
Allele-specific RNA N6-methyladenosine modifications reveal functional genetic variants in human tissues
- Shuo Cao1,3,
- Haoran Zhu1,3,
- Jinru Cui1,3,
- Sun Liu1,3,
- Yuhe Li1,
- Junfang Shi1,
- Junyuan Mo1,
- Zihan Wang1,
- Hailan Wang1,
- Jiaxin Hu1,
- Lizhi Chen1,
- Yuan Li1,
- Laixin Xia1 and
- Shan Xiao1,2
- 1Department of Developmental Biology, School of Basic Medical Sciences, Southern Medical University, Guangzhou 510515, China;
- 2Guangdong Provincial Key Laboratory of Cardiac Function and Microcirculation, Guangzhou 510515, China
-
↵3 These authors contributed equally to this work.
Abstract
An intricate network of cis- and trans-elements acts on RNA N6-methyladenosine (m6A), which in turn may affect gene expression and, ultimately, human health. A complete understanding of this network requires new approaches to accurately measure the subtle m6A differences arising from genetic variants, many of which have been associated with common diseases. To address this gap, we developed a method to accurately and sensitively detect transcriptome-wide allele-specific m6A (ASm6A) from MeRIP-seq data and applied it to uncover 12,056 high-confidence ASm6A modifications from 25 human tissues. We also identified 1184 putative functional variants for ASm6A regulation, a subset of which we experimentally validated. Importantly, we found that many of these ASm6A-associated genetic variants were enriched for common disease–associated and complex trait–associated risk loci, and verified that two disease risk variants can change m6A modification status. Together, this work provides a tool to detangle the dynamic network of RNA modifications at the allelic level and highlights the interplay of m6A and genetics in human health and disease.
The surge of human genome sequencing data has revealed billions of genetic variants in the population. Genetic variants contribute to the phenotypic diversity in the population, and more than 10,000 genetic variants have been revealed to be associated with diseases by genome-wide association study (GWAS) (Maurano et al. 2012; Wang et al. 2021). At the molecular level, genetic variants can affect a number of processes, including DNA methylation, histone modification, gene expression, and post-transcriptional splicing (McDaniell et al. 2010; Shoemaker et al. 2010; The ENCODE Project Consortium 2012; McVicker et al. 2013; Leung et al. 2015; Roadmap Epigenomics Consortium et al. 2015; Schultz et al. 2015; GTEx Consortium 2017; Gate et al. 2018; Yang et al. 2019; Liu et al. 2020c; Amoah et al. 2021). However, the effect of many variants is often subtle, making it challenging to systematically identify and interpret functional variants, leaving a great deal of genetic variants with unknown functions.
N6-Methyladenosine (m6A), the most abundant modification on mammalian mRNA and long noncoding RNA (lncRNA), is catalyzed by the METTL3/METTL14/WTAP methyltransferase complex preferentially at RRACH motifs (Liu et al. 2014; Ping et al. 2014), is removed by the demethylases FTO and ALKBH5 (Jia et al. 2011; Zheng et al. 2013), and has been implicated in the regulation of a series of cotranscriptional and post-transcriptional processes (Dominissini et al. 2012; Batista et al. 2014; Wang et al. 2014a,b, 2015c; Alarcón et al. 2015; Geula et al. 2015; Liu et al. 2015; Meyer et al. 2015; Zhou et al. 2015; Vu et al. 2017; Ke et al. 2017; Xiang et al. 2017; Xu et al. 2017; Yoon et al. 2017; Zhang et al. 2017; Li et al. 2017b; Huang et al. 2018; Liu et al. 2020a). m6A has been widely explored in human tissues via high-throughput sequencing (MeRIP/m6A-seq), revealing an enrichment for expression quantitative trait loci (eQTLs) and GWAS single-nucleotide polymorphisms (SNPs) (Xiao et al. 2019; Liu et al. 2020b; Zhang et al. 2020a). Many human genetic variants may affect m6A levels, leading to changes in gene expression (Zhang et al. 2020b; Xiong et al. 2021). However, accurately measuring the m6A changes in response to genetic variants and determining the interplay between m6A and genetic variation remains challenging.
Allele-specific analyses measure the effects of the imbalance between allelic variants in the same genome, mitigating context-specific effects (McDaniell et al. 2010; Onuchic et al. 2018; Zhou et al. 2018; Yang et al. 2019; Sun and Zhang 2020; Amoah et al. 2021). We hypothesized that an allele-specific m6A modification (ASm6A) analysis may greatly improve the accuracy and sensitivity of conventional MeRIP/m6A-seq assays (McIntyre et al. 2020), providing improved resolution of m6A quantification, especially in human tissue samples.
In this study, we developed a transcriptome-wide ASm6A identification approach to accurately and sensitively detect m6A imbalances between two alleles in human tissues, which could shed light on the exploration of functional genetic variants associated with human health and disease and the trans-factors involved in m6A regulation.
Results
Generation and evaluation of an ASm6A analysis pipeline
To measure the m6A changes in response to genetic variation, we developed a pipeline to identify genome-wide ASm6A from m6A sequencing (MeRIP-seq) data (Fig. 1A). First, high-confidence heterozygous variants were identified by variant calling followed by stringent filtering to minimize the bias introduced through transcription and mapping (Castel et al. 2015). Then the allelic read counts from m6A IP and input sequencing data were calculated, and a Fisher's exact test was performed to assess the modification difference between the two alleles at each heterozygous site. Last, the significant allelic m6A imbalance events within m6A peaks were retained and called as ASm6As (Fig. 1A).
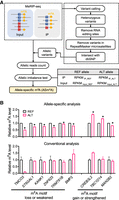
ASm6As analysis pipeline generation and evaluation. (A) Pipeline for ASm6As identification. Allelic variants were identified by variant calling and stringent filtering. Allelic reads were then counted from sequencing data of input and IP, and ASm6As were predicted using a Fisher's exact test. (B) Comparison between allele-specific analysis and conventional analysis. In the m6A motif loss or weakened group, the potential m6A motifs (reference to alternative allele) were found in TMCO3 (AAACT to AAATT), ST6GAL1 (GGACT to GGGCT), KANK1 (GAACT to GAGCT), ZNF623 (GGACC to AGACC), TRMT61B (GAACT to GAGCT), and BMP2 (AGACT to AGTCT). In the m6A motif gain or strengthen group, they were found in CREB3L2 (GCACT to GAACT), TBC1D14 (CGACT to GGACT), and MAN2B2 (GAGCA to GAACA). The relative m6A level was calculated by dividing the reads of IP by input, then normalized to the reference allele. Data are mean ± SD from two independent experiments. In the allele-specific analysis, (*) P < 0.05 in both replicates as assessed by Fisher's exact test.
To compare the performance of ASm6A analysis with conventional MeRIP analysis in identifying genetic variant–associated m6A change, we developed a reporter assay using alleles predicted to affect m6A through changes of the m6A motif. In the “m6A motif loss or weakened” group, the alternative allele disrupted the m6A motif RRACH, such as rs11164135 on TMCO3, which contains a C-to-T SNP in the fourth position of the motif (i.e., AAATT instead of the reference AAACT sequence). In the “m6A motif gain or strengthened” group, the alternative allele contained a stronger m6A motif. We cloned the alternative and reference m6A peak regions into reporter constructs and cotransfected them into HEK293T cells. The RNA was then extracted, fragmented, and subjected to MeRIP and ASm6A analysis. In the conventional analysis group, the REF and ALT reporters were transfected into cells and subjected to the MeRIP procedure separately, and then, the m6A levels on the REF and ALT reporters were compared. We found that the ASm6A analysis showed expected m6A-level changes in eight of nine reporters (Fisher's exact test), even with minor differences between the two alleles, across two replicates (Fig. 1B). In contrast, the conventional MeRIP analysis showed more unexpected m6A changes and required more replicates to analyze the statistical difference in m6A changes between the two alleles. Thus, our ASm6A analysis pipeline can sensitively and accurately detect m6A changes associated with genetic variants.
ASm6A profiling in human tissues
We next used this pipeline to explore the profile of ASm6As across diverse human tissues. We collected 90 reported MeRIP-seq data sets covering 25 tissues from 18 individuals (Xiao et al. 2019; Liu et al. 2020b; Zhang et al. 2020a). After passing these data through our pipeline, we obtained a total of 12,056 ASm6As (Fig. 2A). A representative ASm6A, shown in Figure 2B, had a higher level of m6A on allele T than on allele C across three tissues (brain cerebellum, heart, and liver) from the same individual. To validate these ASm6As, we randomly chose several ASm6As from a kidney sample and performed m6A immunoprecipitation followed by Sanger sequencing. An imbalance in the m6A levels on two alleles was observed (Fig. 2C; Supplemental Fig. S1A), which was consistent with the results from the pipeline analysis.
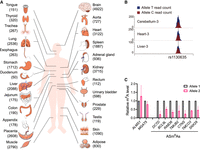
ASm6A profiling in human tissues. (A) Numbers of ASm6As identified in 25 human tissues. (B) ASm6A modification at the rs1130635 locus in three different tissue samples from the same individual. The horizontal coordinate represents the genomic position, and the vertical coordinate represents the number of sequence reads supporting two alleles (T and C) in the IP sample. The T/C read counts in input samples were 40/36, 26/31, and 40/45 in Cerebellum-3, Heart-3, and Liver-3, respectively. (C) MeRIP followed by Sanger sequencing showing the relative m6A level of allele 2 versus allele 1 at genes in the Kidney-2 sample.
We have also applied our method to the m6A methylome generated by the base-resolution and quantitative m6A sequencing method GLORI-seq and m6A-SAC-seq in HEK293T and HeLa cells (Supplemental Fig. S1B; Ge et al. 2023; Liu et al. 2023). A total of 91 ASm6As were identified. Sixty-six of them have at least one nearby m6A site with >10% m6A imbalance between the two alleles. And 27 ASm6As identified by MeRIP-seq in the human tissues were supported by this analysis. Two representative ASm6A sites are shown in Supplemental Figure S1C.
Analysis of the full set of ASm6As in human tissues showed that they were enriched for a GGAC motif (Supplemental Fig. S2A) and preferentially found at the CDS and 3′ UTR, peaking at stop codon regions (Supplemental Fig. S2B,C), properties that are similar to those of typical m6A modifications (Dominissini et al. 2012). As allelic imbalance analysis can capture the effect of rare variants (Sun et al. 2016; Onuchic et al. 2018), we annotated these ASm6A variants with allele frequency from the Genome Aggregation Database (gnomAD) (Karczewski et al. 2020) and found 2123 were rare variants (minor allele frequency [MAF] < 5%). The fraction of rare variants in ASm6A variants ranged from zero to 24.3% in different samples (Supplemental Fig. S2D). We then analyzed the host genes of ASm6As in different tissues and found that these genes were enriched in tissue-specific processes, suggesting a potential tissue-specific role for ASm6A. For example, the ASm6A host genes in the brain were enriched in neuron differentiation, and the ASm6A host genes in the heart were enriched in heart morphogenesis (Supplemental Fig. S2E).
Identification of putative functional variants for ASm6A regulation
We next sought to determine the genetic variants that can regulate the m6A level at the heterozygous sites. We reasoned that if a variant in a heterozygous site was functional in m6A regulation, we would consistently observe an allelic imbalance in the m6A modification across samples carrying it. To identify such sites, we adapted reported methods (Fig. 3A; Amoah et al. 2021; Li et al. 2021). For each ASm6A, the concordance of m6A methylation bias in different samples was analyzed. Genetic variants with high concordance (>90%) were considered as putative functional variants. A total of 1184 variants were identified across samples (Supplemental Table S1). For example, rs1739047003 showed an obvious imbalance in m6A between the reference allele C and the alternative allele T (Fig. 3B). Among these, 217 were rare variants.
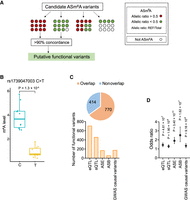
Prediction of functional variants for ASm6A regulation. (A) Diagram showing the identification strategy of putative functional variants from all ASm6A variants. (B) m6A levels at the reference allele C or alternative allele T of rs1739047003 site in 20 samples. (C) The total proportion (top) and numbers (bottom) of putative functional variants that overlap with annotated functional genetic variants, including eQTL, sQTL, ASE, allele-specific RBP binding (ASB), and GWAS causal variants. The odds ratio, confidence interval, and P-value are shown. (D) Enrichment of putative functional variants in five genetic annotations. The error bars indicate the upper and lower bounds of the 95% confidence interval.
In addition, the majority of these putative functional variants (n = 770, 65.0%) overlapped with genetic variants associated with other molecular phenotypes, including all eQTLs, splicing quantitative trait loci (sQTLs) in previous work (GTEx Consortium 2017), allele-specific expression (ASE) in AlleleDB, allele-specific RBP binding (ASB) (Bahrami-Samani and Xing 2019; Yang et al. 2019), or GWAS causal variants in the CAUSALdb database (Fig. 3C; Wang et al. 2020). Moreover, these variants were significantly enriched in the functional genetic variants (Fig. 3D). To test the influence of tissue specificity in the analysis, we have also conducted enrichment analysis using the ASm6As and QTLs derived from the same tissues. The results showed enrichment of ASm6As for eQTLs and sQTLs in most tissues (Supplemental Fig. S3A,B). The ASE data we used did not provide tissue information, so we did not perform tissue-specific enrichment analysis.
Together, these results suggest that the putative functional variants may be involved in the regulation of other molecular phenotypes.
Validation of putative functional variants for ASm6A regulation by reporter assay
To validate our analysis method and the predicted functional variants for ASm6A, we generated 26 pairs of reporters containing ASm6A variants that have different concordance ratios across the samples (ranging from >50% to >90%) and eight pairs of reporter-harboring heterozygous sites not identified as ASm6As in any human tissues (not-ASm6A variants), and cotransfected them into HEK293T cells, followed by MeRIP. We found that with a rising concordance ratio, the validated fraction also augmented. Also, all putative functional variants (concordance ratio > 90%) gave rise to a similar allelic m6A bias, as was observed in the tissue samples (Fig. 4A). In contrast, only one of the eight (12.5%) not-ASm6A variants generated allele-specific modifications in HEK293T cells (Fig. 4A). The allelic ratio of the validated variants, including functional variants, in tissue samples and HEK293T cells showed a positive correlation (R2= 0.74) (Fig. 4B). For example, we found the alternative allele G of functional variant rs117097571 located in the gene TBC1D14 was more likely to be modified by m6A in the tissues. In the validation reporter assay, the m6A level of the G allele was also higher than that of the reference allele C (Fig. 4C). Similarly, functional variant rs370724501, which is a rare C-to-T variant in the protein-coding gene ZNF217 (MAF = 9.848 × 10−5), had a higher level of m6A at the reference allele C than at the alternative allele T in both the tissue samples we analyzed and our reporter assay (Fig. 4D).
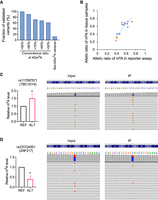
Validation of putative functional variants for ASm6As by reporter assay. (A) Fraction of validated ASm6As in the reporter assay. ASm6A reporters with different concordance ratios across samples are shown. (B) Correlation of the allelic m6A ratio (REF/total) of validated ASm6A variants in tissue samples and HEK293T reporter assay. The orange dots indicate the functional variants. (C,D) Examples of validated variants. (Left) The relative m6A level of ref versus alt. (Right) Captures showing the overall allelic reads and 15 representative reads.
Prediction of trans-factors involved in ASm6A regulation
To further investigate the regulation of ASm6A, we annotated the putative functional variants for ASm6A and the surrounding sequence for RNA-related features (Fig. 5A); 10.85% of the variants were enriched in the m6A consensus motif RRACH (Fig. 5A,B), which may directly mediate the binding of methyltransferases or demethylases to regulate ASm6A.
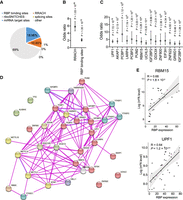
Prediction of trans-factors involved in ASm6A regulation. (A) Proportion of the putative functional variants for ASm6As that overlap with RNA-related features, including RBP binding sites, RRACH sequence, miRNA target sites, riboSNITCHES variants, and splicing sites. (B) Enrichment of the putative functional variants for ASm6As versus random control variants in RRACH sequence and RBP binding sites by Fisher's exact test. (C) Enrichment of the putative functional variants for ASm6As in binding sites of different RBPs by Fisher's exact test. (D) Protein–protein interactions between the RBPs in C and known m6A writers, readers, and erasers. The red sphere indicates the RBPs in C. (E) Correlation between RBP expression and aggregative m6A level of its targets for RBM15 (top) and UPF1 (bottom).
Moreover, we found that 18.98% of the variants were enriched in RBP binding sites (Fig. 5A,B). To find what RBPs are associated with ASm6A, RBP binding sites contained in the ENCODE Project Consortium (The ENCODE Project Consortium 2012) were used. In the ENCODE Project, almost all the RBP binding sites data were from K562 and/or HepG2 cell lines. So, we performed RBP enrichment analysis using functional variants for ASm6A regulation, which is a subset of variants that has high concordance in various tissue samples. We found that the binding sites of 15 RBPs were significantly enriched for putative functional variants (Fig. 5C). We have also analyzed the enrichment of RBP binding sites in HepG2 cells for all the ASm6As in liver samples (Supplemental Fig. S4A) and found that 10/15 of the RBPs enriched for functional variants also enriched in this analysis. In addition, 80 RBPs were found to have allelic binding sites overlapping with functional variants (Supplemental Table S2). These RBPs included known m6A readers such as IGF2BP1 and IGF2BP2 (Fig. 5C), which are involved in the regulation of mRNA stability and translation (Huang et al. 2018), suggesting that ASm6A may facilitate allelic stability and translation of RNA through altering the binding of m6A readers.
It has been reported that the interactions of RBPs with m6A methyltransferases, demethylases, or readers can alter m6A levels (Zhou et al. 2015; Zheng et al. 2017; Wang et al. 2019; An et al. 2020) or facilitate m6A-mediated translation (Alarcón et al. 2015; Meyer et al. 2015; Choe et al. 2018), so we analyzed the interaction network of the enriched RBPs. We found that 59 RBPs were predicted to interact with the methyltransferase complex, demethylases, or m6A reader proteins (Fig. 5D; Supplemental Table S2). And in these RBPs, 35 have a significant correlation between the expression level of RBPs and the m6A levels of the binding regions in the tissue samples (Fig. 5E; Supplemental Fig. S4B; Supplemental Table S2). We speculate that these RBPs may be involved in ASm6A regulation or function. Supporting these findings, among these RBPs, RBM15 is a known m6A regulator on Xist RNA through interaction with METTL3 (Patil et al. 2016), and EIF3H can interact with METTL3 to enhance translation and promote oncogenesis (Choe et al. 2018).
ASm6A is associated with risk variants in common diseases
It has been previously reported that genetic regions corresponding to m6A peaks in human tissues are enriched for disease-associated variants (Xiao et al. 2019; Zhang et al. 2020a). We therefore sought to explore if there is a link between ASm6A and human complex traits and common diseases. To assess this, we considered GWAS risk SNPs collected from the GWAS catalog (MacArthur et al. 2017) and SNPs in linkage disequilibrium (LD) with them. Of all the ASm6As in human tissues, 2560 were also GWAS risk loci (Fig. 6A). Notably, the GWAS risk loci were significantly enriched for all ASm6A variants (odds ratio = 1.58, 95% CI [1.51–1.66], P = 5.04 × 10−75, Fisher's exact test). Furthermore, ASm6A variants in 22 out of the 25 tissues were enriched in GWAS risk loci (Supplemental Fig. S5A). Also, 228 putative functional variants for ASm6A regulation were GWAS risk loci.
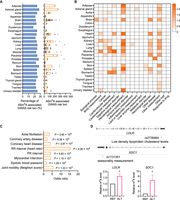
ASm6A analysis reveals m6A-regulating risk variants in common diseases. (A) The fraction of ASm6A variant–associated GWAS risk loci in each tissue (left), and the number of ASm6A variant–associated GWAS risk loci in each sample (right). (B) Enrichment of ASm6As from various tissues in 17 GWAS categories. Fisher's exact test, P < 0.05. (C) Odds ratio showing the enrichment of host gene of ASm6A-associated risk loci from the heart in GWAS traits and diseases. The most significant traits/diseases are shown. (D) Validation of the allelic m6A modifications on GWAS risk variants rs2738464 and rs1131351 by reporter assay.
To interpret the roles of ASm6A variants in human common diseases, the GWAS risk loci were divided into 17 categories (Xiao et al. 2019). We found that ASm6A variants were enriched in cardiovascular measurement, hematological measurement, and neurological disorder categories in a tissue-specific manner (Fig. 6B). Importantly, the host gene of ASm6A-associated risk variants in the heart were enriched in atrial fibrillation and coronary artery disease (Fig. 6C), whereas those in the brain were enriched in schizophrenia and epilepsy (Supplemental Fig. S5B).
We then performed reporter assays to validate if the ASm6A-associated risk loci could regulate m6A variance in cells. rs2738464 in the 3′ UTR of the LDLR gene, a probable GWAS causal variant associated with low-density lipoprotein cholesterol levels and coronary artery disease (Wang et al. 2020), showed higher m6A levels on the alternative allele C (Fig. 6D). We also found that rs1131351 in the 3′ UTR of SDC1 gene, which is associated with seasonality measurement trait, had a higher m6A level on the alternative allele G (Fig. 6D).
We further found that the biological processes of the host genes of ASm6A-associated risk loci in the heart were enriched in cardiocyte differentiation, artery development, and heart morphogenesis (Supplemental Fig. S5C) and that, in the brain, they were enriched in axonogenesis (Supplemental Fig. S5C), suggesting that ASm6As may affect diseases or traits by impacting the genes in which they are located. It is known that m6A can affect the RNA level by affecting transcription and stability (Wang et al. 2014a; Huang et al. 2018; Li et al. 2020; Liu et al. 2020a), and we therefore asked whether ASm6A-associated risk loci can affect host gene expression. We generated the ASE sites from input sequencing data and found that 90 ASm6A-associated risk loci were also ASE sites. This suggested a potential mechanism by which m6As could contribute to disease through regulation of host gene expression.
Discussion
Recent studies have shown that m6A loci are enriched in human genetic variants. Here, we accurately and sensitively detected m6A imbalances between two alleles in human tissues, identified a large number of high-confidence ASm6As, and validated them using a reporter assay. We further investigated the potential for trans-proteins acting on ASm6As. Last, we found that ASm6As are enriched in human common traits and diseases.
Technical and biological variations in sample preparation, immunoprecipitation efficiency, and other experimental conditions render conventional MeRIP/m6A-seq analysis methods highly challenging for the accurate detection of m6A changes (McIntyre et al. 2020). The ASm6A analysis we developed overcomes these limitations by directly measuring the imbalance effect between two alleles in exactly the same cellular environment. Thus, ASm6A enables accurate and sensitive detection of m6A changes, facilitating the systematic identification of functional variants and trans-factors involved in m6A regulation.
Allelic imbalance analysis is performed in a single sample, which is also beneficial for the detection of effects of rare and de novo variants in diseases (Sun et al. 2016; Onuchic et al. 2018). We found 2123 rare variants are associated with ASm6As; 217 rare variants are putative functional variants; and four were validated using a reporter assay. Future applications of our approach may be investigating the rare or de novo genetic mutations causing m6A modification changes in disease contexts, especially developmental diseases or tumorigenesis, in which m6A has been reportedly implicated (Batista et al. 2014; Wang et al. 2014b; Geula et al. 2015; Li et al. 2017b; Vu et al. 2017; Xu et al. 2017; Yoon et al. 2017; Zhang et al. 2017).
A large number of putative variants have been implicated in m6A regulation, which presents a challenge for validating their impact on m6A at scale. To address this challenge, we generated a reporter system to validate the putative functional variants for ASm6A in cultured cells. Most of the tested variants observed in human tissues were recapitulated in the reporter system, and the inconsistency of the remaining variants may be owing to the lack of cell type–specific factors in HEK293T cells or the limited insert size cloned into the reporters, as discussed in other genetic variant reporter assays (Amoah et al. 2021).
Up to now, the number of m6A sequencing data in human tissues is relatively small, and base-resolution and quantitative m6A sequencing data in human tissues have yet to emerge. Although ASm6A analysis is not constrained by the sample size, the discovery of functional variants for ASm6A regulation by concordance analysis performs better when having a bigger sample size. We have collected the available m6A sequencing data in human tissues in this study to identify the functional variants, and validated a small part of them by a reporter assay. However, there still may be false-positive or false-negative variants in this analysis, and the tissue specificity of functional variants was not analyzed owing to the limited sample size from different tissues. With the growth of sequencing data, the accuracy of functional variants discovery would increase, and the tissue specificity of the functional variants would offer more fine regulating details for m6A in human tissues.
In summary, our work provides a new method to detect the dynamic network of RNA modifications at the allelic level. Accompanied with the growing atlas of human omics data, ASm6As analysis will contribute to increased resolution in our understanding of the role of m6A and associated genetic variants in human health and diseases.
Methods
MeRIP-seq data collection and alignment
MeRIP-seq raw sequencing reads were downloaded from the NCBI Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/; accession numbers GSE114150, GSE122744) and Genome Sequence Archive in BIG Data Center (https://ngdc.cncb.ac.cn/gsa/) accession number CRA001315. Trim_galore (version 0.5.0; http://www.bioinformatics.babraham.ac.uk/projects/trim_galore/) was used to trim adaptors and control read quality. Then, the clean reads were mapped to the human genome (hg19) using STAR (version 2.7.3a) (Dobin et al. 2013) with parameters set as ‐‐twopassMode Basic. The hg19 reference genome was used primarily owing to the comprehensive nature of relevant data and tools available at the time. Furthermore, the regions of interest in this study correspond to genic loci that have been thoroughly annotated in hg19, with minimal changes observed in updated reference genomes such as GRCh38. Therefore, the use of alternative reference genomes is unlikely to significantly alter the conclusions made herein. The Picard suite (http://broadinstitute.github.io/picard/) was used to perform “AddOrReplaceReadGroups” and “MarkDuplicates REMOVE_DUPLICATES = true” procedures to add group information and remove duplicate reads. Last, the unique reads were picked out through SAMtools (Li et al. 2009) with “-q 20.”
Variant calling from input sequencing data
The SplitNCigarReads procedure in the GATK suite (Van der Auwera et al. 2013) was used to split reads into exon segments. The recalibration of bases was performed by BaseRecalibrator based on known SNPs and indels in The 1000 Genomes Project Consortium (2015). Then, ApplyBQSR was performed based on the covariate file generated by the previous step to apply base quality score recalibration. Variants were called using the HaplotypeCaller procedure. Only variants passing the filter “-stand-call-conf 20 ‐‐minimum-mapping-quality 20” were kept for downstream analysis. The variants were retained if they matched the criteria “neither found in UCSC RepeatMasker microsatellites (Tarailo-Graovac and Chen 2009) nor in RNA editing sites (RADAR database)” but were contained in the dbSNP database.
Mapping bias correction using STAR-WASP pipeline
SNVs were called based on the input sequencing data alignments using the GATK suite. Samples from the same individual were combined and used for SNV calling. Then, the SNVs were used as an input list to WASP (van de Geijn et al. 2015), which was embedded in the STAR suite through the “‐‐varVCFfile” parameter.
Identification of ASm6As
First, all biallelic heterozygous sites were picked out using VCFtools (Danecek et al. 2011). The ASEReadCounter procedure contained in GATK was executed to count the reads on the two alleles for each SNV. Only those variants that satisfied the minimum mapping reads on both alleles were considered as effective candidate heterozygous sites (each allele ≥ 2, the sum of two alleles ≥ 10). A Fisher's exact test was used to identify ASm6As by comparing the normalized reads count (RPKM) on reference and alternative allele for IP and input samples. All the P-values produced above were supplied to FDR correction using the Python module “fdrcorrection.” Significant ASm6As were retained if their adjusted P-value was less than 0.05.
We then filtered candidate allele-specific events for location within the m6A peaks. Two peak callers, MACS2 (Zhang et al. 2008) and MeTPeak (Cui et al. 2016a), were used to increase the confidence of m6A peak identification. For MACS2, the P-value cutoff was set to 10−5, and -f BAMPE, ‐‐nomodel, ‐‐call-summits was used. For MeTPeak peak calling, the default parameter settings were used. After “MaxMinNormalization” normalization, consensus m6A peaks were constructed from the above two methods by MSPC. To avoid the influence of N6,2′-O-dimethyladenosine (m6Am) modifications, peak regions containing an adenosine at the transcription start site and having more than five reads in both the IP and input spanning −100 to +100 nt of the transcription start site were identified as m6Am peaks and were removed (Schwartz et al. 2014).
To analyze the m6A imbalance events from the base-resolution quantitative m6A sequencing method, the GLORI-seq (GSM6432590, GSM6432591, GSM6432592, GSM6432595 GSM6432596, and GSM6432597) (Liu et al. 2023) and m6A-SAC-seq data (GSM4950342, GSM4950344, GSM4950345, GSM4950347, GSM4950349, and GSM4950350) (Hu et al. 2022) were download. The allelic variants were identified from the untreated data as mentioned above. The reads from GLORI- or SAC-treated data were assigned to the reference or alternative allele file based on the allelic variants generated. The coverage for A + G in GLORI seq data should be 15 or more in both the REF and ALT files, and reads containing four or more unconverted A's were filtered. For m6A-SAC-seq, the read coverage should be greater than five in both the REF and ALT files. Then the conversion rates calculation and m6A site calling were performed as reported (Hu et al. 2022; Liu et al. 2023). The candidate site should be identified as m6A in at least one of either the REF or ALT files. Then a two-tailed Fisher's exact test was performed to assess the reads of m6A in REF and ALT files, and the significant allelic sites were considered as ASm6A sites.
Determination of ASE
Determination of ASE was performed as reported (Ge et al. 2009; GTEx Consortium 2013). For each heterozygous site that satisfied the minimum read number criteria in input, we calculated a binomial P-value by comparing with the expected null ratio (0.5). After FDR correction, significant (FDR < 0.05) ASEs were retained.
Inference of putative functional variants for ASm6A regulation
All of the ASm6A variants were considered as candidate variants. The following criteria were used to identify putative regulating variants for ASm6As: The variants (1) occurred in at least two samples, (2) met the minimal read counts requirement, (3) were identified as ASm6A variants and having concordant allelic direction in at least 90% of the samples overlapping the m6A peaks, and (4) showed no significant difference (P-value > 0.1, Fisher's exact test) of allelic m6A level in the concordant samples (Li et al. 2021).
Motif analysis
For each ASm6A, flanking regions from 4-bp upstream to 4-bp downstream (e.g., all 5-mer positions that include the variant) were extracted from hg19 reference genome by BEDTools “getfasta” (Quinlan and Hall 2010) and then used to search for motifs. The strand of each flanking region sequence was assigned based on the overlapping transcript strand information. For the sets of sequences, the RRACH motifs were searched using HOMER findMotifi.pl.
The metagene profile of ASm6A variant sites was generated using the R package GUITAR (Cui et al. 2016b) with default parameters.
ASm6A validation by Sanger sequencing
The tissue samples (Xiao et al. 2019) were used following the ethical regulations of the medicine ethics committee of Nanfang Hospital, Southern Medical University. The RNA extraction and MeRIP were performed as previously described (Xiao et al. 2019). The input and immunoprecipitated RNA were reverse-transcribed into cDNA using a GoScript reverse transcription system (Promega). The MeRIP-qPCR was performed as previously described (Xiao et al. 2019). The sequences around ASm6A variants were amplified by 30 rounds of PCR, cloned into T-vectors, and transfected into DH5α-competent cells. Single clones were subjected to Sanger sequencing using an ABI 3730XL DNA analyzer, and the reads of each ASm6A variant were counted and calculated. The results are shown in Figure 2C.
Reporter assay
Genomic DNA of HEK293T cells were extracted, and then the genomic regions containing the alleles and the m6A peak were cloned into CS2 vectors via homologous recombination after BamHI and XbaI digestion to linearize the vectors. Alleles different from the extracted genome were introduced by site-directed mutagenesis. The reporter plasmids were pooled and transfected into HEK293 cells. Then, RNA was isolated using TRNzol reagent (Tiangen). After DNase I digestion, the RNA was fragmented and subjected to MeRIP as previously described (Xiao et al. 2019). The immunoprecipitated and input RNAs were subjected to library generation. Sequencing was performed on an Illumina Nova platform. The predicted alleles in Figure 1B were from kidney samples and were located in the m6A peaks in HEK293T cells. Two batches of reporter assay were performed in Figure 1B, and five were performed in Figure 4. The primers used are supplied in Supplemental Table S3.
Trim_galore was used for adapter trimming as well as quality control. The “umi_tools extract” was used to extract UMIs and append to the read name. The RT primer sequence was trimmed by fastp (Chen et al. 2018). Then, these clean FASTQ files were aligned to the human reference genome using STAR. “umi_tools dedup” was used to deduplicate reads based on the mapping coordinates and the UMI attached to the reads. The count of reads from IP and input sample BAM files were obtained using BEDTools “multicov,” and the m6A level of each fragment was quantified as: RPKMIP/RPKMInput. Only the fragments whose m6A level exceeded the average m6A level of POU5F1 and GAPDH were considered to have m6A modifications. Heterozygous variants located within these fragments were regarded as valid variants and used for downstream analysis. For each valid variant, the number of reads on each allele was counted. Loci with reads on any allele below two were discarded. Fisher's exact tests for each heterozygous site were performed to check the significance of m6A modification difference between two alleles. Variants having a significant m6A imbalance and similar modification bias with the majority of the tissue samples in 80% of the replicates were considered validated variants.
For the reporter assay analysis using conventional MeRIP analysis, the REF and ALT allele reporters were transfected to the HEK293T cells separately. Then the two groups of RNA were extracted and subjected to a MeRIP-seq procedure as previously described (Xiao et al. 2019). The m6A level was calculated by RPKMIP/RPKMInput (Wan et al. 2015; Xiao et al. 2019), and then the m6A level of REF and ALT alleles was compared.
Gene Ontology analysis of ASm6A host genes
ASm6As were assigned to genes referring to the human reference annotation (gff3 file). The clusterProfiler (Yu et al. 2012) R package was used to determine the overrepresentation of biological process categories of ASm6A host genes.
Enrichment analysis of ASm6As
The eQTL and sQTL data sets were obtained from the GTEx Consortium (2017). The ASE data sets were obtained from AlleleDB (http://alleledb.gersteinlab.org/). The allele-specific RBP binding data sets were from reported papers (Bahrami-Samani and Xing 2019; Yang et al. 2019). The causal variants were downloaded from CAUSALdb (http://mulinlab.org/causaldb). RBP binding sites were downloaded from The ENCODE Project Consortium (2012). To generate the control variant set, the SNPsnap website (Pers et al. 2015) was used to sample 100 sets of variants, and the MAF, number of variants in linkage disequilibrium (LD buddies), distance to nearest gene, and gene density were considered. To match the location of ASm6A variants, the EAS data set of SNPsnap was divided into promoter-TSS, 5′ UTR, exon, intron, 3′ UTR, TTS, intergenic, and noncoding regions. Then, a two-tailed Fisher's exact test was used to compare the number of ASm6A variants and control variants that overlap with RBP binding sites or other indicated data sets as reported (Zhang et al. 2020b). The overlaps between ASm6As and rare variants and GWAS-related ASm6A and ASE were not significant.
Enrichment of ASm6A variants in GWAS SNP data sets
For GWAS enrichment analysis, the total ASm6A data set was used. Control variants were randomly sampled by SNPsnap. The GWAS SNPs were collected from the NHGRI GWAS Catalog (MacArthur et al. 2017). Only autosomal SNPs were retained. SNiPA (Arnold et al. 2015) was used to obtain the LD mutations within a 500-kb window with a parameter of r2 > 0.8 in the East Asian population for each GWAS SNP. A Fisher's exact test was used to test enrichment of ASm6A variants in GWAS SNP data sets (Zhang et al. 2020b). Enrichr was used in the enrichment analysis of the host gene of ASm6A-associated risk loci in the GWAS traits/diseases (Kuleshov et al. 2016). R v4.2.0 was used (R Core Team 2022).
Enrichment of ASm6A variants in 17 GWAS categories
GWAS SNPs were grouped into 17 categories according to the NHGRI GWAS catalog (MacArthur et al. 2017). One hundred control SNP sets were generated as in the enrichment analysis above. The number of SNPs in each GWAS category and its control SNP set were determined. Then 100 Fisher's exact tests were performed. If the P-value was greater than 0.05 in one test, the corresponding odds ratio was set to zero. The mean odds ratio of 100 Fisher's exact tests was shown (Xiao et al. 2019).
Construction of protein–protein interaction network
Interactions with ASm6A-enriched RBPs and known m6A writers, readers, and erasers (Jia et al. 2011; Zheng et al. 2013; Liu et al. 2014; Ping et al. 2014; Wang et al. 2014a, 2015; Xiao et al. 2016; Li et al. 2017a; Yin et al. 2022) were visualized through the STRINGdb R package (Szklarczyk et al. 2019). Among these active interaction sources, only “experiment” records were shown in the network.
Correlation between RBP expression and m6A level of its targets
The TPM of each RBP in different samples was calculated as was the aggregated m6A score of m6A peaks that overlapped with the RBP-binding sites. Based on the expression of a given RBP and its corresponding aggregated m6A score, a regression analysis was performed, and the P-value was calculated.
Data access
All the raw and processed sequencing data generated in this study have been submitted to the NCBI BioProject database (https://www.ncbi.nlm.nih.gov/bioproject/) under accession number PRJNA794937. The custom codes are provided as Supplemental Code.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
This work was supported by the National Key R&D Program of China (2019YFA0111200, 2021YFA0805400), the National Natural Science Foundation of China (82230053, 32222016, 32170845, 32200454), the Guangdong Basic and Applied Basic Research Foundation (2022B1515020107), and the Science and Technology Program of Guangzhou, China (202201010922).
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.277704.123.
- Received January 15, 2023.
- Accepted June 13, 2023.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see https://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








