
Accurate integration of multiple heterogeneous single-cell RNA-seq data sets by learning contrastive biological variation
Abstract
For most biological and medical applications of single-cell transcriptomics, an integrative study of multiple heterogeneous single-cell RNA sequencing (scRNA-seq) data sets is crucial. However, present approaches are unable to integrate diverse data sets from various biological conditions effectively because of the confounding effects of biological and technical differences. We introduce single-cell integration (scInt), an integration method based on accurate, robust cell–cell similarity construction and unified contrastive biological variation learning from multiple scRNA-seq data sets. scInt provides a flexible and effective approach to transfer knowledge from the already integrated reference to the query. We show that scInt outperforms 10 other cutting-edge approaches using both simulated and real data sets, particularly in the case of complex experimental designs. Application of scInt to mouse developing tracheal epithelial data shows its ability to integrate development trajectories from different developmental stages. Furthermore, scInt successfully identifies functionally distinct condition-specific cell subpopulations in single-cell heterogeneous samples from a variety of biological conditions.
In investigating the gene expression heterogeneity in single cells, single-cell RNA sequencing (scRNA-seq) offers enormous advantages over traditional transcriptome sequencing techniques. It also shows huge potential in many biological and medical fields to shed light on the internal behavior of complex organisms, such as developing brain tissue (Patel et al. 2014; Wang and Navin 2015; Björklund et al. 2016; Wang et al. 2017; Papalexi and Satija 2018; Rosenberg et al. 2018). To get thorough interpretations of a specific biological process, integrative analysis of multiple data sets from various biological and technical conditions is a key step. The characterization of biological heterogeneity across different biological conditions is still difficult, nonetheless, as a result of the confusion of biological and technical variation (also known as batch effects).
To meet the unprecedented computational challenge, several strategies have been developed (Haghverdi et al. 2018; Barkas et al. 2019; Hie et al. 2019; Korsunsky et al. 2019; Stuart et al. 2019; Welch et al. 2019; Polański et al. 2020; Gao et al. 2021; Liu et al. 2021; Zhang and Nie 2021), mostly using two alternative methodologies: removing the difference between identified (i) similar cells and (ii) shared cell types across batches. Mutual nearest neighbor (MNN) (Haghverdi et al. 2018) is a common method that serves as the basis for numerous integration techniques for similar-cells-based methods. It searches MNN pairs across batches and treats these in-pair cells as being in the same cell states so that they can be gathered in the following phase. Scanorama (Hie et al. 2019), Seurat v3 (Stuart et al. 2019), BBKNN (Polański et al. 2020), Conos (Barkas et al. 2019), and the fast version of MNN called fastMNN (Haghverdi et al. 2018) use the same strategy, with searching MNN pairs in dimensionally reduced spaces such as canonical correlation analysis (CCA) or principal component analysis (PCA) space, instead of in the original gene expression space directly. Among these, BBKNN and Conos build global neighborhood graphs across all batches, which are used directly for the downstream analysis. Methods based on the identification of shared cell types include Harmony (Korsunsky et al. 2019), LIGER (Welch et al. 2019), and scMC (Zhang and Nie 2021). Harmony iteratively uses maximum batch diversity clustering and cell-specific linear correction using a mixture model in the PCA embedding. The integrative non-negative matrix factorization (iNMF) method yields a low-dimensional matrix that LIGER uses to perform joint clustering and quantile normalization procedures. scMC assigns the shared cell types across batches and eliminates differences between any two groups of “confident cells” of shared cell types. Incorrect connections between genetically similar cell types, particularly in the integration scenario with imbalanced cell type compositions across batches, could, however, disrupt either strategy. Recent benchmarking studies (Tran et al. 2020; Luecken et al. 2022) also revealed the inferior performance of these approaches on complex integration tasks.
To address these problems, we present an integration model named single-cell Integration based on unified contrastive biological variation learning (scInt). scInt offers several salient features as follows. First, instead of directly calculating the similarities between cells based on Euclidean distance, as most approaches do, scInt uses a similarity measure based on a cluster-specific exponential kernel, capturing both the inter- and intrabatch similarity structures. Second, scInt filters the incorrect connections of the cell similarity relationships using the contrastive principal component analysis (cPCA) method (Abid et al. 2018), a potent tool for identifying the minute differences between two data sets. Third, scInt has developed a unified framework for learning contrastive biological variation that allows for the global integration of multiple scRNA-seq data sets and the knowledge transfer from already integrated reference data to newly arrived query data. We benchmark scInt and other state-of-the-art methods in integration and reference-based mapping tasks using both simulated and real data. We also apply scInt to several integrative analyses on real data sets under various scenarios to show the capability of identifying condition-specific cell subpopulations across multiple heterogeneous data sets.
Results
scInt uniformly learns the contrastive biological variation for data integration
scInt aims to integrate multiple scRNA-seq data sets that may be generated from different conditions, species, or technologies (Fig. 1A,B). Specifically, for each cell, scInt searches the most similar cells in its remaining batches, by computing the similarities based on inter- and intrabatch similarity structures (Fig. 1C; Supplemental Fig. S1; Methods). Then, scInt uses the cPCA technique to distinguish the different genetic states across batches and filter the incorrect cell–cell connections (Fig. 1C; Supplemental Fig. S1; Methods). The difference vectors between all cells and their similar cells are regarded as the invisible data introduced by the technical variation. Thus, the contrastive biological variation can be learned by applying cPCA to the gene expression matrix and the difference matrix. Finally, using this contrastive biological variation, scInt performs global integration for all batches, resulting in a joint embedding served as the input of various downstream analysis tasks (Fig. 1D). In addition, under the same contrastive biological variation learning framework, the reference-based data integration is available to scInt (Fig. 1E; Supplemental Fig. S2; Methods), allowing flexible integrative analysis of multiple scRNA-seq data sets in biological and medical applications.
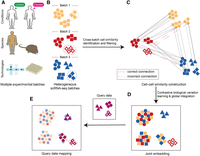
Overview of the scInt method. (A) scInt integrates multiple scRNA-seq batches that may come from different conditions, species, or technologies. (B) Three heterogeneous batches. Batches and cell types are represented by different colors and shapes, respectively. The dashed cell groups represent the same cell types with different transcriptomic states. (C) Cell–cell similarity construction. Similarities based on a cluster-specific exponential kernel are identified and further filtered in the cPCA space. (D,E) Integration and reference-based mapping of multiple batches. A unified contrastive biological variation learning framework is used for integration (D) and reference-based mapping (E).
Benchmark of scInt and other state-of-the-art methods using simulated and real transcriptome data
We first benchmarked scInt against 10 state-of-the-art methods using the simulated and real data under different integration scenarios with ground-truth cell type labels (Fig. 2; Supplemental Figs. S3–S5). We applied the uniform manifold approximation and projection (UMAP) algorithm (Becht et al. 2019) to visualize the integrated single-cell data. To evaluate the performance of the overall batch effect removal and biological signals reservation of integration results, we used two scoring metrics: the adjusted Rand index (ARI) (Hubert and Arabie 1985) and the average silhouette width (ASW) (Büttner et al. 2019; Methods). We also used the inverse Simpson's index (LISI) (Korsunsky et al. 2019) to assess the local separation of cell types and the local mixing of batches (i.e., cell type LISI and batch LISI; Methods).
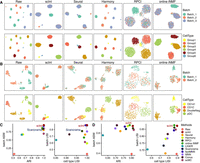
Benchmarking scInt against other methods using simulated and real data. (A) UMAP visualizations of simulation 1 before and after integration by scInt, Seurat, Harmony, RPCI, and online iNMF. Cells are colored by batch labels (top row) and cell type labels (bottom row). (B) UMAP visualizations of the human dendritic data before and after integration by scInt, Seurat, Harmony, RPCI, and online iNMF. (C) The comparison of evaluation metrics, including ARI, batch ASW, cell type LISI, and batch LISI, of the integrated results on simulation 1. (D) The comparison of evaluation metrics, including ARI, batch ASW, cell type LISI, and batch LISI, of the integrated results on human dendritic data.
First, we considered the integration scenario with unbalanced cell subpopulation compositions and rare cell subpopulations across batches. We generated simulation 1 with the Splatter R package (Zappia et al. 2017) (see Supplemental Methods for details). It contains six cell subpopulations and three batches, with five subpopulations in batch 1, four in batch 2, and four in batch 3 (the first column of Fig. 2A). Furthermore, Group5 is the unique cell subpopulation in batch 1, and Group6 is a rare cell subpopulation in batch 3. Cells within the same cell subpopulations across different batches presented distinct clusters in the unintegrated data, suggesting severe batch effects (Fig. 2A). Scanorama and scInt succeeded in integrating all these three batches, and preserved the cellular identities of Group5 and Group6, leading to higher batch removal and cell type conservation scores (Fig. 2C; Supplemental Fig. S3A). The other methods, on the other hand, either failed to capture the rare cell subpopulations, such as Seurat, or failed to integrate several cell subpopulations, such as Harmony (Fig. 2A). Additionally, we also used two simulated data sets (simulations 2 and 3, see Supplemental Methods for details), proposed by a recent benchmark study (Luecken et al. 2022), to further evaluate the performance of scInt against other methods. It showed that scInt outperformed other methods, removing the batch effects correctly, whereas preserving the simulated cell subpopulation heterogeneity (Supplemental Figs. S4, S5). Especially for simulation 3, which contained complex and nested batch effects, only RPCI, MNN, fastMNN, scMC, and scInt successfully integrated the data (Supplemental Fig. S5). Among these five methods, scInt well mixed all batches and achieved the highest evaluation metric scores.
Second, we assessed the integration performance on real data of human dendritic cells (DCs) (Villani et al. 2017). This data consists of unbalanced cell subpopulations: CD141, double negative cells (DoubleNeg), and plasmacytoid DC (pDC) cells in batch 1, CD1C, DoubleNeg, and pDC cells in batch 2, where CD141 and CD1C are biologically similar cell types. Only RPCI, online iNMF, and scInt discriminated CD141 and CD1C cells whereas integrating other shared cell types (Fig. 2B). Other methods, such as Seurat, connected CD141 and CD1C cells improperly (Fig. 2B). For the integration performance scores, scInt achieved the highest level of batch mixing and cell type separability scores (Fig. 2D; Supplemental Fig. S3B).
scInt efficiently maps new data onto already integrated data
Next, we evaluated the ability of scInt to map new data onto already integrated data using two reference-based mapping tasks. We compared scInt with three well-known approaches, including Seurat v4 (Hao et al. 2021), Symphony (Kang et al. 2021), and online iNMF, using both simulated and real data. Our proposed “global” mapping differs slightly from the other three approaches. For each reference-based mapping task, scInt incorporates new contrastive biological variations of the query and learns a new variation matrix. Thus, it differs from the other approaches in that the learned projection matrix and resulting joint low-dimensional space are different each time.
In the first mapping task, we tested if scInt could map multiple query batches onto the reference data whereas successfully mapping the rare cell subpopulation. We selected batches 1–3 of simulation 2 (containing 7450 cells) as the reference data and batches 4–6 of simulation 2 (containing 4647 cells) as the query data. Meanwhile, Group7 is a rare cell subpopulation within batches 2 and 5. Seurat and scInt effectively eliminated the batch effects of the raw data and delivered well-built references (the top row of Fig. 3A). However, online iNMF mixed a small number of Group3 cells with Group1, and Symphony showed a mixture of cells between Group5 and Group7 in the reference integration. For reference-based mapping, online iNMF and scInt precisely mapped the query onto the reference (the bottom row of Fig. 3A). In contrast, Seurat and Symphony well mapped all except Group7 of the cell subpopulations. We used a 5-NN classifier to predict the labels of the query cells in the joint low-dimensional embedding of reference and query data and calculated the prediction F1 score and accuracy (ACC) of each label to evaluate the prediction performance of mapping results (Methods). scInt accurately predicted all labels of query cells (Supplemental Fig. S6A), obtaining a mean cell type F1 score of 1 and an overall accuracy of 1 (Fig. 3B). In contrast, online iNMF and Symphony showed some incorrect label predictions because of the mixture of different cell subpopulations in the reference integration, and Seurat incorrectly connected Group5 and Group7, leading to lower mean cell type F1 and overall accuracy scores.

Benchmarking scInt against other methods in the reference-based integration tasks. (A) UMAP visualizations of the density of integrated reference cells (top row) and the scatters of mapped query cells (bottom row) of simulation 2, from scInt, Seurat, Symphony, and online iNMF. Cells are colored by ground-truth cell type labels, with gray shadows representing the reference. (B) Heatmap comparing the 5-NN predicted labels (columns) and the original labels (rows) of the query. The color bar indicates the proportion of query cells per original cell type label that was predicted to be for each reference label (rows sum to 1). (C) UMAP visualizations of the density of integrated reference cells (top row) and the scatters of mapped query cells (bottom row) of the human pancreas data, from scInt, Seurat, Symphony, and online iNMF. Cells are colored by ground-truth cell type labels, with gray shadows representing the reference. The mast cells are the unique cell type in the query data and are colored dark purple. (D) Heatmap comparing the 5-NN predicted labels (columns) and the original labels (rows) of the query.
To further assess the ability of mapping real scRNA-seq data, we applied scInt to the reference-based mapping task of multiple human pancreas data sets. The pancreas batches 1–4 were profiled with four different plate-based sequencing technologies (Fluidigm C1, CEL-Seq, CEL-Seq2, and Smart-seq2, respectively) (Grün et al. 2016; Muraro et al. 2016; Segerstolpe et al. 2016; Lawlor et al. 2017), and batches 5–8 were profiled with inDrop (Baron et al. 2016), which was a droplet-based technology. Thus, we selected pancreas batches 1–4 (containing 5887 cells) as the reference data and the remaining four batches (containing 8569 cells) as the query data to test if scInt could overcome the interplatform effects. In addition, batches 1–4 contained two rare cell types, including epsilon cells (7 cells) and immune cells (27 cells), which made both the reference integration and mapping tasks more challenging. Seurat, Symphony, and scInt successfully removed the batch effects and detected both rare cell types in the reference integration (the top row of Fig. 3C). However, online iNMF showed a mixture of cells between rare cell types and other cell types. Further, Seurat, Symphony, and scInt succeeded in overcoming the interplatform effects and mapping query cells onto the reference whereas preserving the unique cell identity of mast cells (the bottom row of Fig. 3C). Among these methods, scInt achieved the best performance in the cell type prediction by the 5-NN classifier in the joint embedding (Supplemental Fig. S6B), with a mean cell type F1 score of 0.96 and an overall accuracy of 0.95 (Fig. 3D). We also performed these two reference-based mapping tasks using our alternative mapping models, achieving performance comparable to the default “global” model (Supplemental Fig. S7).
scInt reveals integrated trajectories of mouse developing tracheal epithelial data
We further applied scInt to integrate multiple developmental batches coming from different time points and evaluated its ability in developmental trajectory discovery. In such a scenario, continuous transcriptional state evolution would hinder the identification of similar cells or cell types across time points. Additionally, the shared cell types between batches may be quite limited, resulting in unbalanced cell type compositions.
We analyzed 3508 mouse developing tracheal epithelial cells at six time points from embryonic day E12.5 to E18.5 (Kiyokawa et al. 2021). The raw data showed severe batch effects across time points, with cells separated by time points (Supplemental Fig. S8). After scInt integration, cells were well mixed across time points (Fig. 4A), and the major cell types were revealed by the canonical markers (Fig. 4B): basal cells (Krt5), ciliated cells (Foxj1), club cells (Scgb1a1), NE cells (Ascl1), KRT17+ and KRT17− progenitors (Krt17, Scgb3a2), and proliferative cells (Mki67). We also tested the other methods on this data. Among these methods, MNN, fastMNN, Harmony, and scMC successfully integrated batches across multiple time points, distinguishing most cell types (Supplemental Fig. S8). Other methods, however, showed mixtures of several cell types. We also used Monocle 3 (Cao et al. 2019) to infer the trajectories of the integrated data by scInt and other methods (Supplemental Fig. S9). Compared to other methods, scInt revealed clear developmental trajectories and yielded pseudotime inference results consistent with the actual developmental stages of epithelial cells.
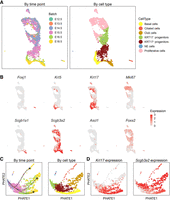
scInt reveals integrated trajectories of mouse developing tracheal epithelial data. (A) UMAP visualizations of the mouse developing tracheal epithelial cells in the scInt-integrated data (from E12.5 to E18.5). Cells are colored by time points (left) and cell types (right). (B) Overlay of the expression patterns of cell type marker genes onto the UMAP space. (C) PHATE visualizations for all cells except proliferative cells. Cells are colored by time points (left) and cell types (right). The arrows indicate the directions of the trajectories. (D) Overlay of the expression patterns of Krt17 and Scgb3a2 onto the PHATE space.
In the original study, Kiyokawa et al. found that Krt17 can serve as a marker for the intermediate cell population in the basal cell lineage, and KRT17+ progenitors contribute to the mature epithelial cells, including basal, club, and ciliated cells. To test whether integration methods could reveal this developmental process, we extracted all cells other than proliferative cells and performed the potential of heat diffusion for affinity-based transition embedding (PHATE) algorithm (Moon et al. 2019) to visualize the trajectory structure of the integrated data. scInt-integrated data could generate a global trajectory structure from E14.5 to E18.5 and presented a clear differentiation process from the KRT17− progenitors to three kinds of mature epithelial cells (Fig. 4C). The expression of the identified marker of intermediate progenitors, Krt17, was abundant in the differentiation of basal cells, indicating its important role in the commitment to mature basal cells (Fig. 4D). In contrast, Scgb3a2 showed abundance mainly in the differentiation of the other two kinds of mature epithelial cells (Fig. 4D). The expression patterns of Krt17 and Scgb3a2 may indicate the binary cell fate decision in the tracheal epithelial maturation. Meanwhile, we performed PHATE visualization on the integrated results of other methods. As shown in Supplemental Figure S10, these methods either failed to preserve the clear differentiation trajectory or mixed distinct cell types incorrectly. In general, scInt successfully integrated the developmental trajectories of mouse developing tracheal epithelial cells, revealing insights into cell lineage evolution.
scInt reveals DiHS/DRESS-specific cell subpopulations
We then tested the capability of scInt for detecting across-condition biological differences. We applied scInt to a scRNA-seq data set containing 20,415 cells from seven human skin samples from a drug-induced hypersensitivity syndrome/drug reaction with eosinophilia and systemic symptoms (DiHS/DRESS) donor and five healthy controls (Kim et al. 2020). Moreover, the samples in this data were assayed with 10x Genomics and prepared using different library construction protocols: 3′ (DiHS/DRESS, HV4, and HV5 samples), 5′ (HV1 replicates 1, 2, HV2, and HV3 samples). The raw data showed discrete populations of most cells across different technologies and conditions (Supplemental Fig. S11).
The original study found that the DiHS/DRESS and healthy lymphocytes were enriched with different signature genes and biological processes, indicating the existence of condition-specific cell subpopulations. To test whether integration methods could reveal the differences between condition-specific cell subpopulations, we integrated the human DiHS/DRESS and healthy skin data using scInt and other methods. scInt separated most cell types annotated by the high expression of typical marker genes, including endothelium (PECAM1), fibroblast (DCN), keratinocyte (KRT1, KRT5), lymphocyte (CD3E), melanocyte (PMEL), myeloid (FCER1G), and pericyte/SM (pericyte/smooth muscle, RGS5), well mixing the cells across technologies and conditions (Fig. 5A–C). In addition to lymphocytes, keratinocytes also displayed condition-specific localization in the UMAP space, indicating the transcriptional differences of keratinocytes across disease statuses. scInt successfully differentiated the DiHS/DRESS and healthy lymphocyte and keratinocyte subpopulations (Fig. 5B). However, for other methods, only scMC could distinguish all four of these condition-specific cell types while well mixing other cell types (Supplemental Fig. S11). We evaluated the performance of these integration results using ARI, batch ASW, cell type LISI, and batch LISI scores, as well as the mean label F1 and label silhouette scores of these four condition-specific cell subpopulations. Compared with other methods, scInt obtained the best overall scores (Supplemental Fig. S12A–C).
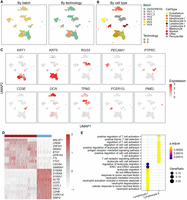
scInt reveals DiHS/DRESS-specific cell subpopulations. (A,B) UMAP visualizations of the human DiHS/DRESS and healthy skin cells in the scInt-integrated data. Cells are colored by batches, technologies (A), and annotated cell labels (B) determined in the scInt-integrated data by examining the expression patterns of known markers. (C) Overlay of the expression patterns of cell type marker genes onto the UMAP space. (D) Heatmap of the top 10 differentially expressed genes of the DiHS/DRESS and healthy lymphocyte subpopulations. (E) The top 10 enriched GO biological processes associated with the DiHS/DRESS and healthy lymphocyte subpopulations.
We then performed differential expression analysis, as well as functional enrichment analysis, on these two groups of condition-specific cell subpopulations (healthy lymphocyte I and DiHS/DRESS lymphocyte II cells, healthy keratinocyte I and DiHS/DRESS keratinocyte II cells) using the clusterProfiler R package (Yu et al. 2012). We found that different genes and biological processes were enriched in lymphocytes specific to DiHS/DRESS and healthy individuals (Fig. 5D,E). The DiHS/DRESS-specific lymphocytes were enriched with genes involved in T cell activation and cell–cell adhesion, indicating the key role of the lymphocyte population in the drug-specific immune response in DiHS/DRESS syndrome, which was consistent with previous observations (Nyfeler and Pichler 1997; Pichler and Tilch 2004; Kim et al. 2020). In addition, neutrophil activation and neutrophil mediated immunity pathways, which play key roles in innate immunity, were enriched in the DiHS/DRESS keratinocytes (Supplemental Fig. S12D). The aforementioned results together show that scInt can reveal condition-specific cell subpopulations and their biological function differences between biological conditions.
scInt reveals inflammation-related mechanisms in the COVID-19 data
Lastly, we showed the potential of scInt in integrating complex real scRNA-seq data from distinct biological conditions. We applied scInt to a single-cell RNA-seq data set with 67,362 peripheral blood mononuclear cells (PBMCs) from three conditions (healthy, remission, and severe) and 12 batches (Supplemental Table S1; Guo et al. 2020). It showed that there were severe batch effects across batches, especially across COVID-19 and healthy samples in the raw data (Fig. 6A).
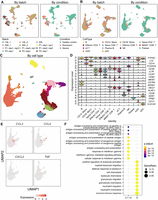
scInt reveals inflammatory-related mechanisms in the COVID-19 data. (A,B) UMAP visualizations of the COVID-19 PBMC data in the raw data (A) and scInt-integrated data (B). Cells are colored by batches and COVID-19 stage conditions. (C) UMAP visualizations of the scInt-integrated data. Cells are colored by identified cell types. (D) Stacked violin plot of the top-important marker gene expression for each cell type. (E) Overlay of the expression patterns of the cytokine storm-related and inflammation-related chemokine genes in the CD14+ monocytes. (F) The top 10 enriched GO biological processes associated with cluster 9 and other CD14+ monocytes.
After scInt integration, cells were well mixed across batches and conditions (Fig. 6B). We performed Leiden graph-based clustering (Traag et al. 2019) onto the scInt-integrated data, identifying 20 clusters (Supplemental Fig. S13). We manually annotated 14 cell types including B cells, plasma B cells, monocytes (CD14+ and CD16+ monocytes), T cells (naive CD4+, effector CD4+, naive CD8+, effector CD8+, MKI67+ CD8+, and γδ T cells), natural killer cells (NK), platelets, myeloid dendritic cells (mDC), and plasmacytoid dendritic cells (pDC), using the known markers (Fig. 6C,D). To investigate the immune responses during COVID-19 statuses, we then quantified the changes in the cell types and cluster proportions in healthy, remission, and severe samples (Supplemental Figs. S14, S15). Several immune cell types were observed to be depleted in the COVID-19 samples, including CD14+ and CD16+ monocytes, naive CD4+ T cells, naive CD8+ T cells, γδ T cells, and DCs (Supplemental Fig. S14). Among these cell types, monocytes play a crucial role in the inflammatory-related mechanisms in COVID-19 patients (Wang et al. 2020; Zhou et al. 2020). Four clusters were identified as CD14+ monocytes, including clusters 0, 7, 9, and 12. Compared with the other three clusters, cluster 9 showed an increase in COVID-19 samples, significant in severe-stage samples. We next explored the distribution of cytokine storm-related and inflammation-related chemokine genes, including CCL3, CCL4, CXCL2, and TNF, in the CD14+ monocyte subpopulation. These genes were primarily expressed in cluster 9 (Fig. 6E), suggesting that cluster 9 may contribute to the cytokine storm in COVID-19 patients. Additionally, the pathway enrichment analysis showed that genes related to cell chemotaxis, neutrophil chemotaxis, and leukocyte chemotaxis were enriched in cluster 9 (Fig. 6F). Thus, cluster 9 was identified as the COVID-19-specific CD14+ monocyte subpopulation associated with the cytokine storm and dysregulated immune responses in COVID-19 patients.
Discussion
We developed a novel method called scInt for the effective integration and reference-based mapping of scRNA-seq data. In both integration and reference-based mapping tasks of multiple heterogeneous data sets, scInt uses a unified contrastive biological variation learning framework. We showed the outperformance of scInt against 10 cutting-edge integration methods using both simulated and real data under several common integration scenarios.
scInt uses a cluster-specific kernel similarity measure to identify similar cells for each cell among all remaining batches, enabling robust similarities construction, especially in the case of complex experimental designs. For calculation of the similarity measure, the preclustering of each batch is performed, aiming to capture similar cell states in the individual batch and determine a common batch effect size factor for each identified cell state. The core assumption is that cells with similar transcription states in a batch receive similar technical effects. By this approach, scInt overcomes the nested batch effects and unbalanced cell group composition designs and successfully identifies the same cell identities across all batches. Indeed, we found that scInt also improves the developmental trajectories integration, which is crucial for tissue development studies. cPCA is used to filter the incorrect cell–cell connections, which are prone to occur in the scenario of similar cell subpopulations. cPCA is sensitive to gene expression differences across batches, such as condition-specific cell types in disease versus healthy tissues. Due to the use of cPCA, scInt can differentiate genetically similar cell types across batches, enabling comparison of single-cell samples from different biological conditions.
With the rapid growth of scRNA-seq data sets, reference-based mapping is turning out to be progressively significant. Recently, several methods have been developed to tackle this problem; however, they generally assumed that the reference could capture all biological variations in the query. Unfortunately, the cell subpopulation composition of the query is always unknown and new biological variations might be introduced. To overcome this obstacle, scInt develops a reference-based mapping method under our unified contrastive biological variation learning framework, enabling the continuous update of new biological variations. scInt learns a new contrastive biological variation matrix in each mapping task, which is the primary distinction from other reference-based mapping methods. To evaluate the capability of reference-based mapping, we have used the simulated and real data to perform the mapping task via scInt and three other current methods.
In addition to the applications to several common scenarios, we also showed the performance of scInt under less typical integration scenarios. We applied scInt to three pathological cases, each with two variants of simulation data, to show the usability and stability of the method in these challenging scenarios. These cases will also help users prepare their data better when using scInt. First, we considered the case when cell subpopulations were only present in a single batch. We applied scInt to variants 1 and 2 of simulation 1 (Supplemental Methods), which contained four and six cell subpopulations only present in a single batch, respectively. It is worth noting that there are no shared cell subpopulations across batches in variant 2, which does not meet the underlying assumption of the vast majority of current data integration methods. scInt successfully integrated variant 1 and kept all cell subpopulations separated on variant 2 (Supplemental Figs. S16, S17). We also used other methods to integrate these two variants. Among them, only MNN could integrate two variants successfully, and Harmony preserved all unique cell subpopulation identities on variant 2 (Supplemental Figs. S16, S17). Second, we considered the case when batches were dominated by a single cell subpopulation. We applied scInt to variants 1 and 2 of simulation 3 (Supplemental Methods), which contained 14 and 16 batches dominated by a single cell subpopulation, respectively. It showed that scInt successfully removed batch effects on variant 1, but failed to integrate variant 2 (Supplemental Figs. S18, S19). Other methods were also applied to integrate variants 1 and 2. On variant 1, RPCI succeeded in removing batch effects, whereas other compared methods showed incorrect alignment of multiple cell subpopulations (Supplemental Fig. S18). Of note, these approaches similarly failed to integrate variant 2 (Supplemental Fig. S19), indicating that this extreme case continued to be difficult for existing tools and our method. Third, we considered the case when rare cell subpopulations were only present in a single batch, because the detection of rare cell subpopulations was also an important purpose of single-cell analysis. We generated simulation 4 by Splatter package and used its variants 1 and 2 to test the ability of scInt to deal with this case (Supplemental Methods). Variants 1 and 2 of simulation 4 contained two and four rare cell subpopulations, respectively, with each rare cell subpopulation only present in a single batch. As shown in Supplemental Figures S20 and S21, scInt successfully removed batch effects and preserved all rare cell subpopulation identities on both variants. In contrast, the integration results of other methods, except MNN and fastMNN, on these two variants showed mixtures of multiple rare cell subpopulations (Supplemental Figs. S20, S21).
In summary, we present a novel method, scInt, for the integration and reference-based mapping of multiple heterogeneous scRNA-seq data sets. scInt is especially suitable for the integrative analysis of multiple heterogeneous data sets coming from different conditions and complex experimental designs, which is illustrated using several published scRNA-seq data sets in this work. Additionally, the contrastive biological variation patterns of other types of single-cell measurements may be easily explored using the proposed framework in the future.
Methods
The scInt algorithm
scInt is proposed to integrate multiple heterogeneous scRNA-seq data sets from distinct experiment batches, biological conditions, species, or technology platforms. Given gene expression matrices from multiple experimental designs, scInt learns contrastive biological variation and projects all batches onto a shared low-dimensional subspace. Also, this variation enables further learning as new data becomes available. Specifically, the scInt algorithm consists of the following main steps:
-
data preprocessing and highly variable features selection;
-
cross-batch cell–cell similarity construction;
-
cell similarities filtering on the cPCA space;
-
global integration by contrastive biological variation learning;
-
integration of already integrated reference data and new data.
Details of each step are described as follows.
Data preprocessing and highly variable features selection
For each scRNA-seq batch, the standard filtering and normalizing processes are used in the raw gene expression matrix. Genes expressed in less than 3 cells and cells with expressed genes of less than 300 are considered to be low-quality genes or cells, respectively, and are filtered. Then, the filtered expression matrices are normalized as follows: gene expression values are divided by the total gene expression value in each cell, multiplied by a scale factor (10,000 by default), and log1p-transformed. Top 2000 highly variable features are selected by default for each batch and then pooled to determine the feature list across all batches used for data integration.
Cross-batch cell–cell similarity construction
The preprocessed gene-by-cell expression matrices X1, · · · , XN, where N is the total number of batches, are used to identify the cell–cell similarity relationships across batches. First, for each
batch, a cell clustering approach is performed to find the cell similarity structure in individual batches. The Leiden algorithm
(Traag et al. 2019) based on the shared nearest neighbor (SNN) graph is used by default, but other clustering algorithms are also available.
We showed that our method was robust over a wide range of resolution parameter values of Leiden clustering algorithm (scInt
parameters). Second, for each batch i, we perform PCA on the expression matrix and then project all batches onto this batch i-specific low-dimensional space. Specifically, for Xi, we compute its gene means μi and standard deviations δi and use these statistics to center and scale all batches: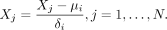 (1)
Then, we learn the truncated left singular matrix Ui of Xi using singular value decomposition (SVD) and use it to project Xj, j = 1, · · · , N onto the PCA space specific to batch i. Third, for each cell is of cluster c in batch i, the minimum cosine distance
(1)
Then, we learn the truncated left singular matrix Ui of Xi using singular value decomposition (SVD) and use it to project Xj, j = 1, · · · , N onto the PCA space specific to batch i. Third, for each cell is of cluster c in batch i, the minimum cosine distance 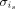 between is and cells in all remaining batches is computed in this low-dimensional space specific to batch i. The median of all distances
between is and cells in all remaining batches is computed in this low-dimensional space specific to batch i. The median of all distances 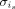 in each cell cluster c identified in batch i is regarded as the unified batch effect size of the cell cluster c. Thus, we let
in each cell cluster c identified in batch i is regarded as the unified batch effect size of the cell cluster c. Thus, we let 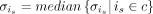 for each is in c. The assumption behind
for each is in c. The assumption behind 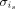 is that cells with similar transcription states in a batch are subject to similar technical effects. In this way, the interbatch
similarities are related to the intrabatch similarities. Then, the cell similarity between each cell is in the batch i and cell jt in the batch j is computed using a cluster-specific exponential kernel:
is that cells with similar transcription states in a batch are subject to similar technical effects. In this way, the interbatch
similarities are related to the intrabatch similarities. Then, the cell similarity between each cell is in the batch i and cell jt in the batch j is computed using a cluster-specific exponential kernel: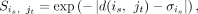 (2)
where d(is, jt) denotes the cosine distance between cell is and cell jt in the batch i-specific low-dimensional space. To reliably select the most similar cells of is, the cells with the top k = 5 similarities are retained. The cell–cell corresponding relationships set of batch i is denoted as
(2)
where d(is, jt) denotes the cosine distance between cell is and cell jt in the batch i-specific low-dimensional space. To reliably select the most similar cells of is, the cells with the top k = 5 similarities are retained. The cell–cell corresponding relationships set of batch i is denoted as 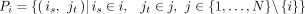 , and the cells in Pi are divided into two subsets including Ci = {is|is ∈ Pi, is ∈ i} and
, and the cells in Pi are divided into two subsets including Ci = {is|is ∈ Pi, is ∈ i} and 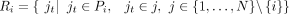 . Note that these batch i-specific sets Pi’s are not directly used for downstream analysis such as clustering analysis, but only to infer the integrated data.
. Note that these batch i-specific sets Pi’s are not directly used for downstream analysis such as clustering analysis, but only to infer the integrated data.
Cell similarity filtering on the cPCA space
This step aims to remove the false similarity connections between different cell populations, which are prone to occur in
the integration scenario with different biological conditions. For batch i, the identified similar cells are embedded in the cPCA space, to capture subtle transcriptional differences. Specifically,
let 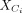 and
and 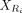 be the expression submatrices for Ci and Ri, respectively. Then
be the expression submatrices for Ci and Ri, respectively. Then 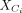 and
and 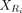 are centered by the mean of
are centered by the mean of 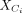 . The goal is to find the contrastive direction of the variation difference between target data
. The goal is to find the contrastive direction of the variation difference between target data 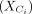 and background data
and background data 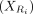 :
: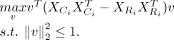 (3)
Note this model is slightly different from the original cPCA model in terms of the parameter α, which we illustrate in the
Supplemental Methods. This optimization problem can be solved by eigenvalue decomposition of
(3)
Note this model is slightly different from the original cPCA model in terms of the parameter α, which we illustrate in the
Supplemental Methods. This optimization problem can be solved by eigenvalue decomposition of  . The top l = 40 contrastive principal components (cPCs) are used to generate the cPCA subspace. When a few similar cells are shared
between batch i and other batches, there may be less than l cPCs identified and used to filter the cell–cell similarities. We applied scInt to variant 2 of simulation 1, which had no
cell subpopulations shared across batches (Supplemental Methods). The results showed that scInt was robust under this integration scenario (Supplemental Fig. S17). Then the cell similarity connections whose cosine similarities are less than T (0.6 by default) are filtered. The preserved similarities are used to construct the similarity matrix S.
. The top l = 40 contrastive principal components (cPCs) are used to generate the cPCA subspace. When a few similar cells are shared
between batch i and other batches, there may be less than l cPCs identified and used to filter the cell–cell similarities. We applied scInt to variant 2 of simulation 1, which had no
cell subpopulations shared across batches (Supplemental Methods). The results showed that scInt was robust under this integration scenario (Supplemental Fig. S17). Then the cell similarity connections whose cosine similarities are less than T (0.6 by default) are filtered. The preserved similarities are used to construct the similarity matrix S.
Global integration by contrastive biological variation learning
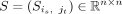 encodes the identified cell–cell similarity relationships, where n is the total cell number of all batches. Also, S is symmetrized, that is, S = (S + ST)/2. Using identified similarities, we construct a matrix Z to represent an invisible data set introduced by technical effects. The idea behind this is that the differences between
the expression vector of each cell is in batch i and its identified similar cells jt’s in batch j are regarded as technical effects. To construct this invisible data set, for each cell, we calculate the difference vectors
between the expression vectors of itself and the weighted averaged sum of their identified similar cells. We use Si,j to denote the submatrix of S representing the cell–cell similarities between batch i and batch j, and column-normalized the non-zero columns of Si,j’s using the SoftMax function. We construct the technical mixing matrix M ∈ ℝn×n:
encodes the identified cell–cell similarity relationships, where n is the total cell number of all batches. Also, S is symmetrized, that is, S = (S + ST)/2. Using identified similarities, we construct a matrix Z to represent an invisible data set introduced by technical effects. The idea behind this is that the differences between
the expression vector of each cell is in batch i and its identified similar cells jt’s in batch j are regarded as technical effects. To construct this invisible data set, for each cell, we calculate the difference vectors
between the expression vectors of itself and the weighted averaged sum of their identified similar cells. We use Si,j to denote the submatrix of S representing the cell–cell similarities between batch i and batch j, and column-normalized the non-zero columns of Si,j’s using the SoftMax function. We construct the technical mixing matrix M ∈ ℝn×n: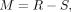 (4)
where R is the diagonal matrix with the diagonal elements representing the column sums of S, that is,
(4)
where R is the diagonal matrix with the diagonal elements representing the column sums of S, that is, 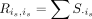 . M encodes all technical mixing coefficients between each batch i and its remaining batches. Then the invisible data set of technical effects can be learned:
. M encodes all technical mixing coefficients between each batch i and its remaining batches. Then the invisible data set of technical effects can be learned: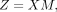 (5)
where X = [X1, · · · , XN] is the stacked gene expression matrix. Because we next learn the covariance matrix of Z, to eliminate the effects of the size of identified similarity relationships specific to each batch and avoid large batches
dominating integration, we divide each column is of M by
(5)
where X = [X1, · · · , XN] is the stacked gene expression matrix. Because we next learn the covariance matrix of Z, to eliminate the effects of the size of identified similarity relationships specific to each batch and avoid large batches
dominating integration, we divide each column is of M by 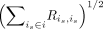 as a scale factor. The cPCA model is used to learn the contrastive biological variation and eliminate the technical effects
of X:
as a scale factor. The cPCA model is used to learn the contrastive biological variation and eliminate the technical effects
of X: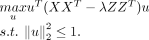 (6)
This optimization problem can be solved by eigenvalue decomposition of the contrastive biological variation D = XXT − λZZT. The result U is the transformation matrix that projects X onto the low-dimensional space that is not affected by cross-batch technical variation, that is, Xcorrect = UTX is the corrected low-dimensional data.
(6)
This optimization problem can be solved by eigenvalue decomposition of the contrastive biological variation D = XXT − λZZT. The result U is the transformation matrix that projects X onto the low-dimensional space that is not affected by cross-batch technical variation, that is, Xcorrect = UTX is the corrected low-dimensional data.
Integration of already integrated reference data and new data
Let Xnew be the gene-by-cell gene expression matrix with nnew cells of the new data. scInt maps Xnew onto already integrated reference data with three optional methods (Supplemental Fig. S2). Primarily, the projection matrix U learned from the previous steps is used to project new data onto the reference data directly, that is, Xupdate = UT[X, Xnew].
For the second method, the cPCA framework is used to integrate the new data. We use the following objective function: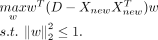 (7)
The top l = 40 cPCs are used to learn projection matrix W to generate the integrated low-dimensional space, that is, Xupdate = WT[X, Xnew].
(7)
The top l = 40 cPCs are used to learn projection matrix W to generate the integrated low-dimensional space, that is, Xupdate = WT[X, Xnew].
The last method, named the “global” model, uses the previous steps to identify the cell–cell similarity relationships between
reference and query data. It constructs a new  by the already gained S of reference data, the identified cell–cell connections between reference and query data, and the Snew of query data if available. Then the new contrastive biological variation
by the already gained S of reference data, the identified cell–cell connections between reference and query data, and the Snew of query data if available. Then the new contrastive biological variation 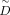 and new projection matrix
and new projection matrix 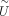 are learned.
are learned. 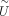 projects X and Xnew onto the same low-dimensional space, that is,
projects X and Xnew onto the same low-dimensional space, that is, 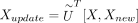 .
.
We note the first two models are suitable for situations where reference data captures all the biological variations of query data, that is, there is no new cell type in the query data. The third model is not subject to this limitation and is more applicable to complex reference-based integration tasks. scInt uses the third model by default.
scInt parameters
In scInt algorithm, there are two main tuning parameters: λ and T. Among them, λ controls the degree of mixing of similar cells across different batches in the integrated low-dimensional space, and T controls the reliability of the retained similarities. The default values for these parameters are as follows: λ = 5 and T = 0.6. scInt was found to be relatively robust to λ and T values within certain ranges (Supplemental Figs. S22, S23). Additionally, we propose a heuristic method to select optimal tuning parameters λ and T from certain sets of candidate values (see Supplemental Methods). We also tested the scInt algorithm under different Leiden clustering resolutions and k (Supplemental Figs. S24, S25) and found that these parameters were robust to the integration results.
Benchmark integration methods
We benchmarked our method with 10 other state-of-the-art integration methods, including MNN and its extension version fastMNN, LIGER and its online version online iNMF, Seurat, Harmony, Scanorama, RPCI, Conos, and scMC. We visualized the integration results in a two-dimensional space using the UMAP algorithm. In the benchmark work, the data sets were filtered and normalized uniformly. For selecting highly variable features, we used the standard “FindVariableFeatures” function implemented in the Seurat R package with the same parameters, except for methods using their specific feature selection functions, such as LIGER. Further, we selected reasonable key parameters of each method as their user guides described, to ensure a fair comparison. For Seurat, Harmony, and scMC, as they were robust to their key parameters in certain ranges, we used their default parameters for integration. When integrating the dendritic data set, as the cell number in each batch was small, we set the “k.filter” parameter from 200 to 40 for Seurat. When integrating the DiHS/DRESS skin data set, we set group.by.vars = c(“Batch”, “Condition”, “Technology”) and tau = c(2, 2, 2) for Harmony. When integrating the simulated and dendritic data sets, as the default clustering algorithm used in scMC failed to find clusters and marker genes, we set the “resolution” parameter to 0.8. For other MNN-based methods, including MNN, fastMNN, Scanorama, and Conos, we set k = 20 in the MNN pair selection uniformly. For RPCI, we set the reference batch as the batch that contained the most cell types, and eigens = 12, 15, 8, 8, 12, and 15 for simulation 1, simulation 2, simulation 3, dendritic, developing tracheal epithelial, and DiHS/DRESS skin data sets, respectively. For LIGER and online iNMF, we set λ = 5 for all data sets, and k = 10, 20, 20, 15, 20, and 20 for simulation 1, simulation 2, simulation 3, dendritic, developing tracheal epithelial, and DiHS/DRESS skin data sets, respectively. The details of parameter selection for RPCI, LIGER, and online iNMF are described in the Supplemental Material (Supplemental Methods; Supplemental Figs. S26, S27). These methods were applied using batchelor (version 1.6.2, for MNN and fastMNN), rliger (version 1.0.0, for LIGER and online iNMF), Seurat (version 4.1.0), harmony (version 0.1.0), Scanorama (version 1.7.1), RISC (version 1.0, for RPCI), Conos (version 1.4.3), and scMC (version 1.0.0). We also benchmarked our method with three other reference-based mapping methods, including Seurat, Symphony, and online iNMF. For Seurat, we used its default parameters to perform the mapping. For Symphony, we set group.by.vars = c(“Batch”, “Donor”) and tau = c(2, 2) for pancreas reference data integration. For online iNMF, we set k = 20 and λ = 5 for reference data integration and mapping for both simulated and real data sets in reference-based mapping tasks. These methods were applied using Seurat (version 4.1.0), symphony (version 0.1.0), and rliger (version 1.0.0), respectively.
Evaluation metrics
To evaluate the performance of our method and other integration tools, we used four benchmark metrics: ARI, batch ASW, cell type LISI, and batch LISI. Additionally, we used the F1 score and ACC to evaluate the prediction accuracy in the reference-based integration tasks.
ARI
ARI evaluates the overlap of the original cell type annotation and the cell clustering results from the integrated data. The ARI value scores range from 0 to 1, where 0 indicates that the two clustering labels are independent of each other, and 1 means that the two clustering labels are the same up to a permutation.
ASW
ASW also measures the cluster preservation. We use it to evaluate the batch mixing score. The batch ASW score for each cell
label p is computed as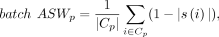 (8)
where s(i) is the silhouette width value of cell i, Cp is the set of cells with the cell label p, and |Cp| denotes the number of cells in the Cp. The final batch ASW scores are averaged:
(8)
where s(i) is the silhouette width value of cell i, Cp is the set of cells with the cell label p, and |Cp| denotes the number of cells in the Cp. The final batch ASW scores are averaged: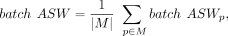 (9)
where M is the set of cell labels.
(9)
where M is the set of cell labels.
LISI
LISI assesses the mixing and separation of a given label in the local distribution. Specifically, LISI computes the inverse
Simpson's index of cell type label (cLISI) or batch label (iLISI) in the nearest neighbors of a cell selected with a fixed
perplexity. For a single cell i with cell label p, a good integration always expects the iLISI is closed to the number of batches the cell label p appears, and cLISI is closed to 1. We modified LISI to make the LISI value between 0 and 1, and used batch LISI and cell
type LISI to measure the integration performance specific to a single batch. For batch b, we compute the batch LISI by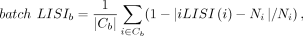 (10)
where Cb denotes the set of cells in batch b, Ni is the number of batches that the cell label of cell i appears, and |iLISI(i) − Ni|/Ni measures the deviation between the batch mixing and the ideal mixing around the cell i ∈ Cb. Similarly, for batch b, we compute the cell type LISI by
(10)
where Cb denotes the set of cells in batch b, Ni is the number of batches that the cell label of cell i appears, and |iLISI(i) − Ni|/Ni measures the deviation between the batch mixing and the ideal mixing around the cell i ∈ Cb. Similarly, for batch b, we compute the cell type LISI by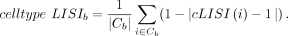 (11)
The final batch LISI and cell type LISI scores are averaged:
(11)
The final batch LISI and cell type LISI scores are averaged: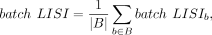 (12)
(12)
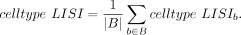 (13)
(13)
F1 scores
The F1 score is a weighted mean of precision rate and recall rate given by the equation: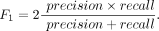 (14)
It returns a value between 0 and 1, where 1 shows the best prediction.
(14)
It returns a value between 0 and 1, where 1 shows the best prediction.
ACC
ACC is calculated by dividing the number of correct predictions by the total number of predictions: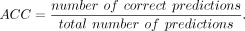 (15)
It shows the prediction performance of the proportion of correct predictions.
(15)
It shows the prediction performance of the proportion of correct predictions.
Data sets
The details of the data sets in this study are listed in Supplemental Table S1.
Software availability
The scInt R package (R Core Team 2020) is available freely at GitHub (https://github.com/Jinsl-lab/scInt). Source codes to reproduce the results in this manuscript are available at GitHub (https://github.com/Jinsl-lab/scInt_reproducibility). Both the R package and source codes are also available as Supplemental Code.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (Grants No. 62271173, No. 11971130, and No. 62172122), the Key Research and Development Program of Heilongjiang (Grant No. 2022ZX01A19), and the Interdisciplinary Research Foundation of HIT (Grant No. IR2021109).
Author contributions: S.J. and Y.Z. designed this project. Y.Z. conceived the idea, developed the model, and designed the analysis. Y.Z., Q.S., J.Q., and J.H. implemented the software and performed the analysis. Y.Z., Q.S., B.Y., and L.W. composed the manuscript. All authors read and approved the final manuscript.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.277522.122.
- Received November 20, 2022.
- Accepted May 3, 2023.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see https://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








