
Genetic control of the dynamic transcriptional response to immune stimuli and glucocorticoids at single-cell resolution
- Justyna A. Resztak1,8,
- Julong Wei1,8,
- Samuele Zilioli2,3,
- Edward Sendler1,
- Adnan Alazizi1,
- Henriette E. Mair-Meijers1,
- Peijun Wu4,
- Xiaoquan Wen4,
- Richard B. Slatcher5,
- Xiang Zhou4,
- Francesca Luca1,6,7 and
- Roger Pique-Regi1,6
- 1Center for Molecular Medicine and Genetics, Wayne State University, Detroit, Michigan 48201, USA;
- 2Department of Psychology, Wayne State University, Detroit, Michigan 48201, USA;
- 3Department of Family Medicine and Public Health Sciences, Wayne State University, Detroit, Michigan 48201, USA;
- 4Department of Biostatistics, University of Michigan, Ann Arbor, Michigan 48109, USA;
- 5Department of Psychology, University of Georgia, Athens, Georgia 30602, USA;
- 6Department of Obstetrics and Gynecology, Wayne State University, Detroit, Michigan 48201, USA;
- 7Department of Biology, University of Rome “Tor Vergata,” 00133 Rome, Italy
-
↵8 These authors contributed equally to this work.
Abstract
Synthetic glucocorticoids, such as dexamethasone, have been used as a treatment for many immune conditions, such as asthma and, more recently, severe COVID-19. Single-cell data can capture more fine-grained details on transcriptional variability and dynamics to gain a better understanding of the molecular underpinnings of inter-individual variation in drug response. Here, we used single-cell RNA-seq to study the dynamics of the transcriptional response to glucocorticoids in activated peripheral blood mononuclear cells from 96 African American children. We used novel statistical approaches to calculate a mean-independent measure of gene expression variability and a measure of transcriptional response pseudotime. Using these approaches, we showed that glucocorticoids reverse the effects of immune stimulation on both gene expression mean and variability. Our novel measure of gene expression response dynamics, based on the diagonal linear discriminant analysis, separated individual cells by response status on the basis of their transcriptional profiles and allowed us to identify different dynamic patterns of gene expression along the response pseudotime. We identified genetic variants regulating gene expression mean and variability, including treatment-specific effects, and showed widespread genetic regulation of the transcriptional dynamics of the gene expression response.
The immune system exerts its function through a delicate and timely balance of pro- and anti-inflammatory processes. An effective response of the immune cells ensures that pathogens are neutralized, yet it is equally crucial that inflammatory processes are timely modulated to avoid pathological states, such as systemic inflammation (Varela et al. 2018), cytokine storm (Fajgenbaum and June 2020), and sepsis (Shankar-Hari et al. 2016). Specificity in recognizing potential pathogens and mounting a proportionate response is important to prevent or resolve infectious diseases; importantly, dysregulation of these processes leads to chronic conditions, including autoimmune, allergic diseases, and asthma. The hypothalamic–pituitary–adrenal axis plays a central role in the balance between pro- and anti-inflammatory processes through regulation of glucocorticoids in the bloodstream. Because of their potent anti-inflammatory effects, synthetic glucocorticoids have widespread pharmacological applications. Steroid anti-inflammatory drugs are used to treat asthma (budesonide) (Jenkins et al. 2020), autoimmune diseases (budesonide, dexamethasone [DEX], prednisone, methylprednisolone) (Snider and Potter 2011; Hughes et al. 2017; Mieli-Vergani et al. 2018; Wang et al. 2018; Mithoowani and Arnold 2019; Strum et al. 2020), and several other inflammatory conditions. DEX is a potent synthetic glucocorticoid that is used to treat leukemia because of its ability to inhibit lymphocyte proliferation (Pui and Evans 2006). Most recently, DEX has been recognized as the first effective drug to prevent severe complications from coronavirus disease 2019 (COVID-19) (Horby et al. 2021). Severe COVID-19 is thought to be a consequence of an exaggerated response of the immune system to SARS-CoV-2 infection (Anka et al. 2021). Elevated production of proinflammatory cytokines results in tissue damage in severe COVID-19 patients and can ultimately be fatal. By activating anti-inflammatory processes, DEX can prevent this “cytokine storm” and improve disease outcome (Horby et al. 2021).
Despite an obviously large contribution of environmental exposures (e.g., allergens, pathogens), genetic and gene–environment interaction effects have a major role in immunological phenotypes (Martin et al. 2021). Genome-wide association studies (GWAS) have successfully identified hundreds of variants associated with autoimmune and allergic disease risk. For example, genetic associations from GWAS explain up to 33% of the heritability of childhood asthma (Pividori et al. 2019). Common genetic variants also explain a large fraction of inter-individual variation in the response to pathogens (Sanz et al. 2018). In addition to large-scale GWAS, in vitro studies of the immune transcriptional response to pathogens have identified thousands of variants that contribute to inter-individual variation (Barreiro et al. 2012; Idaghdour et al. 2012; Çalişkan et al. 2015; Nédélec et al. 2016; Quach et al. 2016; Alasoo et al. 2018; Rotival et al. 2019). These studies have mostly focused on molecular phenotypes, including gene expression, RNA processing, and chromatin accessibility. The same molecular quantitative trait locus (QTL) mapping approaches have also identified genetic variants that modify an individual's response to glucocorticoids (Maranville et al. 2011, 2013), thus highlighting how genetic variation can regulate the important balance between pro- and anti-inflammatory processes.
A key component of the immune system function is the molecular signaling between the different cell types that compose it. This communication is crucial to enable an accurate response to pathogens. However, it is only with the advent of single-cell technology that we can deeply characterize transcriptional responses of individual cell types, while preserving their cross talk through stimulation of the entire PBMC fraction. Single-cell technology also enables the analysis of cell-to-cell transcriptional variability. Gene expression variability plays an important role in biological systems. It has been suggested that in the immune system, gene expression variability increases the ability to respond to immune stimuli (Hagai et al. 2018). In vitro studies show that immune stimuli can modify expression variability in CD4+ T cells (Eling et al. 2018), and variation of gene expression variability is also observed across individuals in human populations (Sarkar et al. 2019; Morgan et al. 2020).
With the great heterogeneity of function between the different cell types comprising the immune system, it is expected that the genetic effects on gene expression will vary across cell types as well. Indeed, multiple studies have leveraged single-cell RNA-seq (scRNA-seq) technologies to map cell type–specific eQTLs across the different subpopulations comprising complex tissues. Studies in peripheral blood mononuclear cells (PBMCs) (van der Wijst et al. 2018), fibroblasts (Neavin et al. 2021), tumor samples (Ma et al. 2022), and pluripotent and differentiating cells (Cuomo et al. 2020; Jerber et al. 2021; Neavin et al. 2021; Elorbany et al. 2022) found several eQTLs that would have been missed in bulk sequencing approaches owing to being active in only one or a few cell types. Although in many single-cell studies most of the detected eQTLs were only found in a single cell type (Kang et al. 2018; Liu et al. 2021; Neavin et al. 2021), this likely overestimates the overall cell type specificity of genetic effects on gene expression owing to incomplete power of eQTL discovery in these data sets (Oelen et al. 2022). Previous approaches have successfully mapped the genetic determinants of responses to infection, drugs, and other stimuli (Barreiro et al. 2012; Idaghdour et al. 2012; Mangravite et al. 2013; Fairfax et al. 2014; Lee et al. 2014; Maranville and Di Rienzo 2014; Siddle et al. 2014; Çalişkan et al. 2015; Moyerbrailean et al. 2016; Nédélec et al. 2016; Quach et al. 2016; Kim-Hellmuth et al. 2017; Knowles et al. 2017, 2018; Manry et al. 2017; Alasoo et al. 2018; Rotival et al. 2019; Huang et al. 2020; Ward et al. 2021) using bulk RNA-seq. However, these genotype-by-environment (GxE) effects are also likely to be cell type–specific (Findley et al. 2021), especially in the context of highly specialized immune responses, as investigated in recent studies of response eQTL (reQTL) mapping using single-cell technology in immune cells exposed to pathogens, bacteria and yeast (Oelen et al. 2022), and influenza A (Randolph et al. 2021). Although different immune stimuli have been studied at single-cell resolution, the response to glucocorticoids has not been previously examined.
Here, we used scRNA-seq to study the dynamics of the transcriptional response to glucocorticoids in activated PBMCs from 96 African American youths with asthma and the genetic basis of variation in gene expression mean and variability across individuals.
Results
Identification of major cell types and gene expression patterns
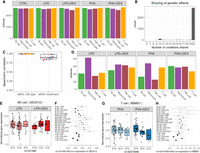
To systematically characterize genetic determinants of transcriptional response to glucocorticoids at single-cell resolution, we studied PBMCs of 96 African American children with asthma. We stimulated the PBMCs with phytohemagglutinin (PHA; a T cell mitogen) or lipopolysaccharide (LPS; a component of the bacterial membrane) and treated with the glucocorticoid DEX for a total of five conditions (including the unstimulated control) (Fig. 1A). We generated a total of 292,394 high-quality cells and detected 46,384 expressed genes from 96 individuals across five conditions (Supplemental Table S1; Supplemental Fig. S1).
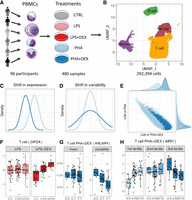
Study overview. (A) PBMCs were collected from 96 African American donors with asthma. Cells were stimulated with phytohemagglutinin (PHA) or lipopolysaccharide (LPS) and treated with glucocorticoid dexamethasone (DEX) for a total of five conditions (including a control), for a total of 480 samples. (B) UMAP visualization of the scRNA data for a total of 293,394 high-quality cells, colored by four major cell types: B cells (green), monocytes (purple), NK cells (maroon), and T cells (orange). (C) Density plot exemplifying changes in mean gene expression between conditions (PHA + DEX vs. PHA). (D) Density plot exemplifying changes in gene variability between conditions (PHA + DEX vs. PHA). (E) Scatterplot representing the low-dimensional manifolds obtained by diagonal linear discriminant analysis (DLDA) in the T cells treated with PHA and PHA + DEX; x-axis denotes the PHA + DEX response pseudotime, and y-axis denotes the PHA response pseudotime. (F) Example of an LPS + DEX response eQTL (reQTL) for DIP2A gene in the T cells. The boxplots depict normalized gene expression levels for the three genotype classes in the two treatment conditions contrasted to identify the reQTL: LPS + DEX (right) and LPS (left). (G) Example of a variability QTL without an effect on the mean gene expression for the ARL6IP4 gene in T cells treated with PHA + DEX. The boxplots depict normalized variability (right) and mean (left) gene expression for the three genotype classes. (H) Example of a DEX response dynamic eQTL for the MRI1 gene in the T cells treated with PHA + DEX.
We clustered the cells and identified four major clusters (Fig. 1B): B cells, monocytes, natural killer (NK) cells, and T cells. T cells formed the largest cluster with 183,289 cells (about 63%), followed by NK cells (47,824, ∼16%), B cells (30,888, ∼11%), and monocytes (30,393, 10%). We confirmed the cell identity using gene expression for cell type–specific genes (Supplemental Fig. S2). Although cells are clustered primarily by cell types in the UMAP plot, we did observe partial separation by treatment when analyzing each major cluster separately (Supplemental Fig. S3).
Using this data set, we will explore the more fine-grained details on transcriptional response to immune treatments, including gene expression (Fig. 1C), variability (Fig. 1D), and dynamics in a short-term span within the treatment (Fig. 1E). Then, we identify the genetic variants regulating gene expression mean and variability (Fig. 1G), including treatment-specific effects (Fig. 1F), and also examine genetic regulation of the transcriptional dynamics of the gene expression response (Fig. 1H).
Cell type–specific gene expression response
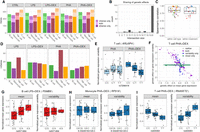
To identify genes differentially expressed in response to the treatments in each cell type, we first aggregated the count data for each cell type–treatment–individual combination. We considered four contrasts in each cell type (Supplemental Fig. S4; Supplemental Table S11): (1) LPS versus CTRL; (2) LPS + DEX versus LPS; (3) PHA versus CTRL; (4) PHA + DEX versus PHA. We detected a total of 6571 differentially expressed genes (DEGs; FDR = 10% and fold change > 1.41) (Fig. 2A; Supplemental Fig. S5). Monocytes showed the strongest gene expression response compared with the other cell types across all treatments (4460 DEGs). LPS induced the weakest gene expression response in all cell types except monocytes. This was expected because LPS stimulates myeloid cells preferentially (Li et al. 2016; Tucureanu et al. 2018). Glucocorticoids induced stronger responses than LPS and PHA across most cell types. In general, we observed that the transcriptional response to glucocorticoids and immune stimuli was highly cell type–specific, such that the majority of DEGs were identified in only one cell type across the four conditions (86% in LPS, 63.7% in LPS + DEX, 66.7% in PHA, and 59.6% in PHA + DEX) (Supplemental Fig. S6).
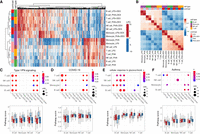
Identification of differentially expressed genes (DEGs). (A) Heatmap of log2 fold change (LFC) of 6571 DEGs (column) across 16 conditions (four contrasts × four cell types; row). (B) Heatmap of Spearman's correlation of LFC across 16 conditions (four cell types × four contrasts). (C–F) Pathway analysis across cell types and treatment conditions for enrichment in DEG (top) and boxplot for average pathway score (bottom) for four pathways: (C) type I interferon (IFN) signaling, (D) coronavirus disease (COVID-19), (E) glucocorticoid stimulus, and (F) asthma.
When we considered the global patterns of treatment effects on gene expression, we observed that effect sizes of DEX and immune stimuli on gene expression were negatively correlated (Fig. 2B), which indicated, as expected, that glucocorticoids reversed the gene expression effects induced by immune stimuli (Fig. 2A; Supplemental Fig. S7). The antagonistic effect of glucocorticoids compared with the immune stimuli was observed also at the pathway level using Gene Ontology (GO) enrichment analysis (e.g., the type I interferon [IFN] signaling pathway, the response to LPS, the cytokine-mediated signaling pathways, and innate immune response [Fig. 2C; Supplemental Figs. S8, S9, S10A–C]). These immune-related pathways shared across cell types appeared to be the molecular basis of how immune stimuli and glucocorticoids affect the immune system transcriptome in opposite directions. To better characterize these gene expression shifts at the pathway level, we derived a pathway-specific score that combines the gene expression changes of all the genes that belong to that pathway term (for more details, see Methods). Here, we considered the type I IFN signaling pathway as an example (Fig. 2C). This pathway was only enriched in genes up-regulated for immune stimuli (LPS or PHA) and down-regulated for the DEX conditions. We also observed that the score of IFN increased after LPS or PHA stimulation, whereas the score in the DEX condition was similar to those of unstimulated cells. Type I IFNs play important roles in the innate and adaptive immune responses not only to viruses but also to bacterial pathogens (Ivashkiv and Donlin 2014; McNab et al. 2015; Boxx and Cheng 2016). Genes enriched in the type I IFN signaling pathway include JAK1, STAT1, STAT2, IRF1, IRF7, IRF8, and OAS1. Treatment with LPS or PHA significantly increased expression of genes in the IFN I pathway, whereas DEX counteracted this effect. We also observed that genes up-regulated in response to immune stimuli or down-regulated in response to DEX were enriched in the COVID-19 pathway, and this enrichment was shared across cell types except monocytes (Fig. 2D).
Moreover, we revealed some DEX-specific pathways, such as the cellular response to glucocorticoid stimulus pathways (FDR < 0.1) (Fig. 2E) and the stress response to metal ion pathways (FDR < 0.1) (Supplemental Fig. S10D), which are both enriched in genes up-regulated in response to treatment with DEX and are consistent with previous findings that glucocorticoids can modulate ion channel activity (Tootell et al. 1983; Hidalgo et al. 1994). In addition, we found that DEX repressed gene expression of the asthma pathway with high cell type specificity, mainly in B cells (FDR < 0.1) (Fig. 2F). B cells play an important role in the induction of asthma by releasing IgE molecules (Romagnani 2000).
Gene expression variability is modified by glucocorticoids and immune stimuli
Beyond looking at the transcriptional response to immune stimuli for each cell type separately, single-cell data provide an unprecedented opportunity to study gene expression dynamics (mean and variability) in response to treatments. Here, we aimed to investigate changes in gene expression variability induced by treatments. We implemented a method based on the negative binomial (NB) distribution to calculate two gene dynamic-related parameters: gene expression mean (μ) and dispersion (ϕ), based on the work of Sarkar et al. (2019). To avoid the mean-variability dependency and detect treatment effects that affect solely variability, we regressed out any residual mean effects on dispersion and used this mean-corrected dispersion as the measure of gene expression variability in the following analyses (Supplemental Fig. S11). Similar to the above differential expression analysis, we compared gene expression variability in four paired contrasts (i.e., LPS, LPS + DEX, PHA, and PHA + DEX) (Supplemental Fig. S12; Supplemental Table S12). We detected a total of 1409 differentially variable genes (DVGs; FDR = 10% and fold change >1.41) across cell types and conditions (Fig. 3A; Supplemental Fig. S13). We identified the most DVGs in monocytes (910) followed by T cells, NK cells, and B cells. DEX induced changes in gene expression variability for 50 to 351 genes, with the most DVGs in monocytes treated with PHA and DEX. When considering gene expression variability, we also observed negative correlations between differential effects of immune stimuli (LPS or PHA) and those of DEX, similar to the findings from differential gene expression analysis (Fig. 3B,C; Supplemental Fig. S14). These findings indicate that DEX dampens the immune response not only by changing gene expression mean but also by altering gene expression variability.
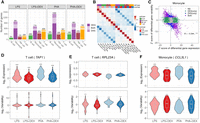
Identification of differentially variable genes (DVGs). (A) Number of the genes with differential mean or variability across cell types and contrasts: genes with significant changes on gene mean only (dark shading), genes with significant changes on gene variability only (medium shading), and genes with significant changes on both gene mean and gene variability (light shading). (B) Heatmap of Spearman's correlation of LFC of gene variability across 16 conditions (four cell types × four contrasts). (C) Scatterplot of the Z-score of differential gene expression (x-axis) and the Z-score of differential gene variability (y-axis) in monocytes across four contrasts. Each dot represents a gene with colors indicating significant (FDR < 10%) changes on gene mean expression only (green), gene variability only (blue), both (purple), and neither (gray). (D) Violin plot of gene expression variability (bottom) and mean expression (top) for the gene (TAP1), an example of a gene that undergoes significant changes only to gene expression in response to immune treatments in T cells. (E) Violin plot of gene expression variability (bottom) and mean expression (top) of the gene (RPL23A), which does not show significant changes in gene expression mean but with significant changes in gene expression variability after immune treatment in T cells. (F) Violin plot of gene expression variability (bottom) and mean expression (top) of the gene (CCL3L1), which shows changes in both gene expression mean and gene expression variability after immune treatment in monocytes.
We then considered the effects of the treatments on mean and variability estimated from the same model. We first confirmed that the fold changes identified with this parametric method are highly correlated with those obtained with DESeq2 (correlation 0.76–0.97) (Supplemental Fig. S15). Among the genes only undergoing changes of mean expression levels, TAP1 was overexpressed in T cells after LPS and PHA stimulation, whereas its expression levels in the DEX conditions were similar to those of unstimulated cells (Fig. 3D). In contrast, we did not observe any changes in gene expression variability of TAP1 across the immune treatments. The TAP1 and TAP2 proteins form a protein complex, called transporter associated with antigen processing (TAP), helping transfer peptides from pathogens into the endoplasmic reticulum (ER) (Eggensperger and Tampé 2015). One example of a gene in which the treatment modifies expression variability without changes in mean expression was RPL23A in T cells following immune stimulation (Fig. 3E). This gene encodes a ribosomal protein, which may be one of the target molecules involved in mediating growth inhibition by IFN (Jiang et al. 1997).
When comparing the effect size of treatments on gene expression mean and variability, we observed genes following two major patterns: independent effect on the mean and the variability, or a negative correlation between the effects on the mean and variability. This last pattern was more pronounced in monocytes (Fig. 3A,C; Supplemental Fig. S16), for 16%–23% genes across contrasts. One such gene is CCL3L1, which encodes a cytokine involved in many inflammatory and immunoregulatory processes, such as inhibiting HIV entry by binding to a protein of CCR5 (Fig. 3F; Dolan et al. 2007). Nevertheless, DEGs and DVGs were largely enriched for similar biological pathways, especially in monocytes and in response to glucocorticoids in all cell types (Supplemental Fig. S17). However, in B cells, NK cells, and T cells, DEGs in response to LPS are enriched mostly in different pathways compared with DVGs in response to the same treatment (Supplemental Fig. S17). Shared enriched pathways included cytokine-mediated signaling, type I IFN signaling, and innate immune response (Supplemental Fig. S18). We also identified several pathways that only appeared to be enriched in DVGs, including ribosome, translation, mRNA catabolic process, protein localization to the ER, and protein targeting to the membrane and to the ER (Supplemental Fig. S19).
Characterizing dynamic responses
Single-cell transcriptomics can also be used to study heterogeneous cell states during important biological processes, such as cell type differentiation during development (Mohammed et al. 2017; Cuomo et al. 2020). Most algorithms developed to study cell type differentiation rely on finding a path (i.e., lower-dimensional manifold) with cells connecting the initial and final states that can be summarized with a trajectory. The gene expression changes of each cell over the trajectory can be mapped to time, or pseudotime. Methods to study gene expression dynamics have focused on cell cycle, differentiation trajectory, and long duration treatments (Cao et al. 2020; Cuomo et al. 2020) and are generally unsupervised. To investigate treatment effects in a short time span, we have used a novel approach based on the diagonal linear discriminant analysis (DLDA) to construct a robust low-dimensional representation of the transcriptional response dynamics for each cell type and treatment. This method is supervised as we know the treatment labels and the two end points in the high-dimensional space where most of the cells (i.e., their centroids) will be. Because this method uses supervised learning and has a well-known solution, the resulting trajectory (i.e., the line connecting the two centroids) should be more stable and reproducible.
We performed resampling analysis to show the robustness and stability of the DLDA compared with two existing methods: Monocle 3 (Qiu et al. 2017) and SCANPY (Wolf et al. 2018). We focused on the T cells treated with PHA + DEX or PHA, as this is the most abundant cell type and the condition with the largest number of DEG. To validate the effectiveness of the novel approach (DLDA), we randomly resampled half the data set for 50 replications. In each replication, we applied our novel approach DLDA and the two other methods to compute response pseudotime.
We do not have the underlying ground truth on how the cells should be exactly ordered according to response pseudotime, yet we can still assess how expression for each of the responding genes correlates with the estimated pseudotime in each iteration across all cells. Ideally, the cells would be sorted similarly in each iteration, and the correlation of the gene expression with the pseudotime would stay constant. If large variation is observed, it indicates that the pseudotime inference is not robust. We computed the variance of correlation coefficient for each gene across 50 replicates and observed that our method shows the lowest variance (Supplemental Fig. S20A).
For each replicate, we can also pick the top 20 most highly correlated genes with the pseudotime variable, which ideally would always be the same. However, across 50 iterations, we identified the following number of most highly correlated genes: 103, 154, and 24 for Monocle, SCANPY, and DLDA, respectively. This result shows that only for DLDA, the most highly correlated genes remain mostly constant across iterations (24 vs. 20 ideally). Not only these genes kept changing for the other methods, but the correlation of these genes with the pseudotime variable was very highly variable (Supplemental Fig. S20B–D). The median value of the Jaccard index for those 20 top genes across all resampling pairs are 0.29, 0.11, and 0.90, for Monocle, SCANPY, and DLDA, respectively (Supplemental Fig. S20E).
In summary, the resampling analysis showed our approach outperforms previous approaches in stability and accuracy of pseudotime and the consistency of the pseudotime-determined genes.
Then, we applied our novel approach to calculate four DLDA axes corresponding to four contrasts for each cell type as follows: LPS (compared with unstimulated), PHA (compared with unstimulated), LPS + DEX (compared with LPS), and PHA + DEX (compared with PHA). As expected, we observed that the first two DLDA axes were able to distinguish cells treated with immune stimuli (LPS or PHA) from the unstimulated group across all cell types (Fig. 4A,B; Supplemental Fig. S21). We observed the clearest separation in monocytes, in line with the strongest gene expression response to stimuli found in monocytes. The latter two DLDA axes clearly divided the cells into two groups (Fig. 4A,B; Supplemental Fig. S21): one treated with DEX and the other treated with immune stimuli. Based on these observations, we defined the first two DLDA axes as immune-stimuli pseudotime and the third and fourth axes as the DEX response pseudotime.
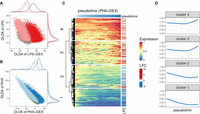
Characterization of dynamic changes along immune response pseudotime. (A) Scatterplot of the DLDA pseudotime for LPS + DEX (x-axis) and the DLDA pseudotime for LPS (y-axis) in T cells. Each dot represents a cell, with the color indicating the treatment condition: gray for unstimulated, pink for LPS, and red for LPS + DEX. (B) Scatterplot of the DLDA pseudotime for PHA + DEX (x-axis) and the DLDA pseudotime for PHA in T cells. Each dot represents a cell, with the color indicating the treatment condition: gray for unstimulated, light blue for PHA, and dark blue for PHA + DEX. (C) Heatmap of relative gene expression for 1617 DEGs (rows) averaged over windows containing 10% of T cells (columns) sliding at a step of 0.1% of T cells along the PHA + DEX response pseudotime. For each gene, gene expression is expressed relative to the highest expression across pseudotime. Red color denotes high expression value, yellow denotes medium expression value, and blue denotes low expression value. The LFC column on the right indicates the LFCs for each DEG between PHA and PHA + DEX. (D) Dynamic patterns of relative gene expression averaged across all genes within each cluster using the same sliding window approach as in C.
We next sought to analyze gene expression dynamic changes along the response pseudotime within each treatment for each cell type separately and identified different dynamic patterns of gene expression. For example, in T cells treated with PHA + DEX, four distinctive gene expression dynamic patterns were identified among 1617 DEGs that were differentially expressed in PHA + DEX (Fig. 4C). Expression of genes within clusters 1 and 2 decreased along DEX pseudotime, and genes in cluster 1 are relatively lower expressed than those of cluster 2. Most of them were down-regulated in the DEX condition compared with PHA (Fig. 4D). In contrast, the genes within clusters 3 and 4 tended to first slightly decrease in expression and then burst at a later point. Most of the genes constituting these clusters were up-regulated. In line with above enrichment analysis for DEGs, the genes within cluster 1 and 2 were mostly enriched in immune-related pathways, such as response to LPS (Supplemental Fig. S22), whereas genes from cluster 4 were preferentially enriched in metal ion–related pathways, such as stress response to copper ion, cellular response to zinc ion, and cell response to cadmium ion (Supplemental Fig. S22). Cluster 4 thus captures the dynamic response that is specific to DEX. Collectively, this novel supervised approach (DLDA) provides a more fine-grained resolution of the dynamics of gene expression response to immune stimuli.
Genetic effects on gene expression across cell types and treatments
To dissect the genetic contributions to the diversity of immune response dynamics, we mapped the cis-regulatory variants affecting gene expression response to immune stimuli across cell types and treatment conditions. First, we performed cis-eQTL mapping using FastQTL (Ongen et al. 2016) in each cell type–condition combination separately, and discovered 5190 unique genes with genetic effects on gene expression (eGenes, 10% FDR) (Supplemental Tables S3, S13; Supplemental Fig. S23). Genetic effect sizes discovered across cell types and conditions were significantly correlated with those discovered in bulk leukocyte gene expression data on a larger sample that included the same individuals analyzed here (Supplemental Fig. S24A; Resztak et al. 2021). Expectedly, genetic effects on gene expression best correlated with those from bulk data from T cells as these are the most abundant PBMC type, thus contributing most to the eQTL signal discovered in bulk data.
We next sought to investigate if the genetic effects were shared or context-specific in PBMCs. For this analysis, we used multivariate adaptive shrinkage (mash) method (Urbut et al. 2019) on eQTL summary statistics. Mash uses an empirical Bayes method to estimate patterns of similarity in effect sizes across conditions and then uses these patterns to improve accuracy of effect size estimates while also increasing power. Here, mash takes advantage of parallel measures of genetic effects across the different cell types and conditions, thus improving the power of eQTL discovery. Indeed, we found approximately an order of magnitude more eGenes in each condition at 10% LFSR (Fig. 5A; Supplemental Fig. S25; Supplemental Tables S3, S14, S15) compared with condition-by-condition analysis at 10% FDR using FastQTL. The increase in power is owing to a high degree of sharing of genetic effects across conditions. Borrowing information across conditions also allows us to increase the precision of genetic effect size estimation. Correlations between genetic effects estimated in each condition and in bulk leukocyte gene expression data are all close to 0.6 (Spearman's correlation, P-value < 0.01) (Supplemental Fig. S24B), which is a marked improvement over correlations between FastQTL estimates in single-cell and bulk data (Supplemental Fig. S24A). Additionally, 41% of the eGenes we discovered were novel compared to eGenes discovered in the bulk leukocyte data set (Supplemental Fig. S26).
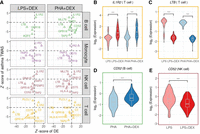
Genetic effects on gene expression. (A) Barplot of the number of genes with eQTLs (eGenes) in each condition significant in multivariate adaptive shrinkage analysis (10% LFSR). (B) Barplot of the number of eGenes (y-axis) with genetic effects in the same direction across the number of conditions shared (x-axis). (C) Boxplot of Spearman's correlations between significant genetic effects on gene expression for each pairwise comparison of two treatment conditions within each cell type and for each pairwise comparison of two cell types within each treatment condition; color reflects cell types (as in A) or treatment: control (gray), LPS (pink), LPS + DEX (red), PHA (light blue), and PHA + DEX (dark blue). (D) Barplot of the number of genes with response eQTLs (reGenes). (E) Boxplot of normalized gene expression for SEC61G gene in NK cells treated with LPS (left) and LPS + DEX (right) across the three genotype classes of the reQTL rs1031486 (x-axis). (F) Forest plot of the estimated genetic effect for rs1031486 minor allele on the expression of the SEC61G gene across all conditions (values reflect slopes as in E). Each line represents the 95% confidence interval around the estimate. (G) Boxplot of normalized gene expression for RBMS1 gene in T cells treated with PHA (left) and PHA + DEX (right) across the three genotype classes of the reQTL rs142470489 (x-axis). (H) Forest plot of the estimated genetic effect of rs142470489 minor allele on the expression of the IRF3 gene across all conditions (values reflect slopes as in G). Each line represents the 95% confidence interval around the estimate.
Genetic effects on gene expression were in the same direction across all 20 conditions for the majority of eGenes (2832) (Fig. 5B; Supplemental Figs. S27–S29). A subset of eGenes (84) shared genetic effects in the same direction across 10 conditions (Fig. 5B), representing a subdivision between NK cells and T cells in one group and monocytes and B cells in the other group. We observed higher correlations of genetic effect sizes across treatment conditions within the same cell type than across cell types for the same treatment condition (Fig. 5C; Supplemental Figs. S30, S31), with higher within cell type correlations for monocytes and B cells (Spearman's ρ > 0.99) compared with NK cells (Spearman's ρ > 0.95) and T cells (Spearman's ρ > 0.97). A similar result was observed when considering eGenes that were shared across pairs of conditions, with the cell type–specific eGenes (6%–51%) representing a larger fraction than the treatment-specific eGenes (1%–5%) in the pairwise comparisons (Supplemental Fig. S29). This result highlights the stronger contribution of cell type effects compared with environmental perturbations on genetic regulation of gene expression, which we have observed previously in other cell types (Findley et al. 2021).
We analyzed pairs of treatment and control conditions within each cell type to discover response eQTLs (reQTLs). Overall, we found 328 genes with reQTLs (reGenes) (Fig. 5D). For the treatments including DEX, the number of reQTLs was roughly similar across cell types. In contrast, among the different cell types stimulated with LPS or PHA, the largest number of reGenes were found in monocytes (LPS and PHA) and T cells (PHA). eGenes were enriched for DEGs across all contrasts and for DVGs in 14/16 contrasts (Fisher's exact test P < 0.05) (Supplemental Table S4). reGenes were enriched for DEGs only in B cells treated with LPS + DEX and for DVGs in three conditions (B cell LPS + DEX, monocyte PHA + DEX, and NK cell PHA + DEX; Fisher's exact test P < 0.05) (Supplemental Table S5).
DEX reGenes varied between 47 in T cells treated with PHA + DEX and 65 in monocytes treated with LPS + DEX. Three of these genes (RETREG1, MS4A7, and BIRC3) are among 26 genes previously reported as DEX reGenes in EBV-transformed B lymphocytes (Supplemental Table S6; Maranville et al. 2011). Additionally, we found a reQTL for the SEC61G gene in NK cells, where the A allele was associated with higher expression after DEX treatment (Fig. 5E,F; Supplemental Fig. S32). SEC61G is a subunit of the SEC61 translocon responsible for translocation of newly synthesized proteins into the ER. This complex is required for replication of flaviviruses in human cells (Shah et al. 2018). Although SEC61G is primarily known as a proto-oncogene in several types of cancers (Lu et al. 2009; Gao et al. 2020; Liang et al. 2021; Meng et al. 2021), it has previously been found to be up-regulated in NK cells upon IL2 stimulation (Dybkaer et al. 2007). To understand the phenotypic relevance of the reGenes, we considered genes associated with diseases in transcriptome-wide association studies (TWAS) and found that 61 reGenes have previously been implicated in immune diseases (Supplemental Table S16). For example, rs142470489 is a reQTL for the RBMS1 gene, which encodes a member of a small family of proteins that bind single-stranded DNA/RNA, involved in the process of DNA replication, gene transcription, cell cycle progression, and apoptosis. TWAS implicated RBMS1 in inflammatory bowel disease (Zhang et al. 2020). The G allele at rs142470489 increases the DEX immunomodulatory effect on expression of RBMS1 in T cells treated with PHA + DEX (Fig. 5G,H; Supplemental Fig. S33).
Genetic effects on gene expression variability
Single-cell data combined with our new modeling approach allowed us to quantify changes across contexts and individuals in gene expression variability independently of the changes in the mean gene expression. To identify genetic effects on gene expression variability that account for inter-individual variation, we used a QTL mapping approach to discover variability QTLs (vQTLs). We discovered 123 genes with vQTLs (vGenes), with highest numbers of vQTLs detected in T cells across all conditions (Fig. 6A; Supplemental Fig. S34A; Supplemental Tables S7, S17–S19). vGenes were highly enriched for ribosomal proteins (GO enrichment for “cytosolic ribosome” P-value = 1.6 × 10−43). However, overall genetic effects across contexts for vQTLs were less correlated than for eQTLs (Supplemental Fig. S35). We observed the highest correlations of genetic effects across treatment conditions within monocytes compared with the other cell types (Supplemental Fig. S36A), as well as an overall decrease in pairwise correlations across cell types in treatment conditions compared with the control (Supplemental Fig. S36B). Similar to eGenes, genetic effects on gene expression variability were shared in the same direction for most of the vGenes (98) across all cell types and treatment conditions (Fig. 6B; Supplemental Fig. S37). In contrast to eGenes, for vGenes we observed similar correlations of genetic effect sizes across treatment conditions within the same cell type than across cell types for the same treatment condition (Fig. 6C). This is confirmed also when considering sharing of genetic effects across conditions (0.05–0.32 for cell type–specific vGenes and 0.06–0.34 for treatment-specific vGenes) (Supplemental Fig. S38).
Genetic effects on gene expression variability. (A) Barplot of the number of genes with eQTLs (eGenes) for which we did not detect genetic effects on variability (dark shading), number of genes with vQTLs (vGenes) for which we did not detect any genetic effects on gene expression (medium shading), and genes with both eQTLs and vQTLs (light shading). (B) Boxplot of the Spearman's correlations between significant genetic effects on gene expression variability for each pairwise comparison of two treatment conditions within each cell type and for each pairwise comparison of two cell types within each treatment condition; color reflects cell types (as in A) or treatment: control (gray), LPS (pink), LPS + DEX (red), PHA (light blue), and PHA + DEX (dark blue). (C) Barplot of the number of vGenes (y-axis) with genetic effects in the same direction across the number of conditions shared (x-axis). (D) Barplot of the number of genes with response vQTLs (rvGenes). (E) Boxplot of normalized gene expression variability for ARL6IP4 gene in T cells treated with PHA (left) or PHA + DEX (right) across the three genotype classes of the vQTL rs7296418 (x-axis). (F) Scatterplot of genetic effects on mean gene expression (x-axis) versus genetic effects on gene expression variability (y-axis) for T cells treated with PHA + DEX; color represents genetic variants with significant effects on mean gene expression only (green), gene expression variability only (blue), both (purple), and neither (gray). (G) Boxplot of normalized mean gene expression (left) and normalized gene expression variability (right) for PSMB9 gene in B cells treated with LPS + DEX, with significant genetic effect of rs2071464 on mean gene expression but no effect on gene expression variability. (H) Boxplot of normalized mean gene expression (left) and normalized gene expression variability (right) for RPS18 gene in monocytes treated with PHA + DEX, with significant genetic effect of rs1197452483 on gene expression variability but no effect on mean gene expression. (I) Boxplot of normalized mean gene expression (left) and normalized gene expression variability (right) for RNASET2 gene in T cells treated with PHA + DEX, with significant genetic effect of rs400063 both on mean gene expression and on gene expression variability.
To identify response vQTLs, we performed a direct comparison of genetic effect sizes across conditions in each cell type. We discovered 61 unique variability response genes (rvGenes) (Fig. 6D), including a total of 47 for DEX response. We identified 16 rvGenes that were previously implicated in immune diseases via TWAS. For example, ARL6IP4 is associated with multiple sclerosis and allergic rhinitis, and we found the T allele at rs7296418 to decrease gene expression variability of this gene in T cells treated with PHA + DEX but not in the PHA condition (Fig. 6E; Supplemental Fig. S39). ARL6IP4 may have a role in splicing regulation (Ouyang 2009) and was reported to inhibit splicing of the pre-mRNA of herpes singlex virus (HSVI) (Li et al. 2002).
Mapping of dynamic eQTLs using response pseudotime
Although reQTLs represent genetic variants that modulate the average response to treatments across cells, our dynamic analysis uncovered more complex cellular response trajectories, which we have quantitatively analyzed through DLDA. Through the characterization of the dynamic response patterns above, we observed that cluster 4, representing late response genes, in T cells treated with PHA + DEX was enriched for genes with genetic effects on gene expression (OR = 2.53, Fisher's exact test P-value = 3.5 × 10−3). This indicated the potential role of genetic effects in the regulation of dynamic gene expression changes. We therefore systematically investigated genetic variants that modulate these response trajectories (dynamic eQTLs) (Supplemental Fig. S44) and discovered a total of 3899 dynamic eQTLs corresponding to 588 eGenes across all the conditions (Supplemental Tables S10, S23). The largest number of dynamic eGenes were identified in T cells, with dynamic eGenes in the immune-stimuli pseudotime (244 for LPS and 225 PHA) and DEX response pseudotime (18 LPS + DEX and 54 for PHA + DEX). We also observed that dynamic eQTLs are enriched among eGenes and reGenes (Supplemental Figs. S45, S46), yet a large number of them are novel. This pattern is particularly noticeable in T cells (Fisher's exact test P-value < 0.01) (Supplemental Fig. S47A,B). To investigate whether these dynamic eGenes are potentially causal genes for immune diseases, we tested their overlap with immune-related genes identified from TWAS (Zhang et al. 2020) and revealed a total of 92 dynamic eGenes (Supplemental Table S10) in T cells that have previously been implicated in 22 immune-related diseases in TWAS (Zhang et al. 2020).
To illustrate genetic effects on dynamic response patterns along pseudotime, we focused on T cells treated with PHA + DEX. We showed a dynamic eQTL for ABO, which was previously reported to be associated with allergic rhinitis in TWAS (Zhang et al. 2020). This gene encodes an enzyme responsible for glycosyltransferase activity to determine the blood group in an individual. This gene has been linked to the strongest signal for severe COVID-19 by GWAS (Niemi et al. 2021; Shelton et al. 2021). We discovered a dynamic eQTL modulating ABO gene expression along DEX response pseudotime such that ABO expression rapidly increased in heterozygous individuals along DEX pseudotime but remained relatively stable in individuals homozygous for the alternate allele (Fig. 7A). As a result, we observed a negative genetic effect of the alternate allele on ABO gene expression in the mid and late pseudotime. Another example was a dynamic eQTL for the HLA-DRB5 gene, which has been implicated in 14 immune-related diseases, including asthma. The protein product of HLA-DRB5 forms the beta chain of MHC II. MHC II molecules are used by antigen-presenting cells to display foreign peptides to the immune system to trigger the body's immune response but have also been found on activated T cells, where they function as signal-transducing receptors (Holling et al. 2004). We observed divergent genetic regulation effects on this gene across the three pseudotime tertiles: negative genetic effects on this gene in early pseudotime, no genetic effects in mid pseudotime, and positive genetic effects in late pseudotime (Fig. 7B). We revealed very distinct patterns of dynamic changes in HLA-DRB5 gene expression along pseudotime for the three genotype classes such that the gene expression sharply decreased in individuals homozygous for A allele at rs116611418, displayed the least gene expression changes in heterozygous individuals throughout the pseudotime, and rapidly increased in the late pseudotime in individuals homozygous for the alternate allele (Fig. 7B). Additionally, we discovered a dynamic eQTL modulating GADD45G expression along DEX response pseudotime such that GADD45G expression appeared to be extinguished most rapidly in individuals homozygous for the G allele at rs11265809, with heterozygous individuals following an intermediate slope, and carriers of two A alleles displaying the least changes to GADD45G expression along the pseudotime (Fig. 7C). As a result, we observed a genetic effect on GADD45G expression only in the first tertile of the DEX response pseudotime. The protein encoded by this gene responds to environmental stresses (Takekawa and Saito 1998; Ying et al. 2005). This gene was reported to be associated with asthma and systemic lupus erythematosus in the TWAS studies (Zhang et al. 2020).
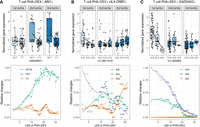
Identification of dynamic eQTLs along immune response pseudotime. For the genes ABO (A), HLA-DRB5 (B), and GADD45G (C), the boxplots on the top represent normalized gene expression in T cells treated with PHA + DEX for each DLDA pseudotime tertile and genotype, and the bottom plots represent the smoothed dynamic gene expression changes along response trajectory pseudotime for each genotype separately (trend lines are fit using locally estimated scatterplot smoothing).
Biological insight into the mechanism underlying asthma
Glucocorticoids are the common drugs used to treat asthma and other autoimmune diseases by suppressing immune response. We wanted to explore how genes associated with asthma respond to these immune treatments. We considered 78 asthma-associated genes identified by a probabilistic transcriptome wide association study (PTWAS) by integration of eQTLs from whole-blood tissue in Genotype-Tissue Expression (GTEx) with asthma GWAS cohorts (Zhang et al. 2020). This type of analysis connects disease risk with gene expression levels. We identified a total of 30 genes that were differentially expressed in at least one condition in our data and were also associated with asthma (Fig. 8A).
The roles of treatments in the asthma-determined genes: (A) Scatterplots of the Z-score of DEX effects (x-axis) on 18 asthma-associated genes against the Z-score from PTWAS (y-axis). Each row and dot color represents the cell type. (B–E) Example genes: (B) IL1R2 in T cells, (C) LTB in T cells, and (D,E) CD52 in B cells and NK cells. Each violin plot summarizes individual average gene expression.
Examples include interleukin 1 receptor type 2 (IL1R2), encoding a cytokine receptor that belongs to the interleukin 1 receptor family (Anderson et al. 2011; Peters et al. 2013). This protein binds interleukin 1 alpha (IL1A), interleukin 1 beta (IL1B), and interleukin 1 receptor type 1 (IL1R1; previously known as IL1RA) and acts as a decoy receptor that inhibits the activity of its ligands. This gene is up-regulated in response to DEX in all cell types (Fig. 8A,B).
The expression of lymphotoxin beta (LTB; previously known as TNFC) is significantly repressed by glucocorticoids via inhibiting the activity of the upstream regulatory factor NF-kB across cell types (Fig. 8A,C; Voon et al. 2004). LTB is down-regulated in response to DEX in all cell types and is also an eGene in T cells treated with DEX (PHA + DEX). The protein encoded by LTB forms a heterotrimer with LTA on the surface of lymphoid cells (Browning et al. 1993), which is involved in a variety of inflammatory responses (Young et al. 2010). This gene was found to decrease gene expression in children with bronchopneumonia following treatment with methylprednisolone in combination with azithromycin (Ye et al. 2021).
CD52 was functionally verified to be a potential candidate causal gene linked to asthma in a previous study (Han et al. 2020). In vivo experiment validated that anti-CD52 antibody reduced the inflammatory response in lymphoid cells and airway epithelium (Han et al. 2020). CD52 displays a cell type–specific transcriptional immune response to DEX, with increased levels in B cells (Fig. 8D) but decreased levels in NK cells (Fig. 8E) after DEX treatment.
Discussion
Single-cell technologies allow the study of all aspects of the gene expression response to stimuli—mean, variability, and dynamics—across different cell types. Here, we used scRNA-seq to study the dynamics of the transcriptional response to glucocorticoids in activated PBMCs. Using this system, we showed that both gene expression mean and variability can be modified by glucocorticoids and immune stimuli. Further, we identified the genetic variants regulating gene expression mean and variability across treatments. We introduced a novel computational approach that allowed us to track the short-term transcriptional response dynamics to treatments at single-cell resolution. Using this method, we identified different dynamic patterns of gene expression along the response pseudotime and showed widespread genetic regulation of these dynamics.
Studies of glucocorticoid response in tightly controlled experimental settings of individual cell types found varied effects of glucocorticoids on different cells (Li et al. 2015; Xing et al. 2015; Tu et al. 2017; Quatrini et al. 2018, 2021; Maeda et al. 2019; Tokunaga et al. 2019). However, glucocorticoid response could not be studied in the context of the cellular complexity of the immune system before the advent of single-cell technologies. Here, we studied the gene expression response of four cell types to glucocorticoids within activated PBMCs. The choice to only focus on four cell types was in order to have enough cells to perform eQTL mapping. We acknowledge that a more fine-grained analysis of different cell subtypes may yield additional insight of cell type–specific responses; however, this is beyond the scope of this study. We revealed an opposite pattern of effect on gene expression between immune stimuli and DEX in all cell types analyzed, which was evident at both the gene level and pathway level. These pathways with antagonistic patterns mainly contained immune-related pathways, such as IFN, response to LPS, cytokine-mediated signaling, and innate immune response, mostly shared across cell types, in line with previous findings (Oelen et al. 2022). Our study also highlighted the specialization in immune response of the different cell types as the majority of DEGs were identified within only one cell type. We observed these cell type–specific patterns on the pathway level, such as the asthma pathway mainly enriched in B cells and T cells and such as granulocyte activation mostly enriched in monocytes. Our results are obtained in a childhood asthma cohort and may not be representative of gene expression levels in healthy children or adult populations, yet the fundamental genetic and biological findings can be most likely extended to healthy individuals. Most importantly, our results provide a unique resource to investigate the molecular underpinnings of immune diseases such as childhood asthma. Many of the DEGs overlap with genes associated with asthma in TWAS and also well-characterized biomarkers. For CD52, we can identify glucocorticoids acting with opposite effects in two different cell types.
Previous studies implied the important role of gene expression variability in many aspects, such as developmental states (Chang et al. 2008; Richard et al. 2016), T cell differentiation (Eling et al. 2018), and aging (Martinez-Jimenez et al. 2017). Our systematic study of gene expression variability in response to immune treatments revealed major effects of immunomodulators on gene expression variability, with a total of 1409 DVGs across cell types and contrasts. We observed the same pattern of negative correlation of effects of immune stimuli and DEX on gene expression variability as for gene expression mean levels. Similarly, these DVGs tended to be enriched in immune-related pathways in an antagonistic way similar to the expression analysis. These findings showed that immune treatments not only affected gene expression but also altered the transcriptional variability of genes involved in the immune system. Although we regressed out the effect of mean on gene expression variability, for a subset of genes, we observed a negative correlation between the effects of each treatment on gene expression variability and mean, respectively. Although this negative correlation could be a consequence of correcting for the mean-variance dependency, our results suggest that for a subset of genes, the treatment affects differently the mean and the variance, thus resulting in different response dynamics. Together, these findings can enhance our understanding of the molecular basis of how individuals respond to immune stimuli and suppress the activated immune system in response to drugs at the cell type level.
Genetic effects or environmental perturbations can affect the average gene expression, but disrupting the cis-regulatory code can also reduce the tightness in the gene transcriptional response, resulting in higher variability among individual cells. The mean gene expression response may not be affected, but the dysregulation in gene expression variability may ultimately result in a modified drug-response phenotype.
Previous single-cell studies in human induced pluripotent stem cells (iPSCs) differentiating into neurons and in response to oxidative stress identified a large fraction (46%) of eQTLs that had not previously been found in the GTEx catalog (Jerber et al. 2021). Similarly, we found that 44% of the eGenes identified in this study were not found in bulk leukocyte data from the same population, which also includes the same individuals and had a larger sample size (Resztak et al. 2021). These additional discoveries were likely owing to cell type–specific genetic effects. However, when we consider genes expressed in all conditions (treatments and cell types) and use a rigorous statistical framework to determine to what extent genetic effects are shared across conditions, we found a high degree of sharing as indicated by the fact that 65% of eGenes were significant in all 20 conditions. This is similar to the findings of the GTEx Consortium that the majority of eQTLs are shared across all 49 surveyed tissues (Aguet et al. 2020). However, we found relatively fewer cell type–specific eQTLs compared with GTEx data across tissues (Ongen et al. 2016), which indicates higher sharing of genetic effects across cell types within the same tissues than across different tissues. This finding is in line with the higher sharing of genetic effects among related brain tissues in GTEx data (Ongen et al. 2016). We previously explored the relative contribution of cell type and treatments in modulating genetic effects on gene expression using allele-specific expression. We found a greater variability of genetic effects across three cell types than across 12 treatments (Findley et al. 2021). Similarly, here we find more variation of genetic effects on gene expression across the four PBMC cell types compared with the five treatment conditions. Importantly, we observed a similar pattern in gene expression variability and mean, although it was weaker for variability.
After filtering all eGenes with shared genetic effects across treatments, we discovered that 25% of eGenes have reQTLs. Although eGenes were more likely to be differentially expressed and differentially variable in the majority of conditions, reGenes were not enriched for DEGs and only enriched for DVGs in two conditions, PHA-activated monocytes and PHA + DEX-treated T cells. This means that genes responsive to environmental conditions are also more likely to harbor genetic variants that affect gene expression levels, whereas genes with GxE effects are less likely to be detected as differentially expressed or differentially variable.
Identification of the genetic variants underlying inter-individual differences in cell-to-cell gene expression variability is technically more challenging than detecting genetic effects on mean gene expression. Previously, researchers have estimated that more than 4000 individuals would be needed to achieve 80% power to detect the strongest genetic effects on gene expression dispersion in iPSCs (Sarkar et al. 2019). This calculation was based on a different technology, in which each cell is processed as an independent library and the number of cells/individual that can be analyzed is very limited. However, with the technology available when we started our study, we were able to interrogate a large number of cells and individuals in the same library, thus reducing technical noise. This likely resulted in greater power to detect genetic effects on gene expression variability. Other factors that may contribute to our ability to discover these vQTLs are the cell types used and our study design. We observed that genetic effects on gene expression variability were most stable across different treatment conditions for monocytes, despite the strongest gene expression response to treatment in that cell type. We also showed that treatments decrease the correlation of genetic effects on gene expression variability across the different cell types, indicating that GxE effects on gene expression variability were cell type–specific. We found high enrichment for ribosomal protein genes within vGenes, which may be owing to higher expression levels of this class of genes, leading to an increased power to detect effects on gene expression variability.
Although we used a measure of gene expression variability that does not have a mean-variance dependency, we found genetic effects affecting both gene expression mean and variability but with opposite direction of effect for 13 genes. Even though eQTLs were enriched in vQTLs, we did not detect significant genetic effects on mean gene expression for most vQTLs. This shows that genetic variants may increase variability of gene expression across cells, without affecting mean gene expression levels. Although eGenes were enriched for DEGs, vGenes were depleted for them. This could be because genes responding to treatments are required to be under tighter genetic control of expression variability. Prior work on variance of gene expression and of its genetic control was performed on bulk RNA-seq data sets, therefore investigating variance in the population. These studies showed that genes with a TATA box have increased noise at both the gene and allelic expression level (Mogno et al. 2010; Sigalova et al. 2020; Findley et al. 2021). TATA box promoters, as opposed to CpG island-associated promoters, have been associated with tissue-specific genes and high conservation across species (Carninci et al. 2006). Findley et al. (2021) showed that genes that are more tolerant to loss-of-function mutations showed greater allelic expression residual variance, indicating that redundancy in gene function allows for less stringent genetic control. This could allow for the evolution of new regulatory elements, resulting in new patterns of gene expression. Conversely, genes that are under negative selection (i.e., low dN/dS) have low residual variance, underscoring the importance of preserving stable expression of these genes. Single-cell studies allow analysis of variability within individuals, as a new molecular phenotype with potentially relevant biological function. Our results suggest that a tighter genetic control of gene expression variability is preserved in specific cell types and conditions. Specifically, vGenes were depleted of DVGs within all contrasts in monocytes but enriched for DVGs in the other cell types following immunostimulation and in T cells treated with DEX. Additionally, we have shown that effects of genetic variation on gene expression variability could vary across environmental conditions. These GxE effects on gene expression variability and the GxE effects on gene expression mean affected genes associated with the same immunological diseases.
Lastly, we introduced a novel computational approach designed to track the short-term transcriptional response dynamics to treatments at single-cell resolution. Our new method is supervised and easy to implement and can be directly applied to studying the dynamics of transcriptional responses to other treatments at the single-cell level to facilitate understanding the diversity in response to environmental stimuli across heterogeneous tissues. Previous studies investigated transcriptional dynamics in experimental designs in which individual cells were sampled at a range of various times (Cao et al. 2020) or collected from various cell type differentiation stages (Cuomo et al. 2020). In these studies, pseudotime was inferred from computational methods (Bergen et al. 2020) or directly measured by experimental design (Cao et al. 2020), but here, we show that the pseudotime trajectory for these unsupervised methods is not very stable when resampling the data. The pseudotime inferred from our novel approach reflects the transcriptional dynamics of the immune response and is robust across resampling. Although our data were sampled at one time point, we still revealed transcriptional response dynamics using this projection, suggesting initial heterogeneity of cells, probably arising from diversity in initial cell states. Alternatively, this heterogeneity of response across cells may be a reflection of the dynamics of the immune system. We purposely used a study design in which all the cell types were cultured together to study immune response to preserve the natural interactions between cells that is key to orchestrate the immune response. This is reflected in the observed gene expression response to stimuli across all the cell types, even though we used immune activators that primarily affect one cell type. Thus, it is likely that in our study, the observed heterogeneity of response across pseudotime reflects the dynamics of secondary signal propagation (cytokine or direct cell-to-cell interaction) across a population of cells. Using this method, we identified different dynamic patterns of gene expression along the response pseudotime and showed widespread genetic regulation of these dynamics. Importantly, we showed genetic effects on the dynamics of ABO expression response to glucocorticoids, which strongly correlated with susceptibility to SARS-CoV-2 infection (Niemi et al. 2021; Shelton et al. 2021).
In summary, our study shows that a full understanding of transcriptional response dynamics requires investigations beyond changes in mean gene expression, which are enabled by single-cell studies and robust computational methods. Elucidating gene expression variability and pseudotemporal response dynamics will contribute to a fuller picture of the heterogeneity within and across cell populations. Studies of transcriptional response dynamics and genetic contributions to their heterogeneity in future experiments of treatment response, tumor microenvironment dynamics, and cell type differentiation across healthy and disease states will uncover new disease mechanisms based on altered control of gene expression heterogeneity.
Methods
Biological samples and genotyping
PBMCs used in this study were collected as part of the longitudinal study Asthma in the Life of Families Today (ALOFT; recruited from November 2010 to July 2018, Wayne State University Institutional Review Board approval 0412110B3F) (Resztak et al. 2021) from children 10–16 yr old and self-reported as Black. PBMCs were extracted using a previously published Ficoll centrifugation protocol (Weckle et al. 2015), cryopreserved in freezing media, and stored in liquid nitrogen until the day of the experiment. All individuals in this study were genotyped from low-coverage (∼0.4×) whole-genome sequencing and imputed to 37.5 million variants using the 1000 Genomes database by Gencove.
Cell culture and single-cell experiments
Cells were processed in batches of 16 donors. For each batch, thawed PBMCs were incubated in starvation media overnight (∼16 h) and subsequently treated with one of the following: 1 µg/mL LPS (Barreiro et al. 2010) + 1 µM DEX (Moyerbrailean et al. 2016), 1 µg/mL LPS + vehicle control alone, 2.5 µg/mL PHA (Moyerbrailean et al. 2016) + 1 µM DEX, 2.5 µg/mL PHA + vehicle control alone, or vehicle control alone (control). After 6 h, cells were pooled across individuals and loaded onto a separate channel of 10x GenomicsChromium machine (10x Genomics), according to the manufacturer's protocol. Library preparation was performed according to the manufacturer's protocol. Sequencing of the single-cell libraries was performed in the Luca/Pique-Regi laboratory using the Illumina NextSeq 500 and 75 cycles high-output kit.
scRNA-seq raw data processing (alignment, demultiplexing, and cell type assignment)
The raw FASTQ files were mapped to the GRCh38.p12 human reference genome using the kb tool (a wrapper of kallisto and bustools) with the argument of workflow setting to be lamanno (Melsted et al. 2021). We removed debris-contaminated droplets using the DIEM R package (Alvarez et al. 2020; R Core Team 2020). The aligned count matrix was transformed into a Seurat object for the subsequent functional analysis. To demultiplex the 16 individuals pooled together for each batch, we used the popscle pipeline (dsc-pileup followed by demuxlet) with the default parameters (Kang et al. 2018). For 96 individuals, we had both control and LPS conditions, whereas the other three conditions (LPS + DEX, PHA, PHA + DEX) were assayed for 64 individuals because of instrument channel failure in batches 2 and 3 for those experimental conditions. This clean data set had a median of 7994 cells (Supplemental Fig. S48), a median of 4251 UMI counts, and 1810 genes measured on average in each cell (Supplemental Fig. S49).
Seurat(V3) was used for preprocessing, clustering, and visualizing the scRNA-seq data (Stuart et al. 2019). We performed standard preprocessing steps (including log normalization and scaling) for all the counts data using the default method in Seurat. Next, we performed linear dimensionality reduction on 2000 highly variable features and obtained 100 principal components (PCs). We also ran RunHarmony with chemistry as covariates to correct chemistry-induced bias (Korsunsky et al. 2019). The top 50 Harmony-adjusted PCs were then used for clustering analysis. Finally, Uniform Manifold Approximation and Projection (UMAP) was applied to visualize the clustering results using the top 50 Harmony-adjusted PCs. Cells from different chemistries, batches, and treatments shared similar clustering patterns (Supplemental Figs. S51–S53).
We identified 13 clusters, then annotated cell clusters based on the following canonical immune cell type marker genes (Supplemental Figs. S2, S55), B cells (MS4A1 or CD79A), monocytes (CD14 or MS4A7), NK cells (GNLY or NKG7), and T cells (CD3D or CD8A). Clusters 0, 4, 5, and 7–12 were annotated as T cells with 183,289 cells; clusters 3 and 6 were annotated as monocytes (30,393); cluster 1 was defined as NK cells (47,824); and cluster 2 was defined as B cells (30,888).
We generated pseudobulk RNA-seq data by summing the spliced counts for each gene and each sample across all cells belonging to each of the four cell types (B cell, monocyte, NK cell, and T cell), separately. We focused on protein-coding genes and filtered out genes with fewer than 20 reads across cells and removed combinations with less than 20 cells. This resulted in a data matrix of 15,770 protein-coding genes (rows) and 1419 combinations (columns) of cell type + treatment + individual, which were used for all subsequent differential expression and genetic analyses.
Differential gene expression analysis
We performed differential gene expression analysis for each of the five batches separately using R DESeq2 package (Love et al. 2014) and then combined the results across five batches using fixed-effect model of meta-analysis. For each batch, we estimated gene expression effects of the treatments using four contrasts: (1) LPS, LPS versus CTRL; (2) LPS + DEX, LPS + DEX versus LPS; (3) PHA, PHA versus CTRL; and (4) PHA + DEX, PHA + DEX versus PHA. To correct for multiple hypothesis testing, we used the qvalue function to estimate the false-discovery rate (FDR) from a list of P-values for each condition, separately. DEGs were defined as those with FDR less than 0.1 and absolute estimated log2 fold change larger than 0.5. We performed GO enrichment analysis (i.e., biological process [BP], molecular function [MF], and cellular component [CC]) for these DEGs identified across conditions using the R clusterProfiler package (V3) (Yu et al. 2012).
Differential gene variability analysis
Based on the computational framework that was established by Sarkar et al. (2019), we adapted an NB distribution to model the count data for each gene j, using two main parameters for the mean(µj) and dispersion(ϕj) in our scRNA data. Then, we derived the variance of gene expression by 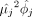 . Similar to what was reported in previous studies (Fan et al. 2016; Eling et al. 2019a,b; Fair et al. 2020), we observed that both gene variance or dispersion linearly depended on mean parameters (Supplemental Fig. S11A,B). To further correct the dependence between dispersion and mean, we defined a residual dispersion, capturing the departure
from the global trend. The residual dispersion was calculated by removing the part of dispersion that could be predicted by
the overall trend of gene mean across genes in each cell type separately. This adjusted dispersion was uncorrelated with the
gene mean value (Supplemental Fig. S11C) across genes.
. Similar to what was reported in previous studies (Fan et al. 2016; Eling et al. 2019a,b; Fair et al. 2020), we observed that both gene variance or dispersion linearly depended on mean parameters (Supplemental Fig. S11A,B). To further correct the dependence between dispersion and mean, we defined a residual dispersion, capturing the departure
from the global trend. The residual dispersion was calculated by removing the part of dispersion that could be predicted by
the overall trend of gene mean across genes in each cell type separately. This adjusted dispersion was uncorrelated with the
gene mean value (Supplemental Fig. S11C) across genes.
To account for the noise from the batch, we implemented the same strategy for differential gene expression analysis for differential gene variability and differential gene mean analyses via fitting models by batch separately, and we meta-analyzed the summary statistics. We first took the log2 transformation of residual dispersion and mean and then fitted models using linear regression in which treatments were considered for each batch, separately. The DVGs and DEGs were defined as those with an FDR less than 0.1 and absolute value of log2 fold change greater than 0.5.
DLDA response pseudotime method
We developed a new pseudotime method based on DLDA to characterize the degree to which single cells respond to immune treatments.
In DLDA, only a subset of genes that are highly differentially expressed between the two conditions are used based on the
following formula (Pique-Regi et al. 2005):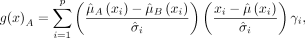 (1)
where the subscripts of A and B represent the two conditions considered in each contrast, respectively,
(1)
where the subscripts of A and B represent the two conditions considered in each contrast, respectively, 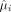 denoting mean gene expression of the ith gene for A, B or across groups and
denoting mean gene expression of the ith gene for A, B or across groups and 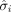 meaning standard deviation of the ith gene across conditions. In this application of DLDA, we only used the DEGs that were differentially expressed in the corresponding
condition (10% FDR by DESeq2) by introducing an indicator variable (γi), assigning to one if the ith gene is a significant DEG in the contrast of A versus B or 0 otherwise.
meaning standard deviation of the ith gene across conditions. In this application of DLDA, we only used the DEGs that were differentially expressed in the corresponding
condition (10% FDR by DESeq2) by introducing an indicator variable (γi), assigning to one if the ith gene is a significant DEG in the contrast of A versus B or 0 otherwise.
Four kinds of DLDA axes were calculated by the following contrasts for each cell type separately: (1) DLDA axis of LPS was estimated in the CTRL and LPS conditions to infer the response pseudotime to LPS; (2) DLDA axis of LPS + DEX was computed in the LPS and LPS + DEX conditions to calibrate the response pseudotime to DEX; (3) DLDA axis of PHA was estimated in the CTRL and PHA conditions to characterize the response pseudotime to PHA stimuli; and (4) DLDA axis of PHA + DEX was estimated in the PHA and PHA + DEX conditions to characterize the response pseudotime to DEX. After the DLDA representing the trajectory pseudotime is calculated for each cell type and treatment, we used a sliding window approach (10% cells in each window, sliding along the defined pseudotime with a step of 0.1% cells), analyzed gene expression dynamic changes along the response pseudotime within each treatment for each cell type separately, and identified different dynamic patterns of gene expression using the k-means algorithm.
Data normalization for genetic analyses
We quantile-normalized the count data using the voom function in the R limma v3 package (Ritchie et al. 2015) and regressed out the following confounding factors: experimental batch, sex, age, and top three PCs of genotype PCs in each cell type treatment combination, separately. For the parameter base models, we also applied the same procedure but starting from the mean and variability estimates instead of the count data.
cis-eQTL mapping
We used FastQTL v2.076 (Ongen et al. 2016) to perform eQTL mapping on gene expression residuals calculated for each cell type and condition as outlined above. For each gene, we tested all genetic variants within 50 kb of the transcription start site (TSS) and with cohort minor allele frequency (MAF) > 0.1. We optimized the number of gene expression PCs in the model to maximize the number of eGenes across all conditions combined. The model that yielded the largest number of eGenes included four gene expression PCs. eQTL discovery for mean and variability data was performed similarly. The model that yielded the largest number of significant genes at 10% FDR included five PCs of residuals for variability and seven PCs of residuals for mean data.
Multivariate adaptive shrinkage
We used the multivariate adaptive shrinkage (mash) method using the mashr package v.0.2.40 in R v4.0 (Urbut et al. 2019). As input, we provided the genetic effect sizes and standard errors from FastQTL. We considered gene–variant pairs with posterior local false sign rate (LFSR) < 0.1 to be eQTLs (Stephens 2017). To analyze sharing of genetic effects across all conditions, we considered the direction of genetic effect across all conditions. Genetic effects are shared across conditions if they have the same sign and are significant in at least one of the conditions considered. To analyze treatment or cell type specificity, we focused on pairwise comparisons of genetic effect sizes. An eQTL is specific to a condition if it is significant at LFSR < 0.1 in either condition and either the direction of effect differs or the difference in the magnitude of the genetic effects is at least twofold. A gene is considered shared/specific if at least one eQTL for that gene is shared/specific across the given set of conditions. To discover reQTLs, we considered the pair-wise union of significant eQTLs in each of the 16 treatment–control combinations. We defined reQTLs as genetic variants whose effect size on gene expression differed by at least twofold between treatment and control conditions. Mash analyses on mean and variability estimates were conducted following this pipeline.
DLDA-eQTL mapping
For mapping dynamic eQTLs that have genetic effects on gene expression interacting with pseudotime, we first equally divided cells in one immune treatment into three tertiles of the corresponding DLDA pseudotime. Then, we mapped eQTLs interacting with these representative tertiles as dynamic eQTLs across cell types in the corresponding immune treatments. We quantile-normalized the data and regressed out confounding factors as described above. To identify genetic variants interacting with each DLDA, we fitted a linear model using the lm function that included both the genotype dosage and the DLDA bin (numerically encoded as 1–3), as well as their interaction. We used the same cis-regions as in the above analysis. We then applied Storey's Q-value method on the P-values for the interaction term using a stratified FDR approach on significant and nonsignificant eGenes (from the FastQTL analysis) (Supplemental Fig. S45).
Integration with TWAS results
We considered the results of probabilistic TWAS (Zhang et al. 2020), where eQTL from whole-blood tissue in GTEx v8 were integrated with the GWAS summary data of asthma. PTWAS identified four, 16, 66, and 72 (union of 78 genes) genes associated with asthma for four GWAS studies: GABRIEL (Moffatt et al. 2010), TAGC (Demenais et al. 2018), and two from UK Biobank (self-reported asthma and asthma diagnosed by doctor) (Pividori et al. 2019). To combine multiple TWAS Z-scores across studies for the same gene, we selected the one with the largest magnitude as representative. We then identified genes that were both associated with asthma risk and differentially expressed in any of the treatment conditions.
Data access
All raw and processed sequencing data generated in this study have been submitted to the NCBI database of Genotypes and Phenotypes (dbGaP; https://www.ncbi.nlm.nih.gov/gap/) under accession number phs002182.v3.p1. Large Supplemental Tables have been deposited in Zenodo (https://doi.org/10.5281/zenodo.7851053). Code and scripts used to make the results are available at GitHub (https://github.com/piquelab/scaip and https://github.com/piquelab/SCAIP-genetic) and as Supplemental Codes 1 and 2, respectively.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
This research has been supported by National Institutes of Health grants from the National Heart, Lung, and Blood Institute (R01HL114097 to S.Z. and R.B.S.; 1R01HL162574 to F.L., R.P.-R., and S.Z.) and the National Institute of General Medical Sciences (R01GM109215 to F.L. and R.P.-R.). We thank members of the Luca, Pique-Regi, Slatcher, and Zilioli laboratories for helpful discussions and comments. We also thank the anonymous reviewers for comments and suggestions that helped to improve the quality of this manuscript. Finally, we also greatly appreciate the ALOFT study participants and their families.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.276765.122.
-
Freely available online through the Genome Research Open Access option.
- Received March 17, 2022.
- Accepted June 8, 2023.
This article, published in Genome Research, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








