
A temporal in vivo catalog of chromatin accessibility and expression profiles in pineoblastoma reveals a prevalent role for repressor elements
- Salam Idriss1,13,
- Mohammad Hallal1,2,13,
- Abdullah El-Kurdi1,3,13,
- Hasan Zalzali4,5,
- Inaam El-Rassi2,
- Erik A. Ehli6,
- Christel M. Davis6,
- Philip E.D. Chung7,8,
- Deena M.A. Gendoo9,
- Eldad Zacksenhaus7,8,10,
- Raya Saab4,5,11,14 and
- Pierre Khoueiry1,3,12,14
- 1Department of Biochemistry and Molecular Genetics, Faculty of Medicine, American University of Beirut, Beirut 1107 2020, Lebanon;
- 2Biomedical Engineering Program, American University of Beirut, Beirut 1107 2020, Lebanon;
- 3Pillar Genomics Institute, Faculty of Medicine, American University of Beirut, Beirut 1107 2020, Lebanon;
- 4Department of Pediatric and Adolescent Medicine, American University of Beirut, Beirut 1107 2020, Lebanon;
- 5Department of Anatomy, Cell Biology, and Physiological Sciences, Faculty of Medicine, American University of Beirut, Beirut 1107 2020, Lebanon;
- 6Avera Institute for Human Genetics, Sioux Falls, South Dakota 57108, USA;
- 7Toronto General Research Institute, University Health Network, Toronto, Ontario M5G 1L7, Canada;
- 8Department of Laboratory Medicine and Pathobiology, University of Toronto, Toronto, Ontario M5S 1A8, Canada;
- 9Centre for Computational Biology, Institute of Cancer and Genomic Sciences, University of Birmingham, Birmingham B15 2SY, United Kingdom;
- 10Department of Medicine, University of Toronto, Toronto, Ontario M5S 1A8, Canada
-
↵13 These authors contributed equally to this work.
Abstract
Pediatric pineoblastomas (PBs) are rare and aggressive tumors of grade IV histology. Although some oncogenic drivers are characterized, including germline mutations in RB1 and DICER1, the role of epigenetic deregulation and cis-regulatory regions in PB pathogenesis and progression is largely unknown. Here, we generated genome-wide gene expression, chromatin accessibility, and H3K27ac profiles covering key time points of PB initiation and progression from pineal tissues of a mouse model of CCND1-driven PB. We identified PB-specific enhancers and super-enhancers, and found that in some cases, the accessible genome dynamics precede transcriptomic changes, a characteristic that is underexplored in tumor progression. During progression of PB, newly acquired open chromatin regions lacking H3K27ac signal become enriched for repressive state elements and harbor motifs of repressor transcription factors like HINFP, GLI2, and YY1. Copy number variant analysis identified deletion events specific to the tumorigenic stage, affecting, among others, the histone gene cluster and Gas1, the growth arrest specific gene. Gene set enrichment analysis and gene expression signatures positioned the model used here close to human PB samples, showing the potential of our findings for exploring new avenues in PB management and therapy. Overall, this study reports the first temporal and in vivo cis-regulatory, expression, and accessibility maps in PB.
Pineoblastoma (PB) is a very rare and aggressive pediatric cancer of grade IV histology, associated with a poor prognosis despite intensive multimodality therapies (Jouvet et al. 2000; Tate et al. 2012; De Braganca and Packer 2013; Parikh et al. 2017). Germline mutations in DICER1 and RB1 genes are known predisposing factors (de Kock et al. 2014, 2020), but also sporadic PB events are frequent with a major role for MYC amplification (Li et al. 2020; Liu et al. 2021). Moreover, DNA methylation profiling and whole-exome sequencing revealed a homozygous deletion in DROSHA, which acts upstream of DICER1 in the miRNA processing cascade, in a subset of human PB tumors (Snuderl et al. 2018). Recent molecular classification identified several PB subgroups with distinct survival and clinical features (Li et al. 2020). Although the oncogenic landscape of PB in each subgroup has been well characterized, the epigenetic events are still ill defined, hence the need to characterize the epigenetic and regulatory landscapes accompanying PB initiation and progression. Elucidating these events is crucial for understanding PB evolution and distinguishing it from other brain tumors, thus paving the way for better targeted therapies for PB.
Cis-regulatory elements (CREs) including promoters, enhancers, and repressors are noncoding DNA sequences harboring binding sites for transcription factors (TFs) required to activate or repress target gene expression (Ong and Corces 2011). Gain or loss of chromatin accessibility triggered by histone tail modifications including acetylation and methylation (Chen et al. 2014a; Bradner et al. 2017; Klonou et al. 2018) affects CREs and gene expression and contributes to the establishment and progression of cancer (Delcuve et al. 2009; Hnisz et al. 2015; Maury and Hashizume 2017; Corces et al. 2018; Jin et al. 2018; Barwick et al. 2021; Stępniak et al. 2021). For instance, 50% of pediatric high-grade gliomas (pHGGs) show mutations in K27M, leading to a reduction of the repressive histone mark H3K27me3 at specific loci affecting gene expression (Bender et al. 2013). Acetylation of N-terminal lysine residues of histones H3 is typically associated with active CREs including enhancers and super-enhancers (SEs) that play an important role in cancer (Hnisz et al. 2013, 2015; Niederriter et al. 2015).
Here, we extracted pineal tissues from mice with targeted overexpression in the pineal gland of the human CCND1 gene in a Trp53-null background (Rbp3-CCND1, Trp53−/−) (Saab et al. 2009) and characterized the accessible genome (ATAC-seq) and transcriptome (RNA-seq) over three key time points spanning the proliferating (postnatal day 10 [P10]), early malignant (P49), and invasive tumorigenic (P90) stages. Additionally, we characterized active enhancers and promoters using H3K27ac ChIP-seq at P90. Our work represents the first time course catalog of in vivo chromatin accessibility and expression during PB progression aiming at identifying new molecular targets for PB and potentially other brain tumors.
Results
Pan-transcriptomic and accessibility maps of PB progression
To investigate gene expression and chromatin accessibility dynamics that occur during initiation and progression of PB, we extracted pineal glands from a well-characterized mouse model in which a human CCND1 transgene under the control of the retinol binding protein 3, interstitial (Rbp3) promoter is selectively expressed in the retina and pineal gland (Liou et al. 1990) in a Trp53-null background (Rbp3-CCND1, Trp53−/−) (Fig. 1A; Skapek et al. 2001; Saab et al. 2009). We investigated three key stages covering PB progression that have been characterized for this model (Saab et al. 2009; Zalzali et al. 2012; Harajly et al. 2016). This includes a proliferating stage, causing pineal gland enlargement (postnatal day 10 [P10]); an early malignant stage, characterized by hyperproliferating pineal cells (P49); and a tumorigenic invasive PB stage (∼P90) (Fig. 1A; Supplemental Fig. S1A). Review of the stained sections of the pineal gland lesions at the different time points by an experienced neuropathologist confirmed these findings. As shown in Supplemental Figure S1A, P10 shows increased cellularity compared with normal pineal gland, with cells showing a high nuclear-to-cytoplasmic ratio, and scattered few mitotic figures. P49 shows a PB confined to the pineal region without infiltration into the surrounding brain, with more cellularity than P10, more atypical cells, and elevated mitotic activity. P90 shows an infiltrative PB invading into the surrounding brain, with dense cellularity, scattered giant tumor cells, areas of necrosis, and brisk mitotic activity.
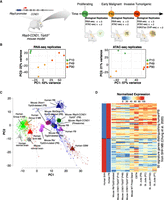
A mouse model recapitulating the evolving PB. (A) The Rbp3-CCND1/Trp53−/− mouse model and timeline representing the three key time points of PB progression, the assays performed, and the number of biological replicates. (B) PCA plots for RNA-seq and ATAC-seq samples used, highlighting the similarity between the biological replicates. (C) GSEA-PCA plot of different mouse model samples of PB and RB, as well as human normal brain, PB, MB, and GBM samples shows that the Rbp3-CCND1/Trp53−/− model is closer to other mouse models of PB, RB, and human PB but is further away from human RB and GBM. For each group, each sample or replicate is represented by a symbol of the same shape and color, albeit more transparent, as the corresponding centroid. Included also are mouse pineal samples with Rbp3-CCND1 that develop hyperplastic pineal glands but no malignant tumors (pinealoma). (D) Heatmap of normalized expression profiles of 92 signature genes distinguishing human PB from SHH MB across multiple human PB and other pineal tumors samples (Methods) as well as different mouse models of PB showing the closer similarity of Rbp3-CCND1/Trp53−/− to the Rb/Trp53-deleted mouse model as well as to human PB samples.
RNA-seq and ATAC-seq data from biological replicates, of single and independent pineal tissue each, clustered together in principal component analysis (PCA; Methods) (Fig. 1B). PCA of RNA-seq samples showed variability among biological replicates while progressing from proliferating (P10) to early malignant (P49) and tumorigenic stages (P90), reflecting an increase in expression divergence with cancer progression. Biological replicates from ATAC-seq were highly similar with correlations of 0.97, 0.93, and 0.79 for P10, P49, and P90, respectively (Supplemental Fig. S1B). RNA-seq and ATAC-seq signals from the same time point showed a decent correlation despite the inherent biological differences of both assays (Supplemental Fig. S1D) and the fact that a gene is often concurrently regulated by multiple accessible elements.
Central nervous system (CNS) tumors in general, and PB in particular, show significant levels of heterogeneity, with different subgroups potentially amenable to specific targeted therapy (Sturm et al. 2016; Capper et al. 2018; Li et al. 2020). We thus wanted to classify the model used in this study in comparison to other mouse models of PB and human samples as described previously (Chung et al. 2020). For this, we conducted a ssGSEA-PCA analysis (Gendoo et al. 2015) across 609 mouse and human brain tumor samples (Chung et al. 2020). Because inter-species clustering using methylation information is currently challenging, we used RNA expression data. The Rbp3-CCND1/Trp53−/− tumors clustered close to human PB and mouse Rb/Dicer1/Trp53−/−, Dicer1/Trp53−/−, and Rb/Trp53 PBs, as well as mouse retinoblastoma (RB), but further away from human RB and GBM samples (Fig. 1C).
Next, we assessed the similarity of the Rbp3-CCND1/Trp53−/− model to human PB by using the 92-gene classifier developed by Chung and colleagues (2020) (Supplemental Table S2), which differentiates human PB from human SHH MB. Indeed, the Rbp3-CCND1/Trp53−/− is very similar to the Rb/Trp53-deleted mouse model and to human PB (Fig. 1D). We also included in our comparison, human pineal gland cancer samples from the CBTN and St. Jude networks (Methods) to further highlight the similarity of the PB mouse models to human PB samples.
In summary, the similarity between the mouse model used here and human PB samples supports the proposed in vivo catalog of accessible regulatory elements and transcriptome presented below and constitutes a unique resource with applications to PB research and treatment.
PB progression is associated with increased chromatin accessibility elements in intergenic regions
To assess the specificity and sensitivity of our Rbp3-CCND1/Trp53−/− samples, we checked the expression of tissue-specific genes (Fig. 2A). The Rbp3 gene (Liou et al. 1990) was specifically expressed in all pineal samples but not in cerebellum tissue obtained from the ENCODE Project Consortium (The ENCODE Project Consortium 2012; Supplemental Methods) (Fig. 2A; Arno et al. 2015). Conversely, we did not detect the expression of cerebellum-specific genes, such as Gabra6 (Wang et al. 2011), in our pineal samples.
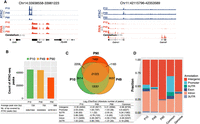
An atlas of PB transcriptome. (A) Genomic tracks of normalized RNA-seq and ATAC-seq profiles for representative loci showing the high expression of Rbp3 specific to pineal tissues (left) and Gabra6 specific to cerebellar tissues (right). Loci coordinates are indicated above. The y-axis scales for both assays are shown in parentheses. (B) Bar plot showing the number of ATAC-seq peaks called at each time point. The table below shows the average peaks size and the peak genome coverage in base pairs. (C) Venn diagram for the number of overlapping OCRs between the three time points. (D) Genomic loci annotation for unique and common OCRs at each time point highlighting the large fraction of intergenic OCRs at P90, whereas common peaks are enriched for promoter OCRs. The genome distribution of the same elements is shown for comparison. The associated table shows the log2 fold change of observed/expected and the absolute number of OCRs between parentheses. Note the increase in the fold change for intergenic elements during progression from P10 to P90 accompanied by a decrease in the log fold change for promoter.
We then called accessibility peaks from ATAC-seq data on all three time points and identified 45,015, 44,280, and 32,617 peaks at P10, P49, and P90, respectively, with an average peak size of 727 bp (Methods) (Fig. 2B; Supplemental Fig. S1C). Overall, we identified 62,831 ATAC-seq peaks that were present in at least one time point. Most of ATAC-seq peaks (21,325) were common to all three time points (Fig. 2C), and more peaks were shared between P49 and P10 (12,081 peaks) than between P90 and P10 (2209 peaks) or P49 (1183 peaks). This suggests that the regulatory genome diverged substantially at the invasive tumorigenic stage with more distinct regulatory elements compared with the proliferating and early malignant stages. Although we called fewer peaks at P90 compared with P10 and P49 (Fig. 2B), the majority of P90 peaks were intergenic with fewer intronic peaks, suggestive of a more prominent role for distal elements, enhancers, or repressors acting at the invasive tumorigenic stage of PB (Fig. 2D). It has recently been shown that tissue-specific enhancers are enriched in intronic regions (Borsari et al. 2021). As PB unfolds, the pineal gland loses tissue specificity as reflected by the increase in intergenic peaks at P90.
Last, we performed Gene Ontology analysis on target genes of time point–specific peaks as well as on peaks common to all three time points. P10-, P49-, and P90-specific peaks were enriched for terms related to axon growth and synapse regulation, reflecting the biology of the pineal gland. Additionally, target genes of P49- and P90-specific peaks, as well as common peaks, are enriched for cell–cell adhesion molecules, reflecting the gradual progression of PB toward the more invasive tumorigenic stage (Supplemental Fig. S2A). In summary, we generated a comprehensive catalog of transcriptome and regulatory elements with putative roles in PB initiation and progression.
The temporal profiles of the accessible genome are related to, and in some cases precede, transcriptional remodeling
To identify regulatory events involved in PB initiation and progression, we called differentially expressed genes (DEGs) and differentially accessible regions (DARs) at absolute log fold change (LFC) > 1 and adjusted P-value (p.adj) < 0.05 for all pairwise time point comparisons (Fig. 3A; Supplemental Tables S3–S8; Love et al. 2014). For DARs calling, we used the union of ATAC peaks identified above (62,831 peaks; Methods). We observed a total of 3621 nonredundant DEGs between any two conditions. The number of DEGs progressively increased at P49 and P90 compared with P10 (Fig. 3A). Despite the continuous postnatal maturation of the pineal gland until ∼2 yr of age in human, which is equivalent to approximately the P10 stage in mouse (Chini and Hanganu-Opatz 2021), the pineal-defining transcriptome is established before birth (Hartley et al. 2015). We thus consider that the observed transcriptional remodeling is primarily related to PB progression.
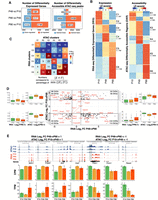
A temporal functional relationship between the regulatory genome and the transcriptome in PB. (A) The number of differentially expressed genes (DEGs; left) and differentially accessible regions (DARs; right) between pairwise time point comparisons. (B) Heatmaps of normalized values (Z-scores) of DEGs (left) and DARs (right). Unsupervised hierarchical clustering segregates the two heatmaps into six clusters each. Numbers underneath cluster names indicate the corresponding number of rows. Target genes of rows in the accessibility heatmap do not match with genes from the expression heatmap. (C) Heatmap with numbers representing the proportion of overlap between OCRs target genes in each DAR cluster (columns; clusters K1 to K6) and genes in each DEG cluster (rows; clusters C1 to C6) using the indicated formula. Color scale represents −log10 of Bonferroni-corrected P-values for the hypergeometric test. (D) Scatter plot for all genes differentially expressed between P49 and P90 and their associated DARs. The plot shows all combinations of cases in which a gene is differentially regulated at P90, whereas its regulatory region is differentially gained or closed earlier, at P49. For each quadrant, the boxplots show, for all gene-regulatory element associations, the distribution of gene expression and of ATAC signal for the accessible elements. (E) Selection of genomic tracks of normalized RNA-seq and ATAC-seq profiles showing expression and accessibility levels for four DARs gained open at P49 as their target genes are up-regulated at P90 and for three DARs gained closed at P49 as their target genes are down-regulated at P90. Loci coordinates are indicated in gray with the y-axis scales of both assays in parenthesis (ATAC-seq; RNA-seq). The CPM and TPM values for the highlighted peaks and their target genes are shown in the bar plots below. Error bars represent standard deviations from the biological replicates.
For accessible regions, the large majority of DARs was observed between P49 and P90 (Fig. 3A), reflecting the considerable change in the regulatory genome occurring during the switch from the early malignant stage (P49) to the invasive tumorigenic stage (P90). As more DEGs were observed at P90 and more DARs at P49, this indicates that the accessible genome is modulated earlier than the transcriptome and can thus be considered to infer early oncogenic events before their manifestation at the expression level. As we progress toward P90, the proportion of DEG–DAR association increases. For instance, 3% of DEGs correlate positively with their associated accessible regions between P10 and P49. This percentage increases to 11% between P49 and P90 (Supplemental Fig. S3A; Supplemental Table S9). Most DEGs have an associated accessible region that does not change significantly (absolute LFC > 1 and p.adj < 0.05) between time points. The same observation is also true when we considered DARs and their associated genes.
We next identified clusters of commonly up- or down-regulated genes and DARs by using k-means clustering on DEGs and DARs and identified, in each assay, six clusters (C1 to C6 for DEGs, and K1 to K6 for DARs) of temporal profiles with distinct functions (Fig. 3B; Supplemental Table S10). The expression clusters C1 (467 genes) and C2 (248 genes) (Fig. 3B, left panel) correspond to genes that are strongly expressed at P10 and enriched for developmental and DNA organization terms (Supplemental Fig. S2B). Genes in C3 (451 genes) showed a higher level of expression at P10 and P49 and were enriched for terms related to visual perception and tissue development. Genes in C4 (467 genes), whose expression was specific to P49, were exclusively enriched for metabolic terms. Genes in C5 (799 genes) displayed a higher expression level at P49 and P90 compared with P10 and were involved in immune response, which might correspond to genes whose expression is a consequence of tumor progression. Cluster C6 (1189 genes) represented genes with gained expression at P90 and involved in neuronal regulation.
ATAC-seq clusters, identified using k-means clustering of all differentially accessible elements, highlighted similar patterns to the expression heatmap with clusters of peaks highly accessible at P10 (K1 and K2), at P10 and P49 (K3), at P49 only (K4), or at P90 only (K5 and K6) (Fig. 3B, right panel). In contrast, there was no ATAC-seq cluster with peaks active at P49 and P90 equivalent to the pattern seen in cluster C5 in the expression heatmap (Fig. 3B, left panel). Gene Ontology analysis performed on closest target genes revealed categories related to neuron development (ATAC clusters K1 and K2), eye development (K3), retina homeostasis (K4), and synapse assembly (K5) (Supplemental Fig. S2B).
To determine if the temporal profiles of both assays are functionally related, we used a previously established metric (Inoue et al. 2019) to compare the normalized percentage of overlaps between target genes of each ATAC-seq cluster and genes from each RNA-seq cluster (Fig. 3C; Supplemental Fig. S3B; Supplemental Table S10). We observed a high degree of overlap between target genes of DARs in each cluster of the accessibility heatmap and genes from the expression heatmap for the equivalent cluster. For example, target genes of ATAC-seq DARs in cluster K3 overlapped significantly with genes in cluster C3 from the expression heatmap, suggesting that K3 DARs and C3 DEGs are functionally related (Fig. 3C). Comparable results were obtained when alternative methods were used (Supplemental Fig. S3C). Furthermore, chromatin accessibility for almost all clusters was found to be acquired first followed by an increase in gene expression of the target gene. For instance, cluster K1 significantly targets genes from clusters C1 and C3; K2 cluster, accessible at P10 and P90, is mostly associated with genes from cluster C6, highly expressed at P90; and K3 cluster targets genes in clusters C3 and C6 (Fig. 3C).
If the accessible genome is often modulated earlier than the transcription, we should observe that for genes that are up-regulated at P90, their associated accessible regions are differentially gained at P49 and similarly for genes that are down-regulated at P90, their associated accessible regions should be gained-close (GC) at P49. To test for that, we compared log2 fold changes of all genes that are differentially regulated between P49 and P90 to the log2 fold changes of their corresponding DARs between P10 and P49 (Fig. 3D,E). Thirty-eight genes that are up-regulated at P90 have their accessible regions gained-open at P49 (Fig. 3D, top right quadrant), and 43 genes that are down-regulated at P90 have their accessible regions gained close at P49 (Fig. 3D, lower left quadrant). The analysis also highlighted cases in which gene expression and their associated regulatory regions do not correlate. As enhancers and repressors coordinate to regulate the expression of their target genes in space and time, the presence of gained-open (GO) or GC regions associated with down- and up-regulated genes, respectively, is expected.
The above shows that temporal profiles identified from ATAC-seq and RNA-seq could be functionally related, allowing us to investigate the gene regulatory network governing PB progression. Major PB-related genes were targeted by DARs (Supplemental Fig. S3D). For instance, two GC DARs at P90 target Drosha are accompanied by a decrease in expression of the gene at P90. Similarly, Pik3r1 is targeted by several GC DARs and is significantly down-regulated at P49 (LFC = −1.67, P-adj = 5.6 × 10−4) and P90 (LFC = −1.17, P-adj = 1.5 × 10−2) compared with P10. Dgcr8 is associated with a GC DAR at its promoter with decreased accessibility at P90 without an impact on the expression of the gene. Last, Dicer1 is targeted by a GO DAR at P90 also without a significant effect on its expression despite a slight increase at P49 compared with P10 (Supplemental Table S12).
Together, the analysis suggests a temporal regulation of the transcriptome by chromatin accessibility and delivers the first and unique resource of genome-wide in vivo regulatory elements during PB development.
PB development is associated with a progressive increase in the frequency of structural variations with distinct events marking PB early malignant and invasive tumorigenic stages
The gradual increase in differential expression profiles during PB progression suggests a substantial deregulation of the transcriptome during cancer progression. Pediatric tumors are characterized by high incidences of structural variants (Chen et al. 2014b), leading to CNV and fusion events. To characterize CNVs and fusion events in PB, we used CaSpER (Serin Harmanci et al. 2020) and called amplification and deletion events at P49 and P90 with P10 as a benchmark. As cancer progresses, the number of amplification and deletion events increased progressively, with a higher number of CNV events observed in P90 replicates indicating a higher degree of DNA dysregulation and structural damage (Fig. 4A). We narrowed down the list of identified CNVs by considering events that were common to replicates from each time point and whose affected genes were differentially expressed (Fig. 4B). All CNV events initiated at P49 were also detected at P90, whereas P90 was characterized by an additional set of unique CNVs. This suggests that common events in P49 and P90 are potential markers for PB initiation (P49) and progression (P90), as recurrent genetic alterations detected across several replicates indicate that these are common driving events for tumor development (García-Chequer et al. 2016).
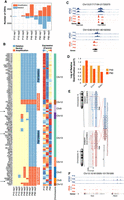
PB is marked by a progressive increase of DNA structural variants. (A) Bar plot highlighting the number of gene amplification (red bars) and deletions (blue bars) for each of the three biological replicates at P10, P49, and P90 time points. (B, left) Heatmap indicating the deletion (blue) and amplification (red) events common between all biological replicates at P49 and/or P90 and associated with differential gene expression of the implicated genes. (Right) Z-score normalized mean expression profiles of the predicted deleted or amplified genes. Rightmost bar indicates the chromosome where the deletion or amplification event took place. Arrows indicate genes that were tested by RT-qPCR. (C) Genomic tracks of normalized RNA-seq and ATAC-seq profiles showing differential down-regulation and decreased accessibility of histone genes H3c10 and H2bc14 and Gas1 in line with predicted deletion events at P49 and P90, respectively. (D) RT-qPCR on three different genes validating the observation from 4B. (E) Graphical representation as plotted by Arriba (Uhrig et al. 2021) of Tc2n-Tc2n tandem duplication in a P90 biological replicate. (F) Genomic tracks of normalized RNA-seq and ATAC-seq profiles of Tc2n showing differential up-regulation of Tc2n at P49 and P90 in comparison to P10.
Furthermore, all deletion events initiated at P49 and common to P90 were located on Chromosome 13 and affected a cluster of 29 histone coding genes covering a locus of 2 Mb (Fig. 4B–D). Down-regulation of histone genes could be related to cancer initiation (Vardabasso et al. 2014) or an altered response to DNA damage (Celeste et al. 2003). We also identified several deletion events at P90, including a 7-Mb deletion affecting the tumor suppressor gene growth arrest specific 1 (Gas1) (Li et al. 2016; Sarkar et al. 2018). Conversely, several amplification events were detected at P49 and P90 and affected genes with known roles in cancer. For instance, Hs1bp3, which negatively impacts autophagy (Holland et al. 2016), a process known to be frequently disabled at the early stages of cancer (White 2012), was targeted by an amplification at P49 same as Rhob, known for its oncogenic roles in glioblastoma (Skuli et al. 2006).
The expression profiles of several other affected genes did not match the observed CNV event. Some genes undergoing amplification events were not up-regulated, and conversely, some genes undergoing deletion events were not down-regulated. In both cases, this is likely owing to the sliding window-based median filtering approach used by CaSpER to call CNV events or owing to autoregulatory feedback loops and redundant mechanisms acting to buffer against loss or gain events of such genes.
Last, we identified gene fusion and tandem duplication events using Arriba (Methods) (Supplemental Table S11; Uhrig et al. 2021) by keeping cases that were common to all replicates at each time point. Only one common and high-confidence tandem duplication was identified at P90 and P49, comprising Tc2n (Tc2n-Tc2n duplication), which was not observed at P10 (Fig. 4E). The duplication breakpoint at P49 and P90 replicates occurs at almost the same location, causing an out-of-frame fusion in the C2 domain in all samples. Tc2n is significantly up-regulated in both P49 and P90 in comparison to P10 with LFC > 5 (Fig. 4F; Supplemental Tables S3–S5). Tc2n can promote cancer by disrupting the TRP53 pathway or modulating NF-kB signaling (Hao et al. 2019a,b; Xu et al. 2021).
PB-associated regulatory elements are enriched for motifs of repressors and significantly overlap with repressive state elements as defined by ChromHMM
To build a gene regulatory network governing PB progression, we first grouped DARs from Figure 3A into GO and GC OCRs at each time point comparison and assessed their degree of overlap (Supplemental Fig. S4A). Most of the regions that are GO at P90, gained their accessibility at P49, as reflected by the increased overlap (63%) between P10 → P90-GO and P49 → P90-GO. The proportion of overlap between OCRs that gained accessibility at P49 (P10 → P49-GO) and those that lost accessibility at P90 (P49 → P90-GC) was high (59%), indicating a putative regulatory role for these OCRs in the switch from the early malignant to the invasive malignant stage. To determine the possible functional role of the GC and GO OCR groups defined above, we identified the TFs acting on them using DNA motif enrichment analysis and identified four main clusters of enriched TF motifs (Fig. 5A,B; Supplemental Table S13). We also performed the same on peaks unique to P10, P49, and P90 and on common peaks (Supplemental Fig. S4D).
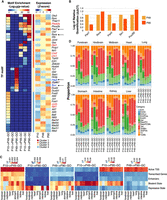
Temporal dynamics of PB genomic accessibility profiles revealing clusters of time point–specific putative transcriptional regulators. (A, left) Heatmap of TF motifs enriched in different GO and GC OCR groups. (Right) Z-score normalized mean expression values of the corresponding TFs at P10, P49, and P90. Red and blue fonts depict differentially up- and down-regulated TF between P10 and P90, respectively. Arrows indicate the list of genes tested by RT-qPCR. (B) RT-qPCR validation for a select list of genes from panel A and from Supplemental Figure S4D (for Pou3f2). (C) Heatmaps of Jaccard indices for the six GO and GC OCR groups in nine ChromHMM annotated mouse tissues from ENCODE (Supplemental Methods) with respect to five ChromHMM categories. (D) Proportion of overlap between each of the GO and GC OCR groups and ChromHMM subcategories. The stacked bar plot shows that P10 → P90−GO and P49 → P90−GO categories have the highest overlap with Heterochromatin state regions (dark green).
The first cluster corresponds to TF motifs enriched in OCRs that gained accessibility at P49 (P10 → P49-GO) and lost accessibility at P90 (P10 → P90-GC and P49 → P90-GC). For instance, 59.3% of OTX2 and 74.7% of RORC motifs found in P10 → P49-GO were also found in P49 → P90-GC (Supplemental Table S13). Despite being differentially down-regulated at P90 compared with P10 (LFC = −0.99, P-adj = 1.16 × 10−8) (Supplemental Table S4), Otx2 remains highly expressed across all time points (Fig. 5A). High expression of Otx2 is specific to Group 3 and Group 4 medulloblastoma (Adamson et al. 2010) in comparison to WNT or SHH subtypes, leading to silencing of its direct downstream target Pax3, known to promote cell differentiation and inhibit tumorigenic properties in Group 3 medulloblastoma (Zagozewski et al. 2020). Pax3 is also differentially down-regulated at P90 (LFC = −4.60, P-adj = 3.60 × 10−19) (Supplemental Table S4), suggestive of a potential role of the Otx2/Pax3 regulatory axis in PB.
The second cluster includes TF motifs enriched in OCRs that lost accessibility at P90 (P10 → P49-GC and P10 → P90-GC) such as PROP1, LHX1, and LHX2, with the latter two being differentially up-regulated (Supplemental Tables S3, S4) and known to be linked to cancer metastasis (Dormoy et al. 2011; Kuzmanov et al. 2014).
The third cluster contains motifs enriched in OCRs with gained accessibility specifically at the invasive tumorigenic stage P90 (P49 → P90-GO), including GLI2, STAT1, and HINFP. For instance, 76.2% of GLI2 and 85.1% of HINFP motifs were specific to P49 → P90-GO OCRs compared with P10 → P90-GO (Methods) (Supplemental Table S13), suggesting a potential role of these TFs as PB progresses. Among the top 10 enriched KEGG pathways for this cluster were “transcriptional mis-regulation in cancer,” “pathways in cancer,” and “PD-L1 expression and PD-1 checkpoint pathway in cancer” (Supplemental Fig. S5), which control both the induction and the maintenance of immune tolerance within the tumor microenvironment (Han et al. 2020) as an adaptive mechanism to evade immune surveillance (Ohaegbulam et al. 2015). Both Pdcd1 and Cd274 (PD-1 and PD-L1 orthologs in mouse, respectively) are differentially up-regulated at both P49 and P90 compared with P10 (LFC of 3.89 and 1.56, respectively) but not between P90 and P49 (Supplemental Tables S3, S4). Additionally, the CD47 immune evasion marker, found on the cancer cell surface of many solid tumors and suggested to be especially enriched in pediatric brain tumors (Gholamin et al. 2017), was also differentially up-regulated at P49 and P90 compared with P10 (LFC of one and 1.4, respectively).
The fourth cluster of TFs enriched motifs corresponds to motifs in OCRs with gained accessibility at P49 (P10 → P49-GO). Among the enriched TF motifs is FOSL2, one of the six master TFs responsible for regulating the mesenchymal gene signature in high-grade glioma (Carro et al. 2010) and whose gene expression, along with Stat3, was differentially up-regulated at P90 compared with P10 (LFC of 1.18 and 1.23, respectively) (Supplemental Table S4).
We observed a positive correlation between target gene expression and promoter-overlapping GO/GC elements (Supplemental Fig. S6). For instance, target genes of P10 → P49-GO elements have higher expression at P49 compared with P10. This correlation was less pronounced when all elements (i.e., including distal elements) were considered possibly owing to the presence of repressive distal elements and to the putative false association of some GO/GC elements with the closest gene as target.
Five out of the nine motifs (YY1, GLI2, HINFP, ZFP867, and SIX1) enriched in P49 → P90-GO regions are known repressors or corepressors, suggesting that accessible regions acquired at P90 correspond to repressive elements. To check for this, we classified our GO and GC regions using previously generated chromatin states from ChromHMM (Ernst and Kellis 2012) on nine mouse tissues (van der Velde et al. 2021), including the midbrain, which represents the most adjacent structure to the pineal gland. All three categories of elements that became accessible as cancer progresses (P10 → P49-GO, P10 → P90-GO, and P49 → P90-GO) were enriched for “repressive state,” particularly the P10 → P49-GO and P49 → P90-GO categories (Fig. 5C). This supports the finding above that P90-acquired OCRs, particularly those acquired at the malignant stage (P49 → P90-GO), correspond mainly to repressive elements enriched for motifs of repressor TFs. Conversely, regions that were gained-close as cancer progresses (P10 → P49-GC, P10 → P90-GC, and P49 → P90-GC) were enriched for “bivalent” and/or “active transcription start site (TSS)” states, possibly helping in maintaining the epigenetic plasticity of the cancer genome as recently proposed (Kumar et al. 2021). When time point–unique or common peaks were considered, P90-acquired OCRs were also enriched for “repressive state” regions, particularly in the midbrain (Supplemental Fig. S4D), whereas P10, P49, and common peaks were enriched for “bivalent,” “enhancer/active TSS,” and “bivalent/active TSS” states, respectively.
As both facultative and constitutive heterochromatins, marked by H3K27me3 and H3K9me3, respectively, constitute the repressive state in ChromHMM, we checked which is the major contributor in our case by calculating the proportion of overlap between each of the GO and GC OCR groups and ChromHMM subcategories (Fig. 5D). For both, P10 → P90−GO and P49 → P90−GO categories, the proportion of overlap with heterochromatin state elements was higher than the remaining categories and for all tissues tested (Fig. 5D, dark green; Supplemental Fig. S4B). In contrast, active TSS and bivalent state elements had the highest overlap in the remaining categories. This suggests that gained repressive state elements correspond mainly to heterochromatin regions that became accessible as cancer progresses. It has been shown that constitutive heterochromatin, which is generally associated with the stable maintenance of gene silencing, is also dynamically regulated in response to stimuli, including cancer (Wang et al. 2016). Additionally, depletion of YY1, whose motif is enriched in P49 → P90-GO regions, is known to alter the structural integrity of heterochromatin by rendering it more accessible (Wu et al. 2009).
Only 6% of P10 → P90−GO or P49 → P90−GO overlapped with ENCODE candidate cis-regulatory elements (cCREs) that are identified by either histone modifications (H3K4me3 and H3K27ac) or CTCF-binding data. This percentage ranged between 48% and 77% for the remaining categories (Supplemental Table S14). As cCREs are combined from all cell types, the enrichment for repressive state of both categories above is most likely specific to PB GO elements.
Last, we determined whether the six categories overlap with histone marks associated with active or repressed chromatin, independent of ChromHMM. For that, we compiled available ENCODE peaks for the following histone marks generated on mouse cell lines: H3K27ac (marks active promoters and enhancers), H3K4me1 (marks enhancer regions), H3K27me3 (repressive mark, usually on gene body), H3K9me3 (marks constitutive heterochromatin), H3K4me3 (marks actively transcribed promoters), and H3K36me3 (marks actively transcribed gene bodies) (Supplemental Table S18). For all the seven cell lines for which these histone marks were available, P10 → P90-GO and P49 → P90-GO regions have the highest overlap with H3K9me3 mark (Supplemental Fig. S4C).
In summary, our findings suggest that as PB progresses, newly gained accessible elements correspond mostly to repressive heterochromatin elements. Corepressors play a major role in conferring rigidity to cancer cells by modulating condensed chromatin states at gene regulatory elements (Battaglia et al. 2010; Czerwińska et al. 2017; Sobocińska et al. 2021).
A PB catalog of in vivo active enhancers and SEs
The increase in intergenic CREs at the tumorigenic stage (P90) (Fig. 2D) suggests that enhancers play a key role in PB progression. As H3K27ac is an established mark for active enhancers, we performed H3K27ac ChIP-seq at P90 and identified active enhancers implicated in PB tumorigenesis (Methods). H3K27ac profile at TSS of expressed genes showed a typical bimodal peak with increased enrichment at +1 nucleosome (Methods) (Fig. 6A). In total, we identified 49,468 high-confidence H3K27ac peaks at P90. Accessible regions unique to P90 lacked H3K27ac signal as opposed to P90 peaks that were shared with other time points (Fig. 6B), potentially owing to the enrichment of GO and P90 unique peaks (not shared with other time points) for repressive elements as shown above.
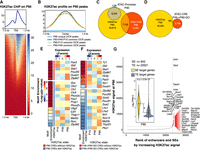
Catalog of PB-related enhancers and super-enhancers (SEs). (A) Aggregated signal and heatmap of P90 H3K27ac ChIP-seq over the TSS of expressed genes showing the expected bimodal profile. (B) Aggregated P90 H3K27ac ChIP-seq signal at P90 unique and common OCRs to P90. (C,D) Venn diagrams showing the overlap between P90 OCRs at promoters and CREs for P90 H3K27ac ChIP-seq peaks and the overlap between P49 → P90-GO CREs and P90 H3K27ac peaks. (E, left) Bar indicates TF motif enrichment in P90 CREs. (Right) Heatmap represents Z-score normalized mean expression values of the corresponding TFs at P10, P49, and P90. (F) P49 → P90-GO CRE peaks were split into those marked with H3K27ac (blue) and those without (red). TF motifs enrichment was performed on each category (left bar). Heatmaps of normalized gene expression for each TF is also shown. (G) H3K27ac peaks at P90 were ranked by ROSE2 to identify TE and SE. Annotated dots correspond to the top 25 identified SEs and PB-specific SEs (red dots). Inset represents violin plots for the normalized expression levels of SE (yellow) and TE (blue-gray) gene targets.
Ninety-eight and one-half percent (98.5%; 8304 out of 8430) of P90 ATAC peaks at promoters were marked with H3K27ac signal (Fig. 6C), whereas only 30.4% (7346 out of 24,187 total CREs; P < 2.2 × 10−16) of P90 ATAC peaks at distal CREs at P90 were marked with H3K27ac. Although other acetylation marks associated with active enhancers can be found enriched on distal regions (Taylor et al. 2013; Pradeepa et al. 2016), our findings suggest that the majority of acquired accessible regions at P90 do not correspond to active enhancers. The set of 7346 CREs corresponding to active distal enhancers at P90 were enriched for TF motifs including three differentially up-regulated TFs at P90, Pax7, Phox2a, and Olig2 (Fig. 6E). The expression of Olig2 is limited to neural progenitor cells and oligodendrocytes and is rapidly down-regulated upon neuronal specification (Ligon et al. 2004). It is an important TF in the development of brain cancers, specifically gliomas (Mehta et al. 2011), and plays thus a potential role in PB development.
When P90 GO regions were considered, a minority of 3.1% (185 out of 5980 regions) had the H3K27ac mark and contained motifs with known oncogenic role such as LHX1/2 (Fig. 6F). Conversely, most P49 → P90-GO regions lack H3K27ac (96.9%, 5795 out of 5980) and were enriched for the repressor or corepressor motifs identified in P49 → P90-GO regions above (Fig. 5A).
Last we called typical-enhancers (TEs) and SEs implicated in PB using the ranked ordering of super-enhancers (ROSE) algorithm (Whyte et al. 2013) and identified 22,521 and 842 TE and SE at P90, respectively (Fig. 6G) (Supplemental Tables S15, S16). To identify PB-specific SEs, we intersected SEs with a set of 281,310 defined ENCODE CREs (Supplemental Methods) and identified 37 intergenic SEs at P90 (Fig. 6G, red dots), targeting genes with known role in the biology of the pineal gland like Crx and Otx2. As expected, SE target genes have higher expression than TE target genes (two-sided Wilcoxon test P < 2.2 × 1016).
Discussion
In this study, we present the first in vivo catalog of regulatory elements and gene expression of PB spanning three key time points in cancer initiation and progression: the proliferating, early malignant, and invasive tumorigenic stages. We have identified clusters of DARs and DEGs with distinct functional annotations that were relevant to each time point and explored the relationships between these clusters. We concluded that the regulatory genome diverges more and often earlier than the transcriptome, a characteristic that is underexplored in cancer biology. Using RNA-seq data, we investigated the extent of copy number and structural variation as PB progressed and identified candidate events implicating genes with known roles in cancer initiation and progression such as histone coding genes, Rhob, and Tc2n.
Eleven percent (11%) of DEGs correlate significantly with their targeting accessible region between P49 and P90. Sheffield and colleagues (2013) showed that only 20% of sites correlated significantly with at least one gene, within 100 kb. Using our conservative approach of linking one accessible region to the closest gene only, we are underestimating the correlation in our case. It has been shown that the closest gene is not always a good predictor for target gene (Snetkova and Skok 2018), and distal regulatory elements are also frequent (Sanyal et al. 2012; Anene-Nzelu et al. 2020).
A slight change in regulatory elements activity (e.g., enhancers and repressors) can have major impact on target genes, mainly TFs. Also, synergistic and additive actions of mammalian enhancers (Choi et al. 2021), distance to target gene, epigenetic effects, and post-transcriptional modifications all contribute to gene expression regulation.
Among the TFs enriched at DARs, several have known oncogenic activity and are highly expressed or up-regulated at P90 compared with P10 and/or P49, including OTX2, FOSL2, and STAT3, with the latter known to promote stemness, survival, and proliferation of cancer cells (Ligon et al. 2004). Additionally, pathway analysis of these clusters of TFs indicated a potential role for the modulation of adaptive immunity in PB that was further supported by the differential up-regulation of Pdcd1, Cd274, and Cd47 at P90, all of which are known therapeutic targets of cancer immune therapy. We also identified key transcriptional repressors enriched at the invasive tumorigenic stage and targeting regions not marked with H3K27ac. We hypothesized that these regions correspond to repressive state elements, which we further assessed using ChromHMM. Many of these TFs correspond to repressors or corepressors (YY1, HINFP, ZFP867). The role of such factors is well known in cancer biology, contributing to transcriptional rigidity and resulting in maintaining a condensed chromatin structure at key promoter and enhancer elements (Battaglia et al. 2010; Czerwińska et al. 2017; Sobocińska et al. 2021). Although chromatin states used to draw these conclusions were defined on non–pineal gland tissues, they serve as a good proxy for predicting states in our samples mostly because we observe the same state in all nine tissues with available ChromHMM predictions in ENCODE, in addition to the high overlap with the H3K9me3 from eight different cell lines with available histone ChIP data from ENCODE. The enrichment for the repressive state in GO accessible regions at P90 may reflect partial DNA methylation as observed previously in breast cancer (Hon et al. 2012) with many affected target genes including tumor suppressors like Dicer1. Conversely, Rb1 and Drosha are targeted by regions that were gained-close at P90, and their expression decreased in line with the decreased expression of both genes observed in PB (Snuderl et al. 2018). The simultaneous GO and GC events at P90 targeting PB-related genes suggest that different epigenetic alterations are involved in the pathogenesis of PB.
The Rbp3-CCND1/Trp53−/−-driven PB model is appealing in addressing epigenetic mechanisms underlying PB progression, because, although none of the genes directly related to PB including Rb1, Drosha, and Dicer1 is genetically targeted, the expression of all three genes was affected to different extents at P90. It has been suggested that dysregulated CCND1 may signal to the Cdkn2a tumor suppressor to induce apoptosis in response to abnormal proliferation signals (Zhu et al. 2013). Indeed, Cdkn2a was overexpressed at P90 versus P10 (LFC of 2.67 and a P-value of 2.1 × 10−5) (Supplemental Table S3).
Although Trp53 in not mutated in primary PB, it is still not clear if its pathway remains intact or disrupted (e.g., by Trp53 deregulation). We showed previously that the progression to full-blown PB is associated with increased TRP53 immunostaining, suggesting a role for a stabilizing Trp53 mutation in human PB (Saab et al. 2009). Moreover, because of genetic compensations, mouse models almost invariably require additional mutations in Trp53 or other genes (often gene homolog) to elicit defects observed in humans. For example, RB loss alone in the retina does not induce RB as in humans and requires additional mutations in mouse orthologs Rbl1 (also known as p107) and Rbl2 (also known as p130) (Macpherson 2008). Likewise, RB loss alone in the pineal gland does not induce PB and requires Trp53 mutation to induce PB (Chung et al. 2020). CCND1 is upstream of pRB, hence the requirement for a similar mutation in Trp53. Thus, the requirement for Trp53 mutation in PB may be species specific (mouse vs. human), or indicative of a role for this tumor-suppression pathway in PB progression.
We were unable to track the dynamics of active enhancers over time owing to the lack of H3K27ac data at P10 and P49. We had anticipated the scarcity of chromatin that could be extracted at P10 and developed a machine learning algorithm to predict the H3K27ac state in time-series experiments using H3K27ac data from two available time points (Hallal et al. 2021). However, we constantly faced unfortunate events in obtaining samples with good enrichment data at P49, which prevented us from using our model. Using available H3K27ac ChIP-seq data, we located and characterized P90 active enhancers marked by both intergenic accessibility and a H3K27ac histone mark and uncovered several up-regulated TFs with enriched TF binding sites in these enhancers, including OLIG2, a neural progenitor marker that is known to be consistently expressed in gliomas and astrocytomas (Ligon et al. 2004).
We anticipate that our findings from gene expression and chromatin accessibility data into tumor-progression mechanisms might be applicable to other cancer models. The model used here showed high similarity to other PB mouse models as well as human PB samples, supporting the applicability of our findings to human PB.
In conclusion, we generated the first in vivo transcriptome and accessibility maps during progression of PB. These data should contribute to the identification of the mechanisms triggering PB progression, yielding new therapeutic targets and approaches for the treatment of PB.
Methods
Mouse studies
Rbp3-CCND1 transgenic mice (Skapek et al. 2001) were obtained as a gift from Dr. Stephen Skapek at St Jude Children's Research Hospital and bred with Trp53−/− mice (Jackson Laboratory, Maine) and maintained in a mixed C57BL/6 × 129/Sv genetic background. PCR for targeted alleles was used to verify mouse genotypes as previously described (Latres et al. 2000; Skapek et al. 2001; Christophorou et al. 2005). Animals were examined at least twice weekly and were euthanized at defined time points (P10, P49, and P90 corresponding to proliferating, early malignant, and invasive tumorigenic stages) or when obviously ill in accordance with the American University of Beirut (AUB) institutional animal care and use committee guidelines; all studies were approved by this committee.
ATAC-seq and ChIP-seq samples preparation
Nuclei were subjected to transposition reaction using a Nextera DNA sample preparation kit (Illumina FC-121-1030) and processed as previously described (Supplemental Material; Buenrostro et al. 2015). For chromatin isolation, tissues were disrupted as for ATAC-seq (Supplemental Material) and cross-linked with 1% formaldehyde on an orbital rotator for 10 min at RT and subsequently quenched by 0.125 M glycine for 5 min at RT with mild rotation. Cross-linked tissues were lysed followed by nuclear lysis to release nuclear chromatin. Chromatin was sonicated in the nuclear lysis buffer into 200- to 700-bp fragments, and chromatin fragment sizes were checked using 1% conventional agarose gel. The detailed H3K27ac ChIP protocol is described in the Supplemental Material. All ChIP steps were performed in a cold room. The efficiency of immunoprecipitation was evaluated by RT-qPCR using positive and negative primers targeting mouse genomic regions (Supplemental Material).
RNA-seq, ATAC-seq, and ChIP-seq data analyses
We performed a quality check on raw reads followed by alignment and removal of duplicates, unmapped reads, secondary, and supplemental alignments for ChIP-seq samples (Supplemental Material).
For ATAC-seq, adapter trimming was performed before alignment. The best two representative samples for each time point P10, P49, and P90 were selected for downstream analysis taking into consideration the pairwise irreproducible discovery rate (IDR) values, TSS enrichment, and the fraction of reads in peaks (FRIP) metric.
For RNA-seq, genes whose sum of raw reads across all nine replicates was greater than 10 reads were considered as expressed. Differential expression analysis was performed using DESeq2 v1.26.0 (Love et al. 2014) in R (R Core Team 2021), and genes with an absolute log2 fold change > 1 and a P-adjusted < 0.05 were considered differentially expressed.
Consensus ATAC-seq peaks at each time point were selected using the following approach: First, alignment BAM files for the selected two replicates representing each time point were merged and peaks called using MACS2 with the same parameters as before. Next, peaks called using the merged BAM file and found in at least one of the two individual replicates were retained in the final consensus peak set for each time point. Consensus peaks selected for each time point were merged using BEDTools (Quinlan and Hall 2010) v2.30.0 to form a union set of peaks for downstream differential accessibility analysis. Raw read counts over the union set of peaks were generated using featureCounts (Liao et al. 2014) v2.0.1.
Data access
All raw and processed sequencing data generated in this study have been submitted to the NCBI Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/) under accession number GSE180868.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
We thank Sara Sinno for the histopathology classification of the mouse pineal tissues and tumors and all members of the Khoueiry laboratory for discussions and comments. This work was technically supported by the AUBFM animal facility and the Pillar Genomics Institute. This project was supported by the American University of Beirut (AUB) (Medical Practice Plan MPP 320154); American University of Beirut (103487 to P.K.); and the National Council for Scientific Research (103509 to P.K.).
Author contributions: P.K. conceived the study. S.I. performed NGS optimization and experiments. H.Z. performed mouse breeding, pineal gland extraction, and RT-qPCR. P.K., M.H., A.E-K., P.E.D.C., and D.M.A.G. performed computational experiments and generated the figures. P.K. and R.S. oversaw the study. All authors interpreted the data and wrote the manuscript.
Footnotes
-
↵14 Co-senior authors.
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.277037.122.
- Received June 17, 2022.
- Accepted January 11, 2023.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see https://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








