
Transposons contribute to the functional diversification of the head, gut, and ovary transcriptomes across Drosophila natural strains
Abstract
Transcriptomes are dynamic, with cells, tissues, and body parts expressing particular sets of transcripts. Transposable elements (TEs) are a known source of transcriptome diversity; however, studies often focus on a particular type of chimeric transcript, analyze single body parts or cell types, or are based on incomplete TE annotations from a single reference genome. In this work, we have implemented a method based on de novo transcriptome assembly that minimizes the potential sources of errors while identifying a comprehensive set of gene–TE chimeras. We applied this method to the head, gut, and ovary dissected from five Drosophila melanogaster natural strains, with individual reference genomes available. We found that ∼19% of body part–specific transcripts are gene–TE chimeras. Overall, chimeric transcripts contribute a mean of 43% to the total gene expression, and they provide protein domains for DNA binding, catalytic activity, and DNA polymerase activity. Our comprehensive data set is a rich resource for follow-up analysis. Moreover, because TEs are present in virtually all species sequenced to date, their role in spatially restricted transcript expression is likely not exclusive to the species analyzed in this work.
In contrast to the genome, an animal's transcriptome is dynamic, with cell types, tissues, and body parts expressing particular sets of transcripts (Pan et al. 2008; Barash et al. 2010; Brown et al. 2014; Söllner et al. 2017). The complexity and diversity of the transcriptome arises from the combinatorial usage of alternative promoters, exons and introns, and polyadenylation sites. A single gene can, therefore, encode a rich repertoire of transcripts that can be involved in diverse biological functions, and contribute to adaptive evolution and disease (e.g., Marasca et al. 2020; Kiyose et al. 2022; Singh and Ahi 2022; Verta and Jacobs 2022). The potential contribution of transposable element (TE) insertions to the diversification of the transcriptome was analyzed soon after the first whole-genome sequences were available (Ganko et al. 2003; Jordan et al. 2003; van de Lagemaat et al. 2003; Franchini et al. 2004; Lipatov et al. 2005). TEs are present in virtually all genomes studied to date and are able to insert copies of themselves in the genome, and although their mutation capacity is often harmful, they also represent an important source of adaptive genetic variation (Volff 2006; Casacuberta and González 2013; Cowley and Oakey 2013; Schrader and Schmitz 2019). Although TEs are a known source of transcriptome diversity, the majority of studies so far rely on incomplete transposon annotations from a single reference genome (e.g., Lipatov et al. 2005). Moreover, methodologies are often specifically designed for particular types of chimeric gene–TE transcripts (e.g., TE-initiated transcripts) (Babaian et al. 2019) or particular types of TEs (e.g., L1 chimeric transcripts) (Pinson et al. 2018) or have been applied to individual cell types or body parts (e.g., Treiber and Waddell 2020; Babarinde et al. 2021). As such, our knowledge on the contribution of TEs to transcriptome diversification is still partial.
Two of the most studied mechanisms by which TEs can generate chimeric transcripts are by providing alternative promoters and protein domains. In human and mouse, 2.8% and 5.2% of the total transcript start sites occur within retrotransposons, respectively (Faulkner et al. 2009). In Drosophila melanogaster, >40% of all genes are expressed from two or more promoters, with at least 1300 promoters contained in TEs (Batut et al. 2013). As well as individual examples of TEs providing protein domains (Tipney et al. 2004; Cordaux et al. 2006; Newman et al. 2008), a comparative genomic analysis of tetrapod genomes revealed that capture of transposase domains is a recurrent mechanism for novel gene formation (Cosby et al. 2021). There is also evidence for the retrotransposon contribution to protein novelty. Approximately 9.7% of endogenous retrovirus open reading frames across 19 mammalian genomes evolve under purifying selection and are transcribed, suggesting that they could have been co-opted as genes (Ueda et al. 2020). Across insects, and depending on the methodology used, the percentage of newly emerged domains (<225 mya) owing to TEs was estimated to be from 1.7% to 6.6% (Klasberg et al. 2018). However, studies that identify and characterize a comprehensive set of gene–TE chimeras to provide a complete overview of their contribution to both transcriptome and protein diversification are still missing.
Besides describing the diverse contributions of TEs to the transcriptome, analyzing the relative contribution of gene–TE chimeras to the total gene expression is highly relevant, as it is informative of the potential functional relevance of the transcripts identified. Studies performed so far suggest that this contribution is related to the position of the TE in the transcript. Transcripts with a TE inserted in the 5′-UTR or internal coding exons show significantly lower mean levels of expression compared with nonchimeric TE–gene transcripts (Babarinde et al. 2021). TEs inserted in 3′ UTRs were associated with reduced gene expression both in humans and mice but with increased gene expression in human pluripotent stem cells (Faulkner et al. 2009; Babarinde et al. 2021). In addition, whether specific TE types contribute to tissue-specific expression has been explored in mammals, in which retrotransposons were found to be overrepresented in human embryonic tissues (Conley et al. 2008; Faulkner et al. 2009). In D. melanogaster, the contribution of TEs to tissue-specific expression has only been assessed in the head, with 833 gene–TE chimeric genes described (Treiber and Waddell 2020). Thus, whether the contribution of chimeric gene–TE transcripts is more relevant in the D. melanogaster head compared with other body parts is still an open question.
Within genes, TEs could also affect expression by changing the epigenetic status of their surrounding regions. In Drosophila, repressive histone marks enriched at TEs spread beyond TE sequences, which is often associated with gene down-regulation (Lee and Karpen 2017). However, there is also evidence that TEs containing active chromatin marks can lead to nearby gene overexpression (Guio et al. 2018). Genome-wide, the joint assessment of the presence of repressive and active chromatin marks has been restricted so far to the analysis of four TE families (Rebollo et al. 2012) and has never been performed in the context of chimeric gene–TE transcripts.
In this work, we performed a high-throughput analysis to detect, characterize, and quantify chimeric gene–TE transcripts in RNA-seq samples from the head, gut, and ovary dissected from the same individuals belonging to five natural strains of D. melanogaster (Fig. 1A; Rech et al. 2022). We implemented a method based on de novo transcriptome assembly that (1) minimizes the potential sources of errors when detecting chimeric gene–TE transcripts and (2) allows the identification of a comprehensive data set of transcripts rather than a focus on particular types (Fig. 1B; Lanciano and Cristofari 2020). Additionally, we assessed the coding potential and the contribution of chimeric transcripts to protein domains and gene expression as proxies for their integrity and functional relevance. Finally, we took advantage of the availability of ChIP-seq data for active and repressive histone marks, H3K9me3 and H3K27ac (Coronado-Zamora et al. 2023), respectively, obtained from the same biological samples to investigate whether the TEs that are incorporated into the transcript sequences also affect their epigenetic status.

Detection of chimeric gene–TE transcripts in five strains of D. melanogaster. (A) Map showing the sampling locations of the five European strains of D. melanogaster used in this study: TOM-007: Tomelloso, Spain (BSk); MUN-016: Munich, Germany (Cfb); JUT-011: Jutland, Denmark (Cfb); SLA-001: Slankamen, Serbia (Cfa); and AKA-017: Akaa, Finland (Dfc). Colors represent the climate zones according to the Köppen–Geiger climate distribution (Peel et al. 2007). (B) Pipeline to detect chimeric transcripts. (C) Contribution of chimeric gene–TE transcripts to the total transcriptome and the body part–specific transcriptome globally and by strain. (All) All the transcripts assembled in the three body parts and the five strains. (D) Schematic of the two groups of chimeric transcripts identified: overlap and alternative splicing insertion group and internal insertion group. Note that these numbers total more than 1931 because some chimeric transcripts can have different insertions in different genomes. Gray boxes represent exons; red boxes, a TE fragment incorporated in the mRNA; and white boxes, a TE fragment that is not incorporated in the final mRNA. The black lines connecting the exons represent the splicing events.
Results
Nine percent of D. melanogaster transcripts, across body parts and strains, are gene–TE chimeras
We performed a high-throughput analysis to detect and quantify chimeric gene–TE transcripts in RNA-seq samples from the head, gut, and ovary in five D. melanogaster strains collected from natural populations (Fig. 1A). The three body parts were dissected from the same individuals, and an average coverage of 32× (22× to 43×) per RNA-seq sample was obtained (three replicates per body part and strain) (Supplemental Table S1; Coronado-Zamora et al. 2023). We de novo assembled transcripts in which we annotated TE insertions using a new D. melanogaster manually curated TE library (Rech et al. 2022). We only considered de novo transcripts that overlap with a known transcript obtained from a reference-guided assembly (Fig. 1B), and we removed falsely fused transcripts, applying the algorithm described by Lima et al. (2017). We also excluded chimeric transcripts with TE fragments whose sequence contains simple repeats in >80% of their length (see Methods) (Benson 1999; Rech et al. 2022). We then used the reference genome of each strain to define the exon–intron boundaries of each transcript and to identify the position of the TE in the transcript (Fig. 1B). The alignment with the reference genome and the accurate TE annotation also allowed us to discard single-unit transcripts, indicative of pervasive transcription when the sequence matched a multiexonic transcript, and TE autonomous expression, which are two important sources of errors when quantifying the contribution of TEs to gene novelty (Fig. 1B; Lanciano and Cristofari 2020).
Overall, considering all the transcripts assembled in the three body parts and the five strains, we identified 1931 chimeric gene–TE transcripts belonging to 826 genes (Supplemental Table S2A; Supplemental Data). Thus, ∼9% (1931/21,270) of D. melanogaster transcripts contain exonic sequences of TE origin. In individual strains, this percentage ranged between 3.9% and 5.2% (549–725 chimeric transcripts per genome), indicating that most of the chimeric gene–TE transcripts are strain specific (1312/1931, 68%) (Fig. 1C). Almost half of the strain-specific chimeric transcripts (48%, 634/1312) were generated through strain-specific TE insertions, whereas the other 52% were through polymorphic (141) or fixed (599) insertions, indicating that differential exon usage is an important mechanism in generating chimeric transcripts. Although the overall contribution of TEs to the transcriptome is 9%, TEs contribute ∼19% (1406/7435) of the total amount of body part–specific transcripts (Fig. 1C).
Although previous studies reported gene–TE chimeras in which the TEs added a transcription start site (e.g., Batut et al. 2013), a transcript termination (e.g., Babarinde et al. 2021), or spliced sites (e.g., Treiber and Waddell 2020), we also identified chimeric transcripts in which the TE was fully annotated inside the UTRs or internal exons. Consequently, we categorized chimeric gene–TE transcripts in two groups (Fig. 1D). The first group contains chimeric transcripts that have a TE overlapping with the 5′ UTR, the 3′ UTR, or introducing alternative splice (AS) sites (overlap and AS insertion group: 766 chimeric transcripts from 415 genes). We found that the majority of the TEs in the overlap and AS insertion group were adding canonical splice site motifs (84.1%, 132/157) (Supplemental Table S2B; Supplemental Materials). The second group contains chimeric gene–TE transcripts in which the TE is annotated completely inside the UTRs or internal exons (internal insertion group: 1451 transcripts from 638 genes) (Fig. 1D). We hypothesized that this group could be the result of older insertions that have been completely incorporated into the transcripts. Indeed, we found that TEs in this group are shorter than those of the overlap and AS insertion group, as expected if the former are older insertions (78% vs. 22.7% proportion of chimeras with TEs < 120 bp; χ² test, P-value < 0.001; see Methods) (Supplemental Fig. S1). Additionally, we found that more transcripts were shared between strains in the internal insertion group than in the overlap and AS insertion group (χ² test, P-value < 0.001) (Supplemental Fig. S2A; Supplemental Table S2C). This observation is also consistent with this group being enriched for older insertions and remained valid when we removed the shorter insertions (χ² test, P-value < 0.001) (Supplemental Table S2C).
To test whether the overlap and AS insertion group and the internal insertion group contribute differently to the diversification of the transcriptome, we performed all the subsequent analyses considering all the chimeric transcripts together and the two groups separately. In addition, because shorter insertions might be enriched for false positives, that is, not corresponding to real TE sequences owing to the difficulty of annotating them, we also performed the analysis with the subset of chimeric gene–TE transcripts that contains a fragment of a TE insertion that is ≥120 bp (638/766 and 672/1451 for the overlap and AS insertion group and the internal insertion group, respectively; see Methods).
Gene–TE chimeric transcripts are more abundant in the head and ovary
Using high-throughput methodologies, 833 chimeric genes were identified in the D. melanogaster head (Treiber and Waddell 2020); however, the relative amount of chimeric gene–TE transcripts across body parts has never been assessed before. We found that the majority of the assembled chimeric gene–TE transcripts across the five strains analyzed were body part specific (72.8%, 1406/1931), with only 9.3% (180) shared across all three body parts (Fig 2A; Supplemental Table S3A). The same pattern was found for the overlap and AS insertion group and for the internal insertion group when considering all insertions and those ≥120 bp (Supplemental Fig. S2B; Supplemental Table S3A).
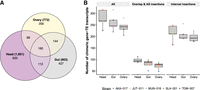
Distribution of chimeric transcripts across body parts and insertion groups. (A) Venn diagram showing the overall number of chimeric transcripts shared across body parts in the five strains. (B) Number of chimeric gene–TE transcripts detected by body part, strain, and insertion group. (All) All chimeric transcripts detected in all body parts and strains.
The head was the body part expressing the most chimeric transcripts (1001) followed by the gut (863) and ovary (772) (Fig. 2A; Supplemental Table S3A). Note that 145 of the chimeric genes identified in this work were previously described by Treiber and Waddell (2020), an expected low number given that many of the detected gene–TE chimeras are strain specific and that the methodologies are different. After accounting for differences in the total number of transcripts assembled in each body part, we observed that the head was expressing the same proportion of gene–TE chimeras as the ovary (6.2% head vs. 6.2% ovary; χ² test, P-value = 1); whereas the head and ovary express more than the gut (5.6% gut; χ² test, P-values = 0.023 and 0.035, respectively) (Supplemental Table S3B). Similar patterns were also found when the overlap and AS insertion group and the internal insertion group were analyzed separately, when we focused on ≥120 bp insertions and at the strain level, except for some comparisons (Fig. 2B; Supplemental Table S3B).
Finally, the head was the body part that expressed the most body part–specific chimeric transcripts (62% head vs. 50% gut; χ² test, P-value < 0.001; and vs. 46.5% ovary, P-value < 0.001), whereas no differences were found between gut and ovary (46.5% ovary vs. 50% gut; χ² test, P-value = 0.25) (Fig. 2A). In the three body parts, these proportions were higher than the total proportion of body part–specific transcripts (23.2%, 15.14%, and 11.25% for head, gut, and ovary, respectively; χ² test, P-values < 0.001 for all comparisons) (Supplemental Table S3B).
Most chimeric transcripts contain TE insertions in the 3′ UTRs
Chimeric gene–TE transcripts are enriched for TE insertions located in the 3′ UTRs in D. melanogaster and in mammals (van de Lagemaat et al. 2003; Lipatov et al. 2005; Babarinde et al. 2021). Consistently, we also found that most of the chimeric gene–TE transcripts contain a TE in the 3′ UTR (963 transcripts from 417 genes) followed by internal exons (569 transcripts from 266 genes) and insertions in the 5′ UTRs (474 transcripts from 273 genes). We also detected 125 chimeric transcripts belonging to 95 genes consisting of a single-unit transcript. Note that 21 of the 5′-UTR insertions detected in this work were experimentally validated in a previous analysis that estimated the promoter TE usage across developmental stages in D. melanogaster (Batut et al. 2013). Indeed, the number of chimeric genes with a TE inserted in the 3′ and 5′ UTRs is higher than expected when taking into account the proportion of the genome that is annotated as UTRs, whereas there is a depletion of TEs in internal exons (χ² test, P-value < 0.001 in the three comparisons) (Supplemental Table S4A). It has been hypothesized that the higher number of insertions in 3′ UTRs could be explained by lack of selection against insertions in this gene compartment (Jordan et al. 2003; Lipatov et al. 2005). We thus tested whether 3′ UTR chimeric transcripts were enriched for TE insertions present in more than one genome, but we found that this was not the case (χ² test, P-value = 0.934) (Fig. 3A; Supplemental Table S4B).
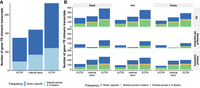
Position and frequency distribution of TEs in chimeric transcripts. (A) Number of gene–TE chimeric transcripts by position and frequency. (B) Number of chimeric gene–TE transcripts by insertion group and body part, according to the insertion position (5′/3′ UTRs or internal exons) and frequency. Each dot represents the number of chimeric gene–TE transcripts according to the frequency: strain-specific (blue), shared across two to four strains (green), and shared across all five strains (orange). These analyses were performed with the subset of chimeric transcripts with only one TE annotated (1634).
In both the overlap and AS insertion group and internal insertion group, TE insertions were also mainly located in the 3′ UTRs (53.9%, 316/586; 42%, 503/1199, respectively). In the internal insertion group, there were more insertions in internal exons than in the overlap and AS insertion group (414 vs. 42; χ² test, P-value < 0.001). This pattern also holds when we only consider ≥120 bp insertions (25 vs. 143; χ² test, P-value < 0.001) (Supplemental Table S4C). Figure 3B shows the number of chimeric gene–TE transcripts globally and by insertion group, body part, and strain (Supplemental Table S4D), where it can be observed that, overall, the previous patterns hold at the body part level.
Retrotransposons and DNA transposons contribute to chimeric gene–TE transcripts
We assessed the contribution of TE families to chimeric gene–TE transcripts. We found that many TE families, 90/146 (62%), were detected in chimeric gene–TE transcripts, as has been previously described in head chimeric transcripts (Supplemental Table S5A; Treiber and Waddell 2020; Rech et al. 2022). From the 90 families contributing to chimeric gene–TE transcripts, 70 are retrotransposons (78%), as expected given that overall there are more retrotransposon families (120/146, 82%, χ2 test, P-value = 0.51) (Supplemental Table S5B). The number of families contributing to the overlap and AS insertion group and the internal insertion group is similar (75 vs. 68, respectively, χ² test, P-value = 0.745) (Supplemental Table S5C). Half of the families (46: 51%) contribute to chimeric transcripts in all body parts, whereas 20 families were body part specific, with 10 being head specific, six gut specific, and four ovary specific (Supplemental Table S5A).
The most common TE families found were roo (38.1%) and INE-1 (26%) (Fig. 4). Indeed, these two families were overrepresented in the chimeric transcripts data set compared with their abundance in the genome: roo in the five strains (χ² test, P-value < 0.0001 for all comparisons) and INE-1 in SLA-001 and MUN-016 (χ² test, P-values < 0.05) (Supplemental Table S5D). roo and INE-1 were also the most common families both in the overlap and AS insertion group (17.3% and 29%, respectively) and in the internal insertion group (48.2% and 28.4%, respectively) (Fig. 4). The same pattern was found when we analyzed only those chimeric transcripts with TEs ≥120 bp (Supplemental Fig. S3; Supplemental Table S5E).
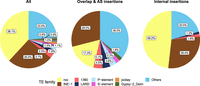
TE family distribution in gene–TE chimeras, globally and by insertion group. Percentage of TE families contributing to gene–TE chimeras in the global data set (All), in the overlap and AS insertion group, and in the internal insertion group. Only TE families found in more than nine chimeric genes are depicted; otherwise, they are grouped in Others.
Because roo insertions were enriched in all the strains analyzed, we further investigated these TE sequences (see Supplemental Material). We found mainly two types of roo insertions: solo LTRs (18 insertions, 16 chimeric transcripts), of which most belonged to the overlap and AS insertion group (16/18), and a short (45-bp to 192-bp) low-complexity sequence mapping to the positions 1052–1166 of the canonical roo element (see Methods). This short roo sequence is more common in the internal insertion group than in the overlap and AS insertion group (805 vs. 127 chimeric transcripts, respectively). Note that a recent analysis by Oliveira et al. (2023) also found this same region of the roo consensus sequence to be the most abundant in chimeric gene–TE transcripts across four D. melanogaster strains. To determine that these low-complexity regions have a roo origin and are not widespread repeats across the genome, we used a strict BLAST search and found that 51 of the low-complexity regions have a roo element insertion as the second best hit and 168 have a roo insertion in the top five hits, suggesting that indeed some of these sequences have a clear roo origin (Supplemental Table S5F). Furthermore, we also tested whether this low-complexity region was present in the roo consensus sequence from a closely related species, Drosophila simulans, and found that this was the case, strongly suggesting that this low-complexity sequence is an integral part of the roo element.
To further investigate why this roo low-complexity region was incorporated into genes, we looked for transcription factor binding site motifs in this region of the canonical roo element. We found a C2H2 zinc finger factor motif repeated six times in this region (MA0454.1). Note that this specific motif is only found once in the roo consensus sequence outside the low-complexity region. A scan in the roo sequences from the chimeras revealed that 90% (778/866) of the transcripts with the low-complexity roo sequence contain at least one sequence of this zinc finger motif, with 25% (196/778) containing three or more (Supplemental Table S5G). Additionally, we also looked for motifs on roo solo LTRs (16 chimeric transcripts) and found that 10 out of 16 transcripts presented at least one motif, all related to DNA-binding domains found in transcription factors, including other C2H2 zinc fingers factor motifs (Supplemental Fig. S4A; Supplemental Table S5H).
Finally, we also performed a motif scan on INE-1 elements (600 transcripts), the second most common family, and on the 20 body part–specific TE families (29 transcripts). Out of 600 of the analyzed transcripts, 471 contained motifs provided by the INE-1 fragment, all of them related to DNA-binding motifs, including the C2H2 zinc finger factors (Supplemental Fig. S4B; Supplemental Table S5H). Out of 29 transcripts with body-specific TE families, 24 contained at least one motif related to 16 different classes, including again C2H2 zinc finger factors (Supplemental Fig. S4C; Supplemental Table S5H). Of the 16 motifs, 62.5% (10/16) were body part specific, with six detected in head chimeric transcripts, two in gut, and two in ovary (Supplemental Fig. S4C).
Chimeric gene–TE transcripts contribute a mean of ∼43% of the total gene expression
Besides identifying and characterizing chimeric gene–TE transcripts, we quantified the level of expression of both chimeric and nonchimeric transcripts genome-wide. We focused on transcripts with ≥1 TPM (transcript per million) in at least one of the samples analyzed (1909 out of 1931 chimeric transcripts, corresponding to 819/826 of the genes; see Methods) (Supplemental Table S6A). We found that chimeric gene–TE transcripts have lower expression levels than nonchimeric transcripts (19,228; Wilcoxon's test, P-value < 0.001) (Fig. 5A). This is in contrast with previous observations in human pluripotent stem cells that reported no differences in expression between chimeric and nonchimeric transcripts (Babarinde et al. 2021). We discarded the possibility that the lower expression of chimeric gene–TE transcripts was driven by the roo low-complexity region identified in 829 of the chimeric transcripts (Wilcoxon's test, P-value < 0.0001) (Fig. 5A). Lower expression of the chimeric gene–TE transcripts compared with nonchimeric transcripts was also found when we analyzed the overlap and AS insertion group and the internal insertion group separately and at the body part and strain levels (Wilcoxon's test, P-value < 0.001 for all comparisons) (Fig. 5A; Supplemental Table S6B).
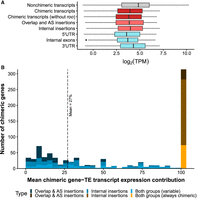
TE insertions within genes affect gene expression. (A) Boxplots for the expression levels, measured as the logarithm of TPM (TPM ≥ 1): for all nonchimeric transcripts of the genome (19,228; in gray), all chimeric transcripts detected in the present study (1909; in dark red), chimeric transcripts without the short internal roo insertion (1073; dark red), all chimeric transcripts belonging to the overlap and AS insertion group (472; light red) and the internal insertion group (1156; light red), and chimeric transcripts divided by position of the insertion (5′ UTR: 354, internal exons: 445, 3′ UTR: 799; cadet blue). (B) Histogram showing the expression contribution of chimeric transcripts to the total gene expression. Blue bars represent the contribution of variable chimeric genes (505 genes), ranging from ∼0.1% to >90% (mean = 27%), and the orange/brown bar represents the genes that always produced chimeric transcripts in all the genomes and body parts in which expression was detected (314 genes). Both groups correspond to genes that generate chimeric transcripts belonging to the overlap and AS insertion group and to the internal insertion group.
We further tested whether TEs inserted in different gene locations differed in their levels of expression compared with nonchimeric TE transcripts. We found that chimeric transcripts had significantly lower expression than nonchimeric transcripts regardless of the insertion position (Wilcoxon's test P-value < 0.001 for all comparisons) (Fig. 5A). Furthermore, insertions in the 3′ UTR appeared to be more tolerated than those in 5′-UTR and internal exons, as their expression level was higher (Wilcoxon's test, P-value < 0.001 for both comparisons) (Fig. 5A). Our results are consistent with those reported by Faulkner et al. (2009), who also found that 3′-UTR insertions reduced gene expression compared with nonchimeric transcripts, and contrast the findings of Babarinde et al. (2021), who found that 3′-UTR chimeric transcripts have significantly higher levels of expression compared with nonchimeric transcripts or with insertions in 5′-UTR and internal exons.
If we focus on the chimeric genes, 38% of them (314 genes) only expressed the chimeric gene–TE transcript (in all the genomes and body parts in which expression of the gene was detected). Most of these genes (76%) contain short TE insertions, and accordingly, most of them belong to the internal insertion group (66%). For the other 62% (505) of the genes, we calculated the average contribution of the chimeric gene–TE transcript to the total gene expression across samples. Although some transcripts contributed on average only ∼0.1% of the total gene expression, others accounted for >90% (mean = 27%) (Fig. 5B). The mean contribution to gene expression of the internal insertion group was higher than that of the overlap and AS insertion group, when considering all the insertions (28.5% vs. 15.7%, respectively; Wilcoxon's test, P-value < 0.001) and when analyzing only those transcripts with ≥120 bp insertions (28.5 vs. 15.7%, respectively; Wilcoxon's test, P-value > 0.0001). Considering only the transcripts that do not contain the roo low-complexity sequence, the mean contribution to gene expression of the internal insertion group was still 25.3%. Overall, taking all expressed chimeric genes into account (819), the average chimeric gene–TE transcripts’ expression contribution to the total gene expression was 43%.
Finally, we evaluated whether there are differences between the expression levels of body part–specific and body part–shared chimeric transcripts. The breadth of expression, measured as the number of tissues in which a gene is expressed, is significantly and positively correlated with the level of expression in Drosophila (Larracuente et al. 2008) and humans (Park and Choi 2010). Consistent with this, we found that body part–shared chimeric transcripts have significantly higher expression levels than do chimeric transcripts expressed in only one body part (Wilcoxon's test, P-value < 0.001) (Supplemental Table S6C) when considering the whole data set and for chimeric transcripts with insertions ≥120 bp (Wilcoxon's test, P-value < 0.001) (Supplemental Table S6C). Because we observed that the head was expressing more body part–specific chimeric transcripts (Fig. 2A), we next assessed if head-specific chimeric transcripts were also expressed at higher levels. However, we observed that the median expression of head-specific chimeric transcripts was similar to those specific to the gut (medianhead = 9.7 TPM [n = 620], mediangut = 10 TPM [n = 427]; Wilcoxon's test, P-value = 0.054) but lower than ovary-specific chimeric transcripts (medianovary = 12.6 TPM [n = 359]; Wilcoxon's test, P-value > 0.0001). However, this is similar to the expression level of transcripts in these body parts (median of gene expression in ovary > gut > head = 21.2 > 15.10 > 12.47).
Finally, chimeric transcripts found in all five strains also have significantly higher expression levels than strain-specific chimeric transcripts (Wilcoxon's test, P-value < 0.001) (Supplemental Table S6D).
17.1% of the TEs within chimeric gene–TE transcripts could also be affecting gene expression via epigenetic changes
We tested whether TEs that produce chimeric transcripts could also be affecting gene expression by affecting the epigenetic marks. We used ChIP-seq data for the three body parts, in each of the five strains analyzed (Coronado-Zamora et al. 2023), for two histone marks: the silencing mark H3K9me3 (Yin et al. 2011; Choi and Lee 2020) and the activating mark H3K27ac, related to active promoters and enhancers (Buecker and Wysocka 2012; Koenecke et al. 2016). We tested whether strains expressing the gene–TE chimeric transcript differed in the gene body epigenetic marks compared with strains that do not express the chimeric transcript (430 genes). For the majority of these genes (266), we did not observe consistent epigenetic patterns across samples expressing or not expressing the chimeric transcripts, and these genes were not further analyzed. Additionally, 42 genes did not harbor any epigenetic marks, whereas 93 genes contained the same epigenetics mark(s) (H3K27ac, H3K9me3, or both marks) in strains with and without that particular chimeric transcript (Supplemental Table S7A). Overall, for 17.1% (72/420) of the genes, we observed a consistent change in the epigenetic status associated with the presence of the gene–TE chimera. This percentage is similar for the overlap and AS group and the internal insertion group (16.9% and 18.4%, respectively). The majority of TEs showing consistent changes in their epigenetic status were associated with gene down-regulation (68%) (Table 1), and this percentage increases when considering chimeric transcripts with insertions in 5′ UTR: 88% (16/18) of the genes are associated with down-regulation (Supplemental Table S7B).
Expression fold-change associated with epigenetic status of strains expressing or not expressing the chimeric transcript
Gene–TE chimeric transcripts are enriched for DNA-binding molecular functions involved in regulation and development
To test whether the biological processes and molecular functions differed across body parts and between the two groups of gene–TE chimeras, we performed a Gene Ontology (GO) clustering analysis (Huang et al. 2009). To analyze the chimeric genes detected in each body part separately, we used the total genes assembled in the corresponding body part as a background. We found that across body parts, chimeric genes were enriched in general cell functions, such as regulation and development (Fig. 6A; Supplemental Table S8A). Some functions were particular to a body part, for example, transcription from RNA polymerase II promoter in the head and cell communication and signaling in the ovary. Note that the overlap and AS insertion group is enriched for biological processes that are body part specific: communication signaling and stimulus in the head, cellular amide and organitrogen compound metabolic processes in the gut and cell projection, assembly, and organization in the ovary (Fig. 6A; Supplemental Table S8A).
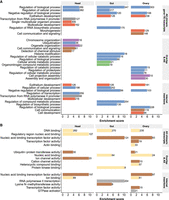
Biological processes and molecular functions of chimeric gene–TE transcripts. (A) Biological processes clustering. (B) Molecular functions clustering. The length of the bars represents the cluster enrichment score. The number in the bars represents the number of genes in each cluster. Names of the annotation clusters are manually processed based on the cluster's GO terms. Colors represent similar annotation clusters. Detailed GO terms of each cluster are given in Supplemental Table S8A,B.
Finally, regarding the molecular function, chimeric genes are enriched for DNA binding processes across body parts (Fig. 6B; Supplemental Table S8B), whereas they are also enriched in the head for regulatory region nucleic acid binding and in ovary for transcription factor activity and actin binding.
Both DNA transposons and retrotransposons add functional protein domains
We next assessed whether TE sequences annotated in internal exons provided functional protein domains. We first confirmed, using the Coding Potential Assessment Tool (CPAT) (Wang et al. 2013) software, that the majority of chimeric protein-coding gene–TE transcripts that have a TE annotated have coding potential (94.3%, 1687/1789) (Supplemental Table S9A). Using Pfam (Mistry et al. 2021), we identified a total of 23 Pfam domains in 33 different chimeric transcripts from 24 genes (Table 2; Supplemental Table S9B,C). These 23 domains were identified in 16 TE families, with nine TE families providing more than one domain. The size of these domains ranged from 9 bp to 610 bp (median = 72 bp) (Supplemental Table S9B). Note that 10 of these 24 chimeric genes have been previously described in the literature (Table 2). We observed similar numbers of transcripts belonging to the overlap and AS insertion group and the internal insertion group (12 and 16, respectively, with five belonging to both). Finally, we found chimeric transcripts adding domains in the three body parts analyzed (Table 2), with an even distribution across them (head: 15; ovary: 13; gut: 12).
Description of the 24 chimeric genes containing a TE providing a protein domain
Both DNA transposons and retrotransposons provide domains (13/24 and 11/24, respectively), and most TEs provided a nearly full domain (22/24, ≥50% coverage), including six TEs adding a full-size domain (Table 2). Chimeric genes were related, among others, to transporter functions (11/24), enzyme functions (nine of 24), and response to stimulus (five of 24) (Supplemental Table S9C). All these chimeric genes have evidence of expression, ranging from 1.43 to 61.69 TPM (median = 18.16 TPM) (Table 2). We did not observe statistical differences between the level of expression of transcripts with complete domains compared with partially/uncompleted domains (Wilcoxon's test, P-value = 0.535). The majority of TEs for which the population TE frequency has been reported (Rech et al. 2022) are fixed or present at high frequencies (eight of 14 TEs) (Table 2).
We assessed if the domains detected in the TE fragments of the gene–TE chimeras were also found in the consensus sequences of the TE family. Because in some cases the same domain was provided by different TE families, in total we analyzed 47 TE fragments. We were able to find the domain sequence in 44 TE fragments from 21 TE consensus sequences (Supplemental Table S9D). Note that for seven of these domains (from seven TEs), we had to lower the Pfam detection thresholds to detect them (see Methods). The three domains that were not identified in the consensus sequences were not detected in the chimeric fragments as full domain sequences.
We performed a Pfam domain enrichment analysis considering domains annotated with nearly full domains and in transcripts expressed with a minimum of 1 TPM using dcGO (Fang and Gough 2013). Overall, seven domains were enriched for the molecular function catalytic activity, acting on RNA (four domains, FDR = 3.44 × 10−4), nucleic acid binding (six domains, FDR = 3.44 × 10−4), DNA polymerase activity (three domains, FDR = 6.81 × 10−5), and RNA-directed DNA polymerase activity (three domains, FDR = 4.66 × 10−7) (Table 3). Five out of seven enriched domains were found in retrotransposon insertions.
Pfam domain enrichment analysis dcGO enrichment results using Gene Ontology with FDR < 0.01
Discussion
TEs contribute to genome innovation by expanding gene regulation, both of individual genes and of gene regulatory networks; enriching transcript diversity; and providing protein domains (for reviews, see Chuong et al. 2017; Modzelewski et al. 2022). Although the role of TEs as providers of regulatory sequences has been extensively studied, their contribution to transcriptome diversification and protein domain evolution has been less characterized. In this work, we have developed a pipeline to identify and characterize chimeric gene–TE transcripts across three body parts and five natural D. melanogaster strains (Fig. 1A,B), and we have quantified their contribution to total gene expression and to protein domains. Although previous studies were hindered by the incomplete annotation of TEs in the genome studied (Lipatov et al. 2005; Treiber and Waddell 2020), in this work, we took advantage of the availability of high-quality genome assemblies, genome annotations, transcriptomes, and histone mark enrichments for five natural strains to carry out an in-depth analysis of gene–TE chimeric transcripts (Rech et al. 2022). Note that all those -omics data can be visualized in the DrosOmics genome browser (Coronado-Zamora et al. 2023). We found that TEs contribute ∼9% to the global transcriptome and ∼19% to the body part–specific transcriptome (Fig. 1C). Contrary to other studies that mostly focus on a single type of chimeric gene–TE transcript, we investigated a comprehensive data set of chimeras. Indeed, we found that besides insertions affecting the transcription start site, transcript termination, and adding spliced sites (overlap and alternative splicing insertions), we also identified a substantial number of TE sequences that were completely embedded within exons (internal insertions) (Fig. 1D). These two types of chimeric gene–TE transcripts shared many properties; for example, they were enriched for body part–specific transcripts (Supplemental Fig. S2B), and they showed lower expression levels than nonchimeric transcripts (Fig. 5A), suggesting that they both should be taken into account when analyzing the contribution of TEs to gene novelty. The internal insertion group contributed more to total gene expression (Fig. 5B); however, we discarded the possibility that this increased expression was owing to shorter TE insertions, which are more likely to be enriched for false annotations compared with longer insertions (Rech et al. 2022). We found, both based on size and frequency, that the internal insertion group is likely to be enriched for older insertions. As such, a higher level of expression of these likely older TEs is consistent with previous observations in tetrapods, suggesting that, over time, gene–TE chimeric transcripts often become the primary or sole transcript for a gene (Cosby et al. 2021). Overall, and taking into account only those gene–TE chimeric transcripts with evidence of expression, we found 102 (5.9%) insertions disrupting the coding capacity, 435 (25.2%) affecting the coding capacity, and 316 (18.3%) and 717 (41.6%) affecting the 5′ and the 3′ end of the gene, respectively, whereas 155 (9%) affected multiple transcript positions (Supplemental Table S6A).
Our finding that TEs contribute to the expansion of the head transcriptome supports the results of Treiber and Waddell (2020) and suggests that ∼6% of genes produce chimeric transcripts in the head owing to exonization of a TE insertion. However, because we also analyzed the gut and ovary, we further show that TEs can significantly contribute to the expansion of transcriptomes of other body parts as well (Fig. 2A). The observation that there are more head-specific chimeric transcripts is consistent with a higher transcriptional complexity in the Drosophila nervous system tissues (Brown et al. 2014). The fact that chimeric gene–TE transcripts tend to be tissue specific could be especially relevant for adaptive evolution as tissue-specific genes can free the host from pleiotropic constraints and allow the exploration of new gene functions (Park and Choi 2010; Rogers and Hartl 2012; Salvador-Martínez et al. 2018).
Finally, we identified a total of 23 TE protein domains co-opted by 24 genes (Table 2; Supplemental Table S9C). Nine of these genes have been previously described as chimeric based on high-throughput screenings or individual gene studies, with some of them, for example, CHKov1 and nxf2, having functional effects (Table 2; Aminetzach et al. 2005; Magwire et al. 2011; Ellison et al. 2020). The majority of the domains were present in the TE consensus sequences (Supplemental Table S9D). Furthermore, the 23 domains identified were enriched for nucleic acid binding, catalytic activity, and DNA polymerase activity molecular functions (Table 3). Although there is evidence for DNA-binding domains being recruited to generate new genes, the previous data come from a comparative genomic approach across tetrapod genomes that focused on DNA transposons as a source of new protein domains (Cosby et al. 2021). The available data for the genome-wide contribution of retrotransposons to protein domains were restricted to endogenous retroviruses in mammals (Ueda et al. 2020). In our data set, which includes both DNA transposons and retrotransposons, the enrichment for DNA-binding domains and for catalytic activity is indeed driven by the retrotransposon insertions (Table 2). Five of the 10 TEs providing protein domains identified in this work for the first time were present at high population frequencies (two) or fixed (three) and are thus good candidates for follow-up functional analysis (Tables 2, 3).
Although we have detected more chimeric transcripts than any prior D. melanogaster study to date, our estimate of the potential contribution of TEs to the diversification of the transcriptome is likely to be an underestimate. First, and as expected, we found that the contribution of TEs to the transcriptome is body part specific (72.8%) (Supplemental Fig. S2B; Conley et al. 2008; Faulkner et al. 2009) and strain specific (68%) (Supplemental Fig. S2A). Thus, analyzing other body parts and increasing the number of genomes analyzed will likely identify more chimeric gene–TE transcripts. Second, although our estimate is based on the highly accurate annotations of TE insertions performed using the REPET pipeline (Rech et al. 2022), highly diverged and fragmented TE insertions are difficult to be accurately annotated by any pipeline and, as such, might go undetected (Gotea and Makałowski 2006; Rodriguez and Makałowski 2022). Still, the combination of an accurate annotation of chimeric gene–TE transcripts, with expression data across body parts, and of the investigation of the protein domain acquisition performed in this work not only significantly advances our knowledge on the role of TEs in gene expression and protein novelty but also provides a rich resource for the follow-up analysis of gene–TE chimeras.
Methods
Fly stocks
Five D. melanogaster strains obtained from the European Drosophila Population Genomics Consortium (DrosEU) were selected according to their different geographical origins: AKA-017 (Akaa, Finland), JUT-011 (Jutland, Denmark), MUN-016 (Munich, Germany), SLA-001 (Slankamen, Serbia), and TOM-007 (Tomelloso, Spain). Further information on collection dates and localities can be found in the work of Rech et al. (2022).
RNA-seq and ChIP-seq data for three body parts
RNA-seq and ChIP-seq data for the five strains were obtained from Coronado-Zamora et al. (2023). A full description of the protocols used to generate the data can be found in the work of Coronado-Zamora et al. (2023). Briefly, the head, gut, and ovary body parts of each strain were dissected from the same adult flies. Three replicates of 30 4- to 6-d-old females each were processed per body part and strain. RNA-seq library preparation was performed using the TruSeq stranded mRNA sample prep kit from Illumina and was sequenced using Illumina 125-bp paired-end reads (26.4 million–68.8 million reads) (Supplemental Table S1). For ChIP-seq, libraries were performed using TruSeq ChIP library preparation kit. Sequencing was performed in an Illumina HiSeq 2500 platform, generating 50-bp single-end reads (22.2 million–59.1 million reads) (Supplemental Table S1). RNA-seq and ChIP-seq raw data are available in the NCBI BioProject database (https://www.ncbi.nlm.nih.gov/bioproject/) under accession number PRJNA643665. The DrosOmics genome browser (http://gonzalezlab.eu/drosomics) compiles all data used in this work and allows for its visual exploration (Coronado-Zamora et al. 2023).
Transcriptome assembly
Reference-guided transcriptome assembly
To perform reference-guided transcriptome assemblies for each body part and strain (15 samples), we followed the protocol that is described by Pertea et al. (2015) using HISAT2 (v2.2.1) (Kim et al. 2019) and StringTie (v2.1.2) (Pertea et al. 2015) and that is available in the work of Coronado-Zamora et al. (2023). Details on the reference-guided transcriptome assembly can be found in the Supplemental Methods.
De novo transcriptome assembly
A de novo transcriptome assembly for each sample was performed using Trinity (v2.15.1) (Grabherr et al. 2011) on the FASTQ files previously processed with fastp (v.0.23.2) (Chen et al. 2018). Trinity used the following parameters: ‐‐seqType fq ‐‐samples_file <txt file with FASTQ directory> ‐‐CPU 12 ‐‐max_memory 78 G ‐‐jaccard_clip. Following the Trinity recommended implementation (Haas et al. 2013), we combined all reads across technical replicates in order to assemble them into a single reference assembly per sample (15 samples: five strains × three body parts). The ‐‐jaccard_clip parameter was used to split falsely fused transcripts derived from gene-dense compact genomes. Next, to keep reliable near full-length transcripts, we used BLASTN (v2.11.0) (Camacho et al. 2009) to assign each de novo transcript to a known D. melanogaster transcript obtained from the reference-guided transcriptome assembly. Next, the script analyze_blastPlus_topHit_coverage.pl from the Trinity toolkit was used to evaluate the quality of the BLAST results, and we followed a conservative approach that only kept a transcript with a coverage >80% with a known D. melanogaster transcript, thus keeping 150,699 assembled transcripts across all samples. Then, we applied the algorithm proposed by Lima et al. (2017) to detect and remove falsely fused transcripts that were not correctly assembled by Trinity. Briefly, we used BLASTN (Camacho et al. 2009) again to assign each transcript to a known D. melanogaster transcript and removed it if it had two or more matches with transcripts from different genes with a coverage of at least 80% over >100 bp. As expected, most of the fusion transcripts were owing to overlapping UTRs between genes. Finally, we tried to minimize other possible sources of confounding errors by excluding transcripts that were not overlapping a known transcript (tagged by StringTie as possible polymerase run-on, intron match on the opposite strand [likely a mapping error], fully contained in a reference intron, or intergenic). The set of final assembled transcripts by Trinity was 131,840.
Identification and characterization of chimeric gene–TE transcripts
We focused on the set of assembled de novo transcripts that passed the previous filters to identify putative chimeric gene–TE transcripts. To annotate TEs in the de novo assembled transcripts, we used RepeatMasker (v4.1.2) (Smit et al. 2013) with the parameters -norna -nolow -s -cutoff 250 -xsmall -no_is -gff) with a manually curated TE library (Rech et al. 2022). Note that RepeatMasker states that a cutoff of 250 will guarantee no false positives (Smit et al. 2013). We excluded transcripts for which the entire sequence corresponded to a TE, that is, were indicative of the autonomous expression of a TE, or excluded TE fragments whose sequence contains simple repeats in >80% of their length, as determined by Tandem Repeats Finder (v4.09) (Benson 1999), with the exception of roo-containing transcripts, which were analyzed separately.
To infer the exon–intron boundaries of the transcript, we used minimap2 (v2.24; with arguments -ax splice ‐‐secondary = no ‐‐sam-hit-only -C5 -t4) (Li 2018) to align the transcript to the gene region obtained from the genome of the corresponding strain from which it was assembled. We excluded single-unit transcripts when they matched a multiexon reference transcript, as it could be indicative of pervasive transcription or nonmature mRNAs. If a single-unit transcript matched a single-exon reference transcript, it was considered as valid. With this process, we obtained the full-length sequence of the gene that encodes for the chimeric transcripts.
We ran RepeatMasker again (same parameters) on the full-length gene to annotate the complete TE sequence and obtain the length of the insertion. We considered that short insertions are those <120 bp (Rech et al. 2022). Finally, we used an ad hoc bash script to define the TE position within the transcript and define the two insertion groups: the overlap and AS insertion group and the internal insertion group. The overlap and AS insertion group has a TE overlapping with the first (5′-UTR) or last (3′-UTR) exon or has overlap with the exon–intron junction and thus introduces AS sites (see “Splice sites motif scan analysis” in Supplemental Methods). The internal insertion group corresponds to TE fragments detected inside exons.
Identification of differential exon usage in strain-specific chimeric transcripts
To assess if the strain-specific chimeras were generated by strain-specific TE insertions or were the result of differential exon usage, we examined whether the genomes for which the chimeric transcript was not identified contained the TE insertion. We used the TE sequence of the strain-specific chimeric transcript as the query in a BLASTN (v2.11.0) (Camacho et al. 2009) search against the corresponding gene region. We applied the following BLASTN parameters: -qcov_hsp_perc 80 and -perc_identity 80. We then categorized the insertions as strain specific or as shared across strains and thus indicative of exon usage.
Expression level estimation
The expression level of the whole set of transcripts assembled was computed by the Trinity package (v2.15.1) (Grabherr et al. 2011), which used salmon (Patro et al. 2017) as the abundance estimation method and applied TPM as expression normalization. Salmon handles multimapped reads and applies an expectation-maximization algorithm that attributes read counts to the most likely transcripts. For the expression analyses, we considered transcripts with a minimum expression level of 1 TPM unless specified. Genes were categorized into three groups: (1) genes that were never detected as producing chimeric isoforms, (2) genes that always were detected as producing chimeric gene–TE transcripts, and (3) genes producing both chimeric and nonchimeric isoforms. For the later type of genes, we calculated the fraction of the total gene expression that comes from the chimeric transcript to compute the contribution of the chimeric transcripts to the total gene expression.
Coding capacity assessment
We assessed whether protein-coding chimeric gene–TE transcripts can produce a protein by using the CPAT (v3.0.4) software (Wang et al. 2013) with default parameters. CPAT has been optimized for the prediction of coding and noncoding isoforms in Drosophila. Thus, we used the coding probability cutoff at 0.39 (Wang et al. 2013).
Pfam scan of domain analysis and enrichment
To scan for Pfam domains (Mistry et al. 2021) in the TEs detected in an internal exon, we extracted the TE sequence from the chimeric transcripts using BEDTools getfasta (v2.29.2) (Quinlan and Hall 2010), translated it to ORF using getorf (EMBOSS:6.6.0.0) (Rice et al. 2000), and scanned it using the script pfam_scan.pl (v1.6) (Finn et al. 2014) to identify any of the known protein family domains of the Pfam database (version 34). We used dcGO enrichment online tool (Fang and Gough 2013) to perform an enrichment of the Pfam domains detected.
We scanned the consensus TE sequences for the domains present in TE fragments detected in the chimeric transcripts using pfam_scan.pl (v1.6) (Finn et al. 2014). If the domain was not detected using pfam_scan.pl default parameters, we lowered the hmmscan e-value sequence and domain cutoffs to 0.05.
ChIP-seq peak calling
ChIP-seq data were processed by Coronado-Zamora et al. (2023), and details on reads preprocessing, alignment, and histone peak calling can be found in the Supplemental Methods. Briefly, we applied the ENCODE ChIP-seq caper pipeline (v2, available at GitHub [https://github.com/ENCODE-DCC/chip-seq-pipeline2]) in histone mode, which used MACS2 peak with default settings to call peaks.
GO clustering analysis
The GO clustering analysis in the biological process (BP) and molecular process (MP) category was performed using the DAVID bioinformatics online tool (Huang et al. 2009). Names of the annotation clusters were manually processed based on the cluster's GO terms. Only clusters with a score greater than 1.3 were considered (Huang et al. 2009).
Statistical analysis
All statistical analyses were performed in the R (v4.1.2) statistical computing environment (R Core Team 2021). Graphics were created using the ggplot2 R package (Wickham 2016).
Data access
The set of chimeric gene–TE transcripts detected in this work is available in GitHub (https://github.com/GonzalezLab/chimerics-transcripts-dmelanogaster) and in the Supplemental Data. Scripts to perform analyses are available at GitHub (https://github.com/GonzalezLab/chimerics-transcripts-dmelanogaster) and in the Supplemental Code.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
We thank Carlos Vargas-Chavez and Simón Orozco for providing the D. simulans REPET TE library. We thank Simón Orozco and Ewan Harney for comments on the manuscript. This project has received funding from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation program (H2020-ERC-2014-CoG-647900) and from grant PID2020-115874GB-I00 funded by Ministerio de Ciencia e Innovacion MCIN/AEI/10.13039/501100011033.
Author contributions: J.G. conceived the project; J.G. and M.C.-Z. designed the analyses; M.C.-Z. performed the analyses; and J.G. and M.C.-Z. wrote and revised the manuscript.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.277565.122.
- Received December 2, 2022.
- Accepted August 8, 2023.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see https://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








