomeres including the rDNA (C), the Suber elements subdivided into three types according to their length (“1.5 kb,” “1.9 kb,” and “2.5 kb”; D), and the Sultan elements (E).
Because we suspected that the aggregate length distribution measurement would mask telomere length dynamics at each extremity, we assigned the reads containing a terminal telomere sequence to a specific chromosome arm based on the presence of specific and unique Sultan elements in the reads and measured telomere length distribution for each Sultan-associated extremity (Supplemental Fig. S3A). Out of the 27 studied class A and B extremities, eight showed telomeres further shortening between tel-m1-0 and tel-m1-1, with large variations in the rate of shortening. They were stabilized or even slightly increased in length between tel-m1-0 and tel-m1-1 in 16 cases. In the remaining three extremities, the very low or even absence of reads in tel-m1-0 and/or tel-m1-1 prevented comparison and suggested the loss of the telomeric repeats or subtelomere sequence. Telomere length distribution at each extremity also displayed variations in tel-m2-1.
Extrachromosomal telomeric DNA molecules, predominantly found as double-stranded or single-stranded circles, are detected in ALT tumors and proposed to be involved in telomere maintenance (Cesare and Reddel 2010). To detect potential circular DNA containing telomere sequences, we performed rolling circle amplification (RCA) assays using the ϕ29 polymerase followed by qPCR to measure telomeric content compared with a control without ϕ29 amplification (Henson et al. 2017). As a positive control, we used a 4.4-kb yeast circular plasmid (pRS306), containing the URA3 gene that we targeted for qPCR, which we spiked into CC-4533 genomic DNA at a relative concentration of 100 plasmid molecules per genome. Although RCA performed on the plasmid led to an approximately eightfold increase in URA3 signal, no ϕ29 amplification of the telomeric content was detected for CC-4533 or the telomerase mutants (Supplemental Fig. S3B), suggesting that RCA templated by telomeric circles is likely not the main telomere maintenance mechanism in telomerase mutants in C. reinhardtii.
To test if a subset of telomere sequences was completely lost, we computed the total number of telomere-containing reads normalized to the sequencing depth of the nuclear genome and found that the average number of telomeres per cell was smaller in the mutants: 22 in tel-m1-0, 18 in tel-m1-1, and 25 in tel-m2-1 versus 36 in CC-4533 (which has an extra mini-Chromosome 1 in addition to the 17 chromosomes) (Fig. 2B).
Overall, telomeres shortened as expected in telomerase mutants, but our data revealed the heterogeneity of shortening pattern at each specific telomere and suggested that telomeres were maintained or newly formed in tel-m1-1 compared with tel-m1-0 in a telomerase-independent manner. In the next sections, we investigate chromosomal rearrangements occurring at subtelomeric regions.
CNVs of subtelomeric elements in telomerase mutants
As a first insight into the stability of repeated subtelomeric elements (rDNA, Suber, and Sultan), we computed their CNVs in telomerase mutants compared with CC-4533.
We used the rDNA sequence as a query to find all reads containing rDNA. We observed an overall decrease of 31%, 36%, and 29% in rDNA copy number in tel-m1-0, tel-m1-1, and tel-m2-1, respectively, compared with CC-4533 (Fig. 2C). We also found that, although in the wild type, it was exclusively found at three subtelomeres as expected (Chaux-Jukic et al. 2021), rDNA could be mapped at several additional regions of the genome in telomerase mutants, implicating rDNA in genome rearrangements (see below). The two major rDNA clusters at subtelomeres 8R and 14R likely lost a large fraction of their rDNA copies.
The same analysis was performed with the Suber element, which is found as three main subtypes of sizes ∼1.5 kb, ∼1.9 kb, and ∼2.5 kb (Hails et al. 1993; Chaux-Jukic et al. 2021), and showed changes specific to each subtype (Fig. 2D). The copy number of the 1.5-kb element was greatly increased in the telomerase mutants compared with the wild type, especially in tel-m1-0, in contrast to the 2.5-kb element, which decreased in copy number in all mutants. The number of 1.9-kb elements only decreased in tel‐m1‐1. Given the organization of Subers in arrays containing only one subtype (Chaux-Jukic et al. 2021), the different changes in copy number depending on the subtype likely reflected duplicated, deleted, and potentially rearranged arrays.
Finally, the Sultan element, the most abundant and widespread repeated element in subtelomeres, was decreased in copy number in both telomerase mutants (Fig. 2E).
Overall, all the main subtelomeric repeated elements showed CNVs in the telomerase mutants, indicating that subtelomeres are involved in chromosome rearrangements.
Contraction, erosion, and expansion of Sultan arrays
To analyze subtelomere structure, we looked for the presence of the Spacer element in all reads because this element is present as a single copy in 29 out of 34 subtelomeres in the wild-type strain, with sufficient sequence differences to uniquely identify the corresponding subtelomere (Chaux-Jukic et al. 2021). Reads containing a Spacer element were thus grouped according to their Spacer and displayed as a schematic representation of all the sequence features they contain (Supplemental Data Set 1).
Although for all Spacers, reads from CC-4533, even after hundreds of generations of culture, were consistent with a subtelomeric structure identical or nearly identical to the one we inferred from the genome assembly of CC-1690 (Supplemental Data Set 1; Supplemental Fig. S1C; Chaux-Jukic et al. 2021), reads from the telomerase mutants revealed many differences compared with CC-4533, including a large variety of rearrangements (Table 1; Supplemental Data Set 1).
Summary of rearrangements classified by types, found at Spacer-containing subtelomeres
We first focused on subtelomeres composed, in CC-4533, of an array of Sultan elements as the only repeated element, which are the vast majority (class A and B, 27 subtelomeres out of 34). In CC-4533, at each class A/B Sultan subtelomere, the reads were consistent with a single population of subtelomeres with a fixed number of Sultans and capped by telomeric repeats, as supported by a majority of reads (Fig. 3A,D). Reads that did not reach the extremity of the chromosome were explained by DNA molecules being physically broken during the extraction and preparation procedure and would be expected to terminate at random position in the Sultan array and within a Sultan element, which we confirmed (Fig. 3B,E). Additionally, in CC-4533, all reads containing simultaneously a specific Sultan element and a telomere sequence could be retrieved, and their number was often close to the number of Spacer-containing reads (Supplemental Fig. S4A), consistent with the fact that Spacer-containing reads did not always reach a telomere sequence because of the physical breakage of the extracted DNA molecule. Both telomerase mutants frequently showed reads that also supported a fixed number of Sultans (Fig. 3A,C). We call such subtelomeres “stabilized Sultan arrays.” However, the number of Sultan elements could differ from CC-4533 (Fig. 3C): In tel-m1-1 and tel-m2-1 combined, we observed a decreased number of Sultans in six cases by two to 32 repeats, and in one case on the contrary, the number of Sultan elements was slightly increased by one. For the rest of the cases (n = 11), no variation of the number of Sultans was observed. We found that stabilized Sultan arrays in telomerase mutants were capped by short telomere sequences (Fig. 3A; Supplemental Figs. S3A, S4A), suggesting that these telomeres were maintained in a telomerase-independent manner.
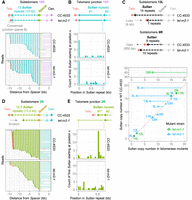
Contraction, erosion, and expansion of Sultan arrays. (A) The stabilized 10R Sultan array of tel-m2-1 capped by a telomere sequence is depicted in comparison to CC-4533. Nanopore reads, colored based on the element (as indicated in the schematic representation; black segments correspond to sequences that did not align to any of the selected queries) and anchored at the Spacer, are shown. (B) For each read, the starting position of the Sultan element adjoining the telomere sequence is recorded, and the overall distribution of counts over the length of a Sultan element is represented. (C) Schematic representation of contraction and expansion of stabilized Sultan arrays (top) and overall changes in Sultan number in stabilized Sultan arrays for tel-m1-1 and tel-m2‐1 (bottom). (D) Same as A with the unstable 2R Sultan array of tel-m2-1. (E) Same as B but for the unstable 2R Sultan array of tel-m2-1. (*) The first Sultan element in CC-4533 is partial.
We then asked whether at the extremities of stabilized Sultan arrays with a decreased number of Sultan elements (n = 6), the telomeres could be established de novo after complete loss or whether Sultan elements were excised internally independently of the initially present telomere sequence. To address this point, we identified the junction between the telomere sequence and the first Sultan element and compared it between CC-4533 and the mutants at the level of individual reads (Fig. 3B; Supplemental Fig. S4B). In two out of six shorter stabilized Sultan arrays (i.e., 4L and 5L in tel-m2-1), we found that the telomere sequence transitioned into the Sultan element at a different position within the Sultan compared with CC-4533, suggesting that the telomeres were added de novo on truncated Sultans (Supplemental Fig. S4A,B). In the remaining four cases (15R in tel-m1-1 and 8L, 13L, and 9L in tel‐m2-1), telomeres were found at the same position of the first Sultan in CC-4533 and the mutants, suggesting that changes in the number of Sultan elements of the array could also be because of excision of internal Sultan elements, without affecting the first Sultan.
At some subtelomeres (three for tel-m1-1 and six for tel-m2-1), the reads were not consistent with a stabilized number of Sultan elements, and the vast majority had different counts, suggesting a heterogeneous population of cells in which that specific extremity shortened progressively. These Sultan arrays were qualified as “unstable.” This situation is exemplified by subtelomere 2R in tel-m2-1 (Fig. 3D). Consistent with a progressive erosion of the extremity, the first Sultan element ended at different positions of the Sultan element in different reads and was often not capped by telomere sequences (Fig. 3E; Supplemental Fig. S4A,B). To rule out that these reads containing a variable number of Sultan elements were not simply owing to physically broken DNA molecules, we searched for all reads containing simultaneously the same Sultan elements and telomeric repeats and compared their number with the number of reads containing the corresponding Spacer (Supplemental Fig. S4A). In contrast to CC-4533, unstable Sultan arrays in the mutants showed fewer telomere-containing reads than Spacer-containing ones, indicating telomere loss and supporting the idea that these extremities truly ended with Sultan elements. The progressive loss of telomeric repeats and Sultan elements was consistent with the overall decrease in the number of telomeres and Sultan copy number (Fig. 2B,E). Erosion of Sultan arrays would eventually lead to their complete loss, a situation that we could clearly evidence for subtelomere 7L (Supplemental Fig. S5A): In tel-m1-0, the subtelomere contained only three Sultans compared with eight in CC-4533, but in tel-m1-1, the extremity was eroded beyond the Spacer and a new telomere was formed with the additional translocation, in a subpopulation of reads, of a sequence from Chromosome 11. The length distributions of the telomere sequence in these two subpopulations were consistent with their position as a terminal telomere sequence and ITS (Supplemental Fig. S5B). Analysis of the junctions revealed a 4-bp microhomology with the telomere sequence (Supplemental Fig. S5C). An analogous comparison between CC-4533, tel‐m1‐0, tel-m1-1, and tel-m2-1 for the class C subtelomere 15L tended to suggest that Suber arrays could also be progressively eroded until completely lost (Supplemental Fig. S5D).
Finally, we asked whether there might be extrachromosomal circular Sultan-containing molecules preferentially enriched in telomerase mutants, which could act as templates to maintain Sultan subtelomeres or limit their erosion. We thus measured the Sultan content by qPCR in RCA products in CC-4533 and the telomerase mutants but found no ϕ29-dependent increase in any strain (Supplemental Fig. S3B).
Complex rearrangements within and between subtelomeres
In telomerase mutants, we observed a large fraction of telomere-containing reads with unusual combinations of subtelomeric elements (Table 1; Supplemental Data Set 1). To better characterize the structure of these rearranged subtelomeres, we again used the Spacer-based analysis to help anchor the reads to a given chromosome extremity.
Fusion between subtelomeric elements
Fusions of elements from different subtelomeres were the most frequent genome rearrangement occurring at subtelomeres (Table 1). The simplest type corresponded to the juxtaposition of two or more arrays of different Sultan elements, either in the same or in reverse orientations, such as subtelomere 6R in tel-m1-0 (Fig. 4A) or subtelomere 15R in tel-m2-1 (Supplemental Fig. S6A). Multiple subtelomeric elements of different types, such as rDNA and Sultan in subtelomere 6L of tel-m1-0 and tel-m1-1, were also observed (Fig. 4B; Table 1). Because rDNA and Sultan do not share sequence homology, we speculate their fusion stemmed from NHEJ-dependent translocation events or end-to-end fusion.
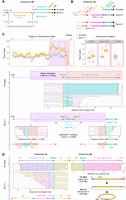
Complex genome rearrangements at subtelomeres. (A) Schematic representation of subtelomere 6R in which, in tel-m1-0, 2 additional Sultan arrays were fused to the initial Sultan array in inverted orientation. (B) Schematic representation of complex rearrangements at subtelomere 6L in tel-m1-0 and tel-m1-1, including Sultan elements from different subtelomeres and rDNA sequences. In tel-m1-1, two subpopulations of reads reveal two distinct structures stemming from the initial rearrangement found in tel-m1-0. (C) Signature of a BFB event at subtelomere 9R in tel-m1-1. (Top) Duplication of the last 170 kb of the chromosome end. Read depth is computed on the indicated regions of Chromosome 9. (Middle) Individual reads supporting the loss of telomeres and 36 Sultan elements, as well as the end-to-end fusion of the 9R sister chromatids. (Bottom) Reads showing the recruitment of >11 kb of rDNA sequences at the new 9R extremity, disrupting an array of MSAT-4B satellite sequences. (D) Representation of reads supporting the fusion of subtelomeres 4L and 4R in tel-m1-1, after complete loss of telomeres and a total of 27 Sultan elements. (Bottom right) Scheme of the inferred circularization of Chromosome 4, also supported by de novo genome assembly (Supplemental Fig. S6E,F). Black segments correspond to sequences that are not aligned to any of the selected queries.
The complexity of the observed genome rearrangements suggested that they formed through a multistep process. To test this hypothesis, we compared the rearrangements found in tel-m1-0 to those in tel-m1-1, as these two samples were collected at different times. We could find intermediate rearrangement states in the earlier tel-m1-0 sample, suggesting that at least some complex rearrangements were formed in multiple steps over time (Fig. 4B; Supplemental Figs. S5A, S6B). For example, at subtelomere 6L, the already complex structure found in tel-m1-0 further changed in tel-m1-1 and diverged into two structurally distinct subpopulations (Fig. 4B). As another example, subtelomere 17L displayed a reduced number of Sultan elements in tel-m1-0 compared with the wild type with a subpopulation of reads capped by telomere sequences, representing a shortened stabilized Sultan array, but contained in tel-m1-1 additional sequences from a (CA)n microsatellite, from Chromosome 15 and from 5R Sultan elements, and fused to the remaining 17L Sultans (Supplemental Fig. S6C).
Signature of BFB events
The read coverage mapped to the assembly of tel-m1-1 genome identified a duplicated region at the extremity of Chromosome 9R (Fig. 4C, top). Closer inspection of the reads anchored by their Spacer element revealed a pattern of two inverted 9R Sultan arrays fused to each other in a head-to-head manner (Fig. 4C, middle). Each array was adjacent to its own 9R Spacer element and downstream 9R-specific sequences. Overall, this configuration was indicative of a fusion between sister chromatids followed by a breakage in anaphase, an event that initiated a canonical BFB cycle until the extremities were stabilized. We then looked for the new 9R extremity in tel-m1-1 and identified the reads that mapped close to the 1×/2× coverage boundary. Reads that mapped only to the 2× side continued with at least two rDNA units over >11 kb (Fig. 4C, bottom). The rDNA sequence disrupted a RTEX-1 long interspersed nuclear element (LINE) retrotransposon framed by two arrays of MSAT-4B satellites, as shown in Figure 4C. What lay beyond the rDNA array remained unknown, in particular whether the extremity was capped by additional telomere sequences. In another example, the structure of the 11R extremity of tel-m1-0 was consistent with at least two cycles of BFB, based on the multiple inverted repeats (Supplemental Fig. S6D). We found a total of five events consistent with BFB in tel-m1-1 (Table 1), indicating that BFB cycles were a common mechanism at play once telomeres were lost.
Circularization of Chromosome 4
We report the head-to-head fusion of subtelomeres 4L and 4R in tel-m1-1, after complete loss of the telomere sequences and partial loss of Sultan elements (from 15 to two for 4L and from 21 to seven for 4R) (Fig. 4D). The de novo assembly of Chromosome 4 in tel-m1-1 without relying on scaffolding with a reference sequence indicated that Chromosome 4 was made of a single contig with no extremity and had therefore a circular structure (Supplemental Fig. S6E,F). Circularization of the chromosome bypasses the need for functional telomeres to stabilize the extremities of linear chromosomes.
New telomeres outside canonical subtelomere regions lead to drastic chromosome rearrangements and karyotype alterations
We next investigated terminal telomere–containing reads not associated with the canonical subtelomeric elements, with the aim of revealing the formation of new extremities outside their normal subtelomeric contexts. Using TeloReader, we gathered all reads containing telomere sequences that were not associated to known subtelomeric elements (Sultan, Spacer, Suber, Subtile, or rDNA). We mapped these reads on the genome of CC-4533 and found such subtelomere-less telomeres at three and two locations in tel-m1-1 and tel-m2-1, respectively. Consistent with the genome assembly, we also detected the new telomere-capped extremity of the duplicated mini-Chromosome 1 from CC‐4533, which transitioned directly into the Chromosome 1 sequence through a microhomology and without a subtelomeric element or other exogenous sequence (Supplemental Fig. S1B).
At subtelomeres 14L in tel-m2-1 and 7L in tel-m1-1 (Fig. 5A; Supplemental Fig. S5A–C), the telomere sequences connected to the chromosome arm several kilobases away from the expected telomere location in CC-4533, and the corresponding arrays of Sultan repeats were lost, but without loss of annotated genes. At these novel telomere junctions, the chromosome arm showed no sequence homology with telomeric repeats, suggesting that the telomere was recruited at these sites by NHEJ rather than homology-mediated mechanisms.
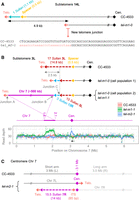
New extremities without canonical subtelomeric elements. (A) Scheme of the loss of 4.9 kb of sequence and formation of a new telomere at subtelomere 14L of tel-m2-1. The new junction sequence is shown. (B) Scheme of the complex new 3L extremity in tel-m1-0 and tel-m1-1. The sequencing depth for Chromosome 7 is shown, revealing the duplicated 500-kb region in tel-m1-1. The sequences of Junction A, B, and C are shown in Supplemental Figure S7, A through C. (C) Scheme of the Robertsonian fission that occurred on Chromosome 7 of tel-m2-1. The new centromere-proximal extremities were stabilized by telomere sequences, with in addition a Sultan array and an ITS for the 7R long arm.
The other new telomeres were involved in more complex rearrangements. The subtelomere 3L of tel‐m1‐0 had 13 Sultan elements remaining after loss of the telomere sequence and four Sultan elements, followed by a short array of 13R Sultan elements and telomere sequence (Fig. 5B). Because it was present in a subpopulation of tel-m1-0 reads and in tel-m1-1, this structure likely represented the first rearrangement step. Subtelomere 3L was further altered by the loss of the telomere and the fusion of 500 kb of duplicated sequence originally located at 2.1 Mb from the end of the left arm of Chromosome 7, which was then capped by a new telomere (Fig. 5B). Analysis of the junctions between these rearranged segments suggested that microhomology-dependent mechanisms were at play for the last step: junctions to telomeres (junction A) and to the 13R Sultan array (junction B) indeed involved microhomologies of 2–4 bp (Supplemental Fig. S7A,B). In contrast, the Sultans from 13R and 3L were connected with the insertion of a short sequence of unknown origin (junction C) (Supplemental Fig. S7C) but without homology or truncation of the neighboring Sultan elements, suggesting an NHEJ-dependent translocation that simultaneously incorporated a short piece of DNA.
On Chromosome 12 of tel-m1-1, the 2-Mb distal part of the right arm was translocated to the subtelomere 5L, and the new right end of Chromosome 12 carried a terminal telomere sequence preceded by a tandem duplication consisting of a fragment from Chromosome 12, nine telomeric repeats forming an interstitial array, and a short Sultan array from subtelomere 7L (Supplemental Fig. S7D,E).
In tel-m2-1, new telomeres were found at two close locations around the centromere of Chromosome 7, at the extremities of a 90-kb region displaying a 2× sequencing depth, indicating that the centromere was duplicated and the two arms of Chromosome 7 split into two telocentric chromosomes (Fig. 5C), thus constituting a Robertsonian fission, the reciprocal event of a Robertsonian fusion whereby two telocentric chromosomes are combined through their long arm with the loss of the very short arms and one centromere (Robertson 1916; Jones 1998). On the left end of the right arm, the new telomere capped an array of 7R Sultan lacking Spacer and a 65-bp ITS. On the right end of the left arm, the new telomere directly transitioned into the centromere.
Overall, duplications, nonreciprocal translocations, deletions, and more complex rearrangements created new extremities outside canonical regions and, in some cases, even altered the karyotype of the telomerase-mutant strains. For each new extremity, stability seemed to be ensured by newly recruited telomere sequences.
DNA methylation is maintained at chromosome extremities and at displaced Sultan arrays
We previously showed that Sultan arrays and, to a lesser degree, Suber arrays were hypermethylated, whereas the two major rDNA clusters were hypomethylated except for a few telomere-proximal repeats (Chaux-Jukic et al. 2021). Sultan subtelomeres are also associated with the heterochromatin mark H3K9me1 (Strenkert et al. 2013). Because subtelomeres in telomerase mutants were heavily rearranged, we asked whether DNA methylation remained associated with the Sultan elements that were no longer close to chromosome extremities and whether new extremities would acquire hypermethylation.
We thus base-called 5-methylcytosine (5mC) at CpG sites using Nanopolish (Simpson et al. 2017) and focused on rearranged loci. We first investigated the complex rearrangement found at extremity 3L of tel-m1-1, where a duplicated 500 kb of Chromosome 7 capped with telomeric repeats formed the new extremity and the original 3L Sultan elements were thus located much more internally (>500 kb) in the chromosome (Fig. 5B). To analyze 5mC content of the new extremity, we selected reads that aligned to the 500 kb of Chromosome 7 and at the same time contained the terminal telomere sequence and, in a second control group, reads that aligned to the 500-kb region but also continued beyond on Chromosome 7 (Fig. 6A; Supplemental Fig. S8A). The new 3L extremity was methylated over a length of ∼12 kb, whereas the same sequence on its original locus on Chromosome 7 was unmethylated and so was the same locus in the wild-type strain (Supplemental Fig. S8A). The 3L Sultan elements as well as the neighboring 13R Sultans in tel-m1-1 maintained their high methylation status despite being now >500 kb away from the extremity (Fig. 6A).
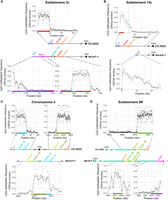
Methylation frequency of new extremities and displaced Sultan elements. (A–D) Analysis of 5mC frequency at CpG sites using reads that unambiguously spanned the indicated regions, in different illustrative cases. (A) At the rearranged 3L chromosome arm of tel-m1-1. Methylation frequency was plotted over different regions of subtelomere 3L in tel-m1-1 and in CC-4533 as depicted. For the methylation frequency at Chromosome 7 in CC-4533 and tel-m1-1, see also Supplemental Figure S8A. (B) At the broken 14L subtelomere of tel-m2-1, where the internal genomic sequence was then capped by a telomere sequence, compared with CC-4533. (C) At the fused 4L and 4R Sultan arrays of tel-m1-1, compared with the two arrays in CC-4533. (D) At the head-to-head fused 9R Sultan arrays and the transition to rDNA in tel-m1-1, compared with the 9R extremity of CC-4533.
That new extremities were hypermethylated even though they did not contain canonical subtelomeric sequences was confirmed at the truncated 14L extremity of tel-m2-1 (Figs. 5A, 6B) and 7L of tel‐m1‐1 (Supplemental Fig. S8B). Other examples of Sultan elements no longer located at subtelomeres but still methylated included the 4L and 4R Sultan elements of the circularized Chromosome 4 in tel-m1-1 (Figs. 4D, 6C) and the head-to-head fused 9R Sultan elements, which resulted from the BFB event we described in a previous section (Figs. 4C, 6D). In the latter example, the Sultan elements were located at least 185 kb away from the new extremity. The reads mapping to the rDNA sequence at the 9R subtelomere showed that the rDNA was methylated (Fig. 6D), which in CC-1690 was only true for the rDNA of subtelomere 1L and a few telomere-proximal rDNA sequences at 8R and 14R subtelomeres, suggesting that the rDNA might constitute the new subtelomere of 9R in tel-m1-1. Subtelomere 16R in tel-m2-1 provided another example of translocated rDNA, now directly between a terminal telomere sequence and Sultan elements, which was also hypermethylated (Supplemental Fig. S8C).
Overall, the Sultan elements that were no longer near chromosome extremities maintained their hypermethylation level. This observation was verified in all cases in tel-m1-1 and tel-m2-1 for Sultan arrays separated from the extremity by up to >500 kb of other nonmethylated sequences. Conversely, sequences that became capped by telomeres and formed new extremities acquired a hypermethylation pattern.
Discussion
Telomeres and subtelomeres are extensively implicated in genome instability induced by telomere shortening or dysfunction, as exemplified in post-telomere-crisis tumors (Maciejowski and de Lange 2017). Many types of telomere-related rearrangements have been studied in experimental systems of different model organisms and in tumor samples from patients. However, a direct assessment and comprehensive picture of the repertoire of genome rearrangements that functional telomeres protect from have rarely been achieved. Long-read sequencing has recently been used for genome assembly of C. reinhardtii and to assess the spectrum of SVs in various strains (Liu et al. 2019; O'Donnell et al. 2020; Craig et al. 2023; López-Cortegano et al. 2023; Payne et al. 2023). Here, we took advantage of long-read Nanopore sequencing to access telomere-induced SVs that would have escaped detection from short-read sequencing or at least would have been difficult to identify, particularly in repeated regions. We chose to sequence heterogeneous populations of telomerase-negative cells containing a complex mixture of subclonal genome rearrangements and to perform our analyses mainly at the level of reads, revealing mosaic rearrangement patterns that would have been missed in assembled genomes. A limitation of this approach is that we are unable in principle to assign specific rearrangements at different loci to the same cell or population of cells. However, most mosaic rearrangements we detected affect a substantial fraction of reads (typically > 10%–20%), and some are thus likely to co-occur in the same cell. On the other hand, rearrangements observed for all reads should be present in the vast majority of the population.
Alternative telomere maintenance
We previously showed that telomeres in C. reinhardtii are maintained by telomerase and that mutants of TERT1, the gene encoding its catalytic subunit, displayed an “ever-shorter telomere” phenotype, which led, in some telomerase-negative cultures derived from backcrosses, to growth defect and cell death consistent with senescence (Eberhard et al. 2019). Because they appear to maintain their telomeres at a short equilibrium length and do not display obvious growth defects, the tel-m1 and tel-m2 mutants are possibly already in a postsenescence state, although this remains to be shown formally. In this work, analysis of chromosome extremities in the long-term cultures of telomerase mutants showed that overall ∼60% were still capped by short telomere sequences, reminiscent of type I postsenescent survivors in S. cerevisiae (Lundblad and Blackburn 1993; Teng and Zakian 1999). For at least a subset of them, the telomere sequence was likely recruited de novo on a truncated subtelomere or on a new chromosome extremity without canonical subtelomere by nonreciprocal translocation, as shown before for cancer cell lines (Sabatier et al. 2005). In all cases, telomeres were maintained, albeit at a short length distribution, which implied that an alternative maintenance mechanism was used, relying, for example, on homologous recombination or BIR but most likely without using extrachromosomal telomere circles.
A number of chromosome extremities completely lacked telomere sequences, and their lost telomeres might have been translocated to the new telomere sites we described. Individual read analysis of such telomere-less extremities showed that the Sultan array composing the subtelomere displayed a length distribution at the population level consistent with progressive loss of sequence. This observation leads to the intriguing possibility that the array of Sultan elements might directly function in end protection instead of telomeric repeats. Consistently, in end-to-end fusion events involving Sultan arrays (e.g., Fig. 4C,D), fewer than 10 Sultan elements were left when fusion occurred, which might suggest that larger Sultan arrays could confer some partial telomere protection. The Sultan element must then ensure telomere protection by, for example, binding a yet-unknown factor, which might be achieved through the short telomere-like sequence at the beginning of the element (Chaux-Jukic et al. 2021) or through another intrinsic binding site. Because Sultan elements are associated with heterochromatin, indirect binding of factors on modified histones might also provide sufficient protection, as proposed for Drosophila melanogaster and some telomerase-negative Schizosaccharomyces pombe survivors (Gao et al. 2010; Jain et al. 2010). Consistently, even when the chromosome extremities are not composed of Sultan elements, but of rDNA repeats or even other sequences, the DNA sequence is highly methylated (see discussion below).
Another strategy to maintain chromosome integrity without telomerase consists in chromosome circularization, which we observed for Chromosome 4 in tel-m1-1. Although we encountered only one such circularization event, the fact that we did not detect another subpopulation with another structure for Chromosome 4 suggests that this circular chromosome conferred some selective advantage (e.g., telomerase-independent genome integrity) or at least was not counter-selected. A similar circularization strategy in response to telomerase absence or disruption of telomere protection was also described in other eukaryotes (Naito et al. 1998; Nakamura et al. 1998; McEachern et al. 2000; Wu et al. 2020; Baumann and Cech 2001) and therefore appears to be relatively well tolerated across evolution.
Overall, although they came at the cost of wide-spread genome rearrangements, the diversity of alternative strategies to maintain chromosome integrity in telomerase mutants found in a single organism is remarkable and illustrates genome plasticity at short timescales.
Mechanisms of genome rearrangements in telomerase mutants
Because the approach used in this work consists in a direct visualization of chromosome sequences on a large scale with as little source of bias as possible, we were able to document a large variety of rearrangements. The fact that most rearrangements involved telomeres and subtelomeres suggested that critically short or dysfunctional telomeres preferentially induced local instabilities, which can propagate through various mechanisms such as BFB cycles involving dicentrics, as reported in yeast and human tumors (Hackett et al. 2001; Hackett and Greider 2003; Pobiega and Marcand 2010; Beyer and Weinert 2016; Maciejowski and de Lange 2017; Umbreit et al. 2020).
Short or dysfunctional telomeres, at the senescence stage or during crisis, can lead to end-to-end fusions as observed across evolution (Chan and Blackburn 2003; Mieczkowski et al. 2003; Heacock et al. 2004; Pardo and Marcand 2005; Capper et al. 2007; Lowden et al. 2008), although telomere sequences themselves may be completely absent at fusion sites in telomerase-negative cells (Blasco et al. 1997; Naito et al. 1998; Nakamura et al. 1998; Hackett et al. 2001). Despite many instances of end-to-end fusions, we found no remaining telomere sequences at fusion sites in our read data sets, suggesting that in C. reinhardtii, even very short telomeres might be sufficient to inhibit fusions. Instead, we detected many subtelomere–subtelomere fusions, the most frequent involving Sultan arrays. They can occur between different subtelomeres, as in the case of Chromosome 4 circularization, or between sister chromatid subtelomeres. Sister chromatid fusions were followed by cycles of BFB, propagating genome instability over multiple divisions and over more internal genomic regions, as for extremity 9R in tel-m1-1. Of note, we did not find any signature of chromothripsis resulting from the aberrant repair of the fragmentation of a chromosomal region, despite chromothripsis being associated with BFB cycles and telomere crisis in cancer (Garsed et al. 2014; Li et al. 2014).
Most rearrangements implicated the repeated elements that are found at the subtelomeres, which could promote homology-mediated mechanisms for amplification, deletion, and translocation. The activation of these mechanisms could contribute to the alternative maintenance strategy, but these rearrangements could also have been induced during senescence and crisis before the emergence of postsenescence survivors. We note, however, that if we assume that tel-m1-0 already represented a postsenescent state, rearrangements were still ongoing between tel-m1-0 and tel-m1-1, thus suggesting that they were not limited to the senescence stage. When different types of elements were found juxtaposed, the transition sequence often involved microhomologies of a few base pairs (e.g., Fig. 5B; Supplemental Fig. S7A,B), suggesting that mechanisms such as microhomology-mediated break-induced replication (MMBIR) (Payen et al. 2008; Hastings et al. 2009) or microhomology-mediated end-joining (MMEJ) (Sfeir and Symington 2015) might be involved. Even when no homology or microhomology was detected at junctions, canonical subtelomeric elements were still frequently found in rearrangements. We speculate that these subtelomeric elements were frequently excised from their native locus owing to terminal instability and were therefore available in subsequent fusion events. Nevertheless, other internal genomic regions were sometimes duplicated and translocated to chromosome ends, supporting the idea of a genome-wide increase in genome instability in telomerase-negative cells (Blasco et al. 1997; Lee et al. 1998; Chin et al. 1999; Hackett et al. 2001; Coutelier et al. 2018).
New chromosome extremities establish heterochromatin
We found new chromosome extremities that were capped by telomere sequences, suggesting that, in C. reinhardtii, telomeric repeats by themselves are sufficient for end protection in a context devoid of canonical subtelomeric elements. These new telomere-capped chromosome extremities formed by sequences originating from internal genomic regions showed high levels of 5mC. We therefore speculate that telomeres can establish new DNA methylation domains in C. reinhardtii, likely to be associated with heterochromatin. Although the heterochromatic nature of telomeres and subtelomeres is conserved in eukaryotes (except for Arabidopsis thaliana where it is less well established) (Gottschling et al. 1990; Baur et al. 2001; Koering et al. 2002; Pedram et al. 2006; Vrbsky et al. 2010; Vaquero-Sedas et al. 2011; Elgin and Reuter 2013; Matsuda et al. 2015), whether and how heterochromatin forms at a new telomere are less well known across organisms. The detection of new extremities in our work thus provides an additional piece of evidence for heterochromatin formation spreading from the telomere sequence being a property of chromosome extremities. Because many chromosome extremities were not capped by telomere sequences but directly by the hypermethylated Sultan arrays, one can speculate that heterochromatin might be a functional feature of a protected extremity in telomerase mutants of C. reinhardtii.
We note that Sultan elements that are displaced from their normal subtelomeric location retain a high level of 5mC, even when found at >500 kb of the extremity of the chromosome. Because we previously showed that Sultan elements are exclusively found at subtelomeres in wild-type strains, we propose that their sequence coevolved with their function at subtelomeres, so that they might recruit methyltransferases to actively contribute to heterochromatin maintenance and spread. This property would allow them to remain hypermethylated even when displaced from subtelomeres. In most cases, the Spacer sequence still acted as the boundary of the hypermethylated domain even when not located at subtelomeres.
To conclude, our results show that C. reinhardtii telomerase-negative cells use a variety of strategies to protect chromosome extremities, including the establishment of DNA methylation, and undergo diverse and complex rearrangements, highlighting a remarkable plasticity of the genome. This work shows the potential of long-read sequencing to provide a comprehensive view of complex genome rearrangements even in mosaic populations of cells at the level of subclones, an analysis that could be applied to tumor genome heterogeneity.
Methods
Strains and growth conditions
Wild-type CC-4533 and tel-m1 (CLiP library identifier: LMJ.RY0402.077111; tert1-1 allele) and tel-m2 (CLiP library identifier: LMJ.RY0402.209904; tert1-2 allele) mutant strains were obtained from the Jonikas laboratory (Li et al. 2016). They were maintained on plates in TAP medium (Harris 2009) at 25°C under low light (5 µE/m2/sec) and restreaked in bulk without subcloning. Before DNA extraction, cells were grown in 200 mL liquid TAP medium until they reached ∼2 × 107 cell/mL and collected by centrifugation at 5000g for 5 min. We previously confirmed for each mutant strain that the single insertion of the paromomycin resistance gene in TERT1 (gene identifier: Cre04.g213652_4532) disrupted the RNA-binding domain (tert1-1) or the catalytic domain (tert1-2), both essential for telomerase function (Eberhard et al. 2019). This was performed by PCR with primers flanking the gene, in the gene, and/or in the paromomycin resistance gene, followed by sequencing of the PCR products. The single insertion was verified by backcrossing to a wild-type strain and, after meiosis and tetrad dissection, the 2:2 segregation of the insertion, of the resistance phenotype, and of the short telomere phenotype. The single insertion was also confirmed in the genome assemblies of the mutants.
DNA extraction and size selection
To extract genomic DNA while preserving high-molecular-weight fragments, we developed a modified version of a Joint Genome Institute protocol (https://www.pacb.com/wp-content/uploads/2015/09/DNA-extraction-chlamy-CTAB-JGI.pdf), as described by Chaux-Jukic et al. (2022). The extraction is followed by a clean-up using magnetic beads and size selection based on solid-phase reversible immobilization (Stortchevoi et al. 2020) using the SRE kit (Circulomics).
RCA assay
RCA followed by qPCR detection and analysis was performed as described previously (Henson et al. 2017), with minor adaptations. Briefly, for each sample, 80 ng of genomic DNA in 10 µL of Tris (pH 7.6) was mixed with 10 µL of the ϕ29 reaction buffer with or without 7.5 units of ϕ29 (Thermo Fisher Scientific). The reaction was then performed for 10 h at 30°C , followed by ϕ29 inactivation for 20 min at 70°C. As a positive control, 0.32 ng of the yeast plasmid pRS306 containing the URA3 gene was spiked into 80 ng of CC-4533 genomic DNA, representing 100 plasmid molecules per genome. For detection of the reaction product, each sample was diluted fourfold, and 4 µL was used in a 20-µL qPCR reaction with Fast SYBR Green (Applied Biosystems) following the manufacturer's instructions and run in a CFX96 real-time PCR detection system (Bio-Rad). Technical qPCR triplicates were performed for each sample. We designed the following qPCR primers: for URA3, 5′‐ATGTCGAAAGCTACATATAAGG-3′ (oZX180) and 5′‐TAGTAAACAAATTTTGGGACCT-3′ (oZX568); for telomere sequences, 5′‐GGTATTTGTCAGGGTGTTAGGGTGTTAGGGTGTTAGGGT‐3′ (oZX576) and 5′‐TCCCGACTATATCCCGAAAACTCTAAATCCCTATAACCCTA‐3′ (oZX578); for the Sultan element, 5′-GGCTGCGTGGCTGGACTGCTGCACT-3′ (oZX579) and 5′‐CATTTCTGACATGTCACACTTTTCAAA-3′ (oZX572); and for ATPC, 5′‐TCGTTCATTGCTCAGGAGTC‐3′ (oZX574) and 5′‐AGCTTGAAGATCTCGTCGTC‐3′ (oZX575). ATPC is a single-copy nuclear gene, which we used for normalization. To design the primers for telomere detection, we adapted a strategy previously developed (Cawthon 2002) for the human telomere motif to C. reinhardtii’s: We added 6 nucleotides not matching the telomere sequence at the 5′ end of each primer, and we introduced several additional mismatches that do not impede annealing to the telomere sequence and PCR but prevent primer dimerization.
Library preparation and sequencing
Sequencing libraries were prepared from SRE-treated high-molecular-weight DNA (except for tel-m1‐1, which was sequenced in two runs, one without SRE treatment and one with), following Oxford Nanopore Technologies (ONT) protocols for genomic DNA without preamplification. Kits LSK109 and LSK110 were obtained from ONT and companion module NEBNext from New England Biolabs (NEB). As a minor modification, we started the preparation with 3 µg of DNA. The second tel-m1-1 library was barcoded using barcode NB07 from kit NBD104 (ONT).
DNA libraries were sequenced on R9.4 or R10.4 Nanopore flow cells in a MinION Mk1C sequencer with default parameters on the MinKNOW operating software, except for the base-calling with Guppy, which was set to real-time high accuracy, and for the MUX scan, which was decreased to 1 h. For each run, 500 ng of DNA was loaded and sequenced for 8–12 h. The flow cell was then washed using the wash kit (ONT) and reloaded with the same sample, up to four times.
Genome assembly
Reads in FASTQ files and with quality > 7 were processed using Porechop with default parameters (https://github.com/rrwick/Porechop), for removing adapters/barcodes and splitting artifactual chimeras. Reads were assembled using Canu (Koren et al. 2017), SMARTdenovo (Liu et al. 2021), and NextDenovo (https://github.com/Nextomics/NextDenovo) using default parameters, and all chromosomes were successfully represented by one main contig. These draft assemblies were polished using Racon (Vaser et al. 2017; https://github.com/lbcb-sci/racon) and Medaka (ONT; https://github.com/nanoporetech/medaka) with the same reads and then scaffolded on the CC-1690 reference genome using RagTag (Alonge et al. 2022; https://github.com/malonge/RagTag). These assemblies were compared with each other and with references (CC-1690, CC-4532) using D-genies (Cabanettes and Klopp 2018; https://github.com/genotoul-bioinfo/dgenies) to assess contiguity. To generate a genome model for each strain, we chose for each chromosome the assembly giving the most colinear chromosome with CC-1690.
To test if Chromosome 4 of tel-m1-1 was circular at the assembly level without relying on a reference genome for scaffolding, we used the assembler Flye (Kolmogorov et al. 2019; https://github.com/fenderglass/Flye), as it generated contigs long enough for this purpose. Visualization of the circular Chromosome 4 was performed using Bandage (Wick et al. 2015; https://rrwick.github.io/Bandage/).
Telomere sequence detection
To detect telomere sequences in individual Nanopore reads, we developed TeloReader, a Python (v3.9.12) script with only three dependencies (pandas [v1.4.3], numpy [v1.19.5], and Matplotlib [v3.5.1]). TeloReader scans each read in both directions, from 5′ to 3′ and from 3′ to 5′, to search for the C-rich telomere motif and the G-rich one, respectively. In the first step of TeloReader, the DNA sequence is transformed into a series of scores corresponding to the best alignment score (using pairwise2 from the package Biopython [v1.79]) of each 8-mer against all eight circular permutations of the telomere motif (CCCTAAAA for the C-rich and TTTTAGGG for the G-rich). This score is between zero and eight, but all scores lower than four are replaced by four to reduce the impact of sequencing errors in the following step. The second step defines the sequence in a sliding window of 15 bp (size_window) as telomeric if the average score is greater or equal to seven (min_mean_window). In a third step, consecutive overlapping telomeric sliding windows are merged to form a single telomere sequence. We added other constraints and rules to ensure the specificity and sensitivity of TeloReader: The minimal length of a telomere sequence is set at 16 bp (min_len); a telomere sequence must contain at least eight scores of eight; and a configuration in which a nontelomeric sequence is found between two telomere sequences (as defined after the third step) can be considered as a telomere sequence if the nontelomeric sequence is <20 bp (max_size_gap) and if the average score of the whole sequence encompassing the two telomere sequences and the nontelomeric sequence in-between is greater than 6.5 (min_mean_telo). A telomere sequence is considered as terminal if it is found at <50 bp from the extremities of the input sequence (3′ extremity for a C-rich telomere and 5′ extremity for a G-rich telomere). See Data access section below.
5mC detection
To detect 5mC in a CpG context, we used nanopolish (Simpson et al. 2017; https://github.com/jts/nanopolish) on the FAST5 files and their associated FASTQ files. Reads were aligned to their corresponding genome assembly using minimap2 (Li 2018; https://github.com/lh3/minimap2). Alternatively, for detection of 5mC in subpopulations, reads were first selected using fast5_subset (https://github.com/nanoporetech/ont_fast5_api) and aligned to a consensus sequence obtained through a multiple alignment using ClustalW (Larkin et al. 2007). The frequency of methylation for each base was obtained using nanopolish call-methylation and the script calculate_methylation_frequency.py.
Bioinformatic analysis
For genome-to-genome comparison, genome models were aligned against references genomes (CC-1690 and CC-4533) using minimap2 (Li 2018) and visualized with Circos plots (Krzywinski et al. 2009). The SV caller MUM&Co (O'Donnell and Fischer 2020; https://github.com/SAMtoBAM/MUMandCo) was used for structural variant detection and classification. For read analysis, reads were mapped to genomes using minimap2 with the following parameters: -a -x map-ont -K 5M -t 3. We used Integrative Genome Viewer (IGV) (Robinson et al. 2017; https://igv.org/) and Tablet (Milne et al. 2013; https://ics.hutton.ac.uk/tablet/) to visualize read mapping. To calculate sequencing depth, the genomes were divided into windows using “makewindows” from BEDTools (Quinlan and Hall 2010; https://bedtools.readthedocs.io/en/latest/index.html), and then depth was averaged over the windows using “bedcov” from SAMtools (Li et al. 2009; https://www.htslib.org/) with the following parameters: -g SUPPLEMENTARY with or without -q 60.
For read-level analysis, subtelomeric elements (Spacer, Sultan, rDNA, and Suber) were searched in reads using BLASTN (Altschul et al. 1990) with the following parameters: -max_target_seqs 10000 -evalue 0.001 -outfmt “6 qaccver qlen saccver slen pident length mismatch gapopen qstart qend sstart send evalue bitscore.” Multiple hits at close positions were filtered using a custom bash script: Hits were sorted by decreasing order of bitscore (sort -rgk 14), and overlapping matches were eliminated. Reads containing subtelomeric elements or telomere sequences (detected by TeloReader) were further blasted against a library of repeated elements (Craig et al. 2021) and against reference genome, both of which were then filtered as above to exclude multiple matches. Annotations from all BLAST hits were then plotted on reads using R statistical software (R Core Team 2021). All lists of filtered BLAST hits lists were merged with “bind_rows”; reads of interest were identified (e.g., match to a given Spacer or Sultan) with “filter”; and all hits on this read subset were extracted with “semi_join.” To order these reads, first, an anchor was selected, most often a specific Spacer sequence, allowing setting an origin and the orientation of each read. From these two pieces of information, new coordinates were recalculated for each hit along the reads and for read extremities. Hits were then plotted with distinct reads along the y-axis and coordinates along the x-axis (e.g., Figs. 3A,D, 4C,D). Because of the error rate of Nanopore sequencing at the raw read level together with rare artifactual chimeric reads (Delahaye and Nicolas 2021), we only describe rearrangements structurally supported by at least two reads.
Computations were mostly run on the cluster of the French Institute of Bioinformatics (https://www.france-bioinformatique.fr/en/home/).
Data access
All sequencing data, which include raw Nanopore FAST5 files and read FASTQ files, and genome assemblies (as FASTA files) generated in this study have been submitted to the European Nucleotide Archive (ENA; https://www.ebi.ac.uk/ena/browser/home) under project accession number PRJEB59713. The code for TeloReader is available as Supplemental Code and at GitHub (https://github.com/Telomere-Genome-Stability/Telomere_2023/tree/main/TELOREADER).
Competing interest statement
The authors declare no competing interests.
Acknowledgments
We thank Olivier Vallon for his critical reading of the manuscript, Maria Teresa Teixeira for reagents and plasmids, and Oana Ilioaia for her technical assistance. Research in G.F.’s laboratory was supported by the French National Research Agency (“ANR” grants ANR-16-CE 12-0019 and ANR-18-CE12-0004). Research in Z.X.’s laboratory was supported by ANR grant “AlgaTelo” (ANR-17-CE20-0002-01), by Ville de Paris (Programme Émergence[s]), and by the French National Cancer Institute (grant INCa_15192). S.E. was supported by the “Initiative d'Excellence” program of the French State (“DYNAMO,” ANR-11-LABX-0011-01). F.C. is currently supported by a Marie Skłodowska-Curie Actions Postdoctoral Fellowship (101064365 Cocco-Next).
Author contributions: Investigation was by F.C., N.A., C.G., and S.E. Conceptualization was by F.C., G.F., S.E., and Z.X. Methodology was by F.C., N.A., and C.G. Software was by F.C. and C.G. Formal analysis was by F.C., N.A., C.G., and S.E. Supervision was by G.F. and Z.X. Writing of the original draft was by F.C. and Z.X. Reviewing and editing were by all authors.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.278043.123.
-
Freely available online through the Genome Research Open Access option.
- Received April 27, 2023.
- Accepted August 9, 2023.
This article, published in Genome Research, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








