
De novo transcriptome assembly of mouse male germ cells reveals novel genes, stage-specific bidirectional promoter activity, and noncoding RNA expression
Abstract
In mammals, the adult testis is the tissue with the highest diversity in gene expression. Much of that diversity is attributed to germ cells, primarily meiotic spermatocytes and postmeiotic haploid spermatids. Exploiting a newly developed cell purification method, we profiled the transcriptomes of such postmitotic germ cells of mice. We used a de novo transcriptome assembly approach and identified thousands of novel expressed transcripts characterized by features distinct from those of known genes. Novel loci tend to be short in length, monoexonic, and lowly expressed. Most novel genes have arisen recently in evolutionary time and possess low coding potential. Nonetheless, we identify several novel protein-coding genes harboring open reading frames that encode proteins containing matches to conserved protein domains. Analysis of mass-spectrometry data from adult mouse testes confirms protein production from several of these novel genes. We also examine overlap between transcripts and repetitive elements. We find that although distinct families of repeats are expressed with differing temporal dynamics during spermatogenesis, we do not observe a general mode of regulation wherein repeats drive expression of nonrepetitive sequences in a cell type–specific manner. Finally, we observe many fairly long antisense transcripts originating from canonical gene promoters, pointing to pervasive bidirectional promoter activity during spermatogenesis that is distinct and more frequent compared with somatic cells.
Mammalian spermatogenesis is the process by which germ cells undergo progressive differentiation to generate mature male gametes. In the mouse, a pool of undifferentiated spermatogonia is recruited with regular periodicity into this process (de Rooij and Russell 2000). Once committed to differentiation, these spermatogonia undergo, as a pool of transit amplifying cells, six highly regulated mitotic divisions, thereby greatly increasing the number of sperm produced from each stem cell. Following these divisions, differentiated spermatogonia undergo a final (premeiotic) DNA replication to produce preleptotene spermatocytes. These cells then progress through a lengthy meiotic prophase, spending a protracted time in pachytene (Oakberg 1956b). Following a relatively rapid completion of the two meiotic divisions, four haploid round spermatids (RSs) are formed from each spermatocyte. These cells then undergo an extensive differentiation process (known as spermiogenesis), during which nuclei elongate, generating so-called elongating spermatids (Oakberg 1956a). These cells then further differentiate, leaving the testis for the epididymis as immature spermatozoa.
During postreplicative spermatogenesis, a wide variety of processes occur requiring distinctive transcriptional programs and regulation (Fig. 1A). In early meiotic prophase, DNA double-strand breaks (DSBs) are formed as a necessary intermediate for meiotic recombination and the subsequent reductional chromosome division during meiosis I (Keeney 2008). In somatic cells, regions surrounding DSBs are known to undergo transcriptional silencing via recruitment of Polycomb group proteins (Shanbhag et al. 2010; Ui et al. 2015), although recent studies suggest that this response depends on the specific site of the lesion relative to transcription initiation (Vitor et al. 2019). Chromatin surrounding persistent DSBs is associated with the presence of H2AK119ub1 and SUMOylated H2A.Z (Huen et al. 2007; Mailand et al. 2007; Kalocsay et al. 2009), but these markers are not found around meiotic DSBs in leptotene or zygotene spermatocytes (Inagaki et al. 2010).
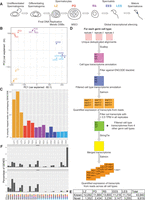
Overview and generation of novel spermatogenic transcriptome. (A) Schematic of spermatogenesis in the adult mouse testis. Key events involving transcriptional regulation are highlighted under the graphic. The following cell types were analyzed in this study: leptotene/zygotene (LZ)- and pachytene/diplotene (P/D)-stage spermatocytes and round (RS), early elongating (EES), and late elongating (LES) spermatids. (B) PCA plot using log2(RPKM) values for genes annotated in GENCODE. (C) Bar plot showing the fraction of uniquely mapped reads that are not overlapping regions previously annotated as genes in GENCODE. (D) Schematic overview of the de novo transcriptome assembly approach. (E) Table showing number of genes found in each cell type. Known indicates any overlap with a previously GENCODE annotated gene, and novel genes do not possess any such overlap. (F) Bar plot showing expression patterns of known and novel genes in spermatogenesis. Colored balls under plot indicate cell type(s) in which the genes are expressed (with gray representing not expressed).
Early prophase spermatocytes also show increased expression of retrotransposons, although different tubules within the same testis show highly variable levels of activity (Branciforte and Martin 1994; Soper et al. 2008; Brown et al. 2010). Transcription of these genetic elements is generally strongly repressed by several mechanisms (Di Giacomo et al. 2013) to prevent their transposition, which could cause increased mutation rate (Gardner et al. 2019). Why germ cells express these elements and whether their expression is important for spermatogenesis are unknown.
Later during meiotic prophase, in pachynema and diplonema, the sex chromosomes, lacking partners for their homologous synapsis, undergo global transcriptional silencing, a process known as meiotic sex chromosome inactivation (MSCI) (Turner 2007). MSCI is dependent on several proteins involved in DNA damage response (Turner 2007), and the chromatin state established during this process has been found to persist even after meiosis, resulting in partial repression of the X and Y Chromosomes in spermatids (Namekawa et al. 2006). Some work has suggested that some X-linked transcripts (particularly microRNAs) can escape from MSCI (Song et al. 2009), although more recent work has argued against this (Royo et al. 2015).
Following meiosis, RSs express a variety of transcripts unique to the male germ line. Some of these transcripts remain translationally repressed for several days to weeks (Schäfer et al. 1995; Cullinane et al. 2015), a process that is essential for completion of spermatogenesis (Lee et al. 1995). As these spermatids differentiate into elongating spermatids, these transcripts become translationally activated (Kleene 1989, 2013). This activation is thought to be required because as spermatids undergo nuclear elongation, they also undergo genome-wide transcriptional repression (Kierszenbaum and Tres 1975). LES nuclei are characterized by a highly compacted structure with a chromatin composition very different from all other eukaryotic cells, dominated by protamines, a class of spermatogenesis-specific DNA packaging proteins, with only few histone proteins retained. It is hypothesized that this dense chromatin structure is inhibitory for transcription and causes the silencing observed in these cells.
Globally, the mammalian testis has been shown to express the highest number of different transcripts out of all examined adult tissues (Ramsköld et al. 2009), with the bulk of these transcripts being found in postreplicative germ cells (Soumillon et al. 2013). This transcriptional diversity has been suggested to provide fodder for the rapid evolution of new genes (Kaessmann 2010). Another hypothesis is that wide-spread transcription in germ cells serves to strengthen transcription-coupled DNA repair pathways to decrease germ line mutation rates (Xia et al. 2020), although a recent analysis of the types of mutations found in human germ cells suggests that this may not be a major driver (Moore et al. 2021).
The catalog of transcripts known to be expressed during spermatogenesis reflects multiple historical features. Early approaches to gene identification used low-throughput sequencing of cDNA libraries derived from various tissues (Adams et al. 1991). In the context of murine spermatogenesis, these libraries were largely generated from whole adult testes (Kerr et al. 1994). Given the differential duration of various stages of spermatogenic differentiation, some cell types, for instance, RSs, are expected to be highly overrepresented in these libraries, whereas rarer populations may be underrepresented (Oakberg 1956b). Second, many gene prediction algorithms rely on conservation to identify putative genes (Mironov et al. 1998; She et al. 2011; Keilwagen et al. 2018). Thus, recently evolved transcripts are expected to be underrepresented among known genes. Finally, most gene prediction programs also emphasize the presence of spliced sequences (Burge and Karlin 1997) and an absence of overlaps with repetitive sequences (Yandell and Ence 2012). Altogether, these features suggest that the existing transcriptome annotation may miss genes that (1) are specifically expressed in relatively rare cell types, (2) are not conserved, (3) are monoexonic, and (4) are overlapping repeat elements. A recent study (Gamble et al. 2020) generated an expanded testicular transcriptome annotation using RNA-seq generated from whole juvenile (25-d postpartum [dpp]) testes; however, this study did not examine cell type–specific expression (particularly excluding elongating spermatids, as these cells are not present at 25 dpp) or the contribution of repetitive sequences to the spermatogenic transcriptome.
We set out to create a more complete catalog of genes expressed during postreplicative spermatogenesis by isolating several specific cell types, performing a deep-coverage, paired-end RNA-seq analysis and generating a de novo annotation for genes expressed in these cell types.
Results
Global transcriptional profiling of postreplicative germ cells
Using our newly developed FACS-based isolation strategy (Gill et al. 2022), we collected one to 10 million cells from 3-mo-old C57BL/6J mice from five distinct stages of postreplicative development (Fig. 1A): leptotene/zygotene (LZ) spermatocytes (which are in the early stages of meiotic prophase), pachytene/diplotene (PD) spermatocytes (which are in the late stages of meiotic prophase), RSs (which are early, transcriptionally active postmeiotic cells), and early and late elongating spermatids (EES and LES, respectively; which are postmeiotic cells undergoing global transcriptional silencing and histone-to-protamine exchange). We isolated total RNA from these cells and performed a deep-coverage, paired-end RNA-seq experiment (Supplemental Fig. S1A). After aligning, deduplicating, and filtering for uniquely mapped reads, we quantified expression levels at the gene level of the comprehensive GENCODE v19 annotation. Principal component analysis (PCA) showed a clear separation of cell types, with the exception of the two elongating spermatid populations (EES and LES). These cell types undergo global transcriptional silencing (Kierszenbaum and Tres 1975), which likely influences their total RNA complement and may explain their transcriptional similarities (Fig. 1B; Kierszenbaum and Tres 1975).
To further validate our comprehensive spermatid RNA-seq results, we isolated round and elongating spermatids using alternative methodologies and performed a single-end RNA-seq experiment. For RSs, we isolated haploid germ cells (stained with Hoechst 33342) by FACS from juvenile mice at 23 dpp, an age at which elongating spermatids have not yet formed. For elongating spermatids, we FACS isolated from adult animals a mixture of EES and LES spermatids expressing a Prm1-promoter-driven EGFP transgene (Haueter et al. 2010). We also downloaded and processed several published RNA-seq data sets that examined similar cell populations (isolated using different approaches) (Erkek et al. 2013; Gan et al. 2013; da Cruz et al. 2016; Lesch et al. 2016; Gaysinskaya et al. 2018; Wang et al. 2020). Comparing the expression levels of GENCODE annotated genes in these additional data sets showed high cell type–specific correlations among nearly all the experiments, despite variation in the cell isolation techniques and RNA sequencing technologies used (Supplemental Fig. S1B).
De novo transcriptome annotation identifies many previously unannotated genes
When we examined the fraction of deduplicated, uniquely mapped reads in our data that fell within the GENCODE annotation (defined as containing even a 1-bp overlap), we observed that a substantial fraction of our reads did not map to known transcripts (ranging from 41.4% in LZ2 to 4.5% in EES1) (Fig. 1C). We wondered whether such reads could represent previously unannotated transcripts, expressed during postreplicative spermatogenesis. To investigate this possibility, we performed a de novo transcriptome assembly (Fig. 1D). We first used Scallop (Shao and Kingsford 2017) to assemble transcripts (without a reference) from each of the cell types using our uniquely mapped, deduplicated reads. We then filtered out all transcripts overlapping ENCODE Blacklist regions (Amemiya et al. 2019). We next quantified transcript levels using Salmon (Patro et al. 2017) and filtered out those with an expression value of <0.5 transcripts per million (TPM) in all samples. We combined the transcriptome annotations of all five cell types using StringTie (Pertea et al. 2015) and finally quantified the expression level of each transcript in all cell types with Salmon.
Our de novo annotation resulted in the discovery of between 1352 and 3239 new transcripts in each cell type examined (Fig. 1E). In total, the number of nonredundant, novel, expressed transcripts was 9919. In comparison, 43,940 transcripts overlapping the GENCODE annotation were found to be expressed in our RNA-seq data (with the same expression threshold requirements). The 9919 novel transcripts identified are generated via alternative splicing from 7653 distinct genomic loci (in comparison to 19,922 loci producing the 43,940 known transcripts). Thus, the newly identified genes represent 27.7% of all loci expressed in spermatocytes and spermatids. We next examined in which cell types the known and newly identified loci were expressed. Thirty percent of known genes were expressed in all five cell types profiled. In contrast, novel genes were generally expressed only in one or two specific cell types, with an approximately twofold overrepresentation in spermatocytes and early spermatids (Fig. 1F).
To confirm that these novel transcripts could be found not only in our RNA-seq data, we examined their expression in our spermatid validation and published data sets (Erkek et al. 2013; Gan et al. 2013; da Cruz et al. 2016; Lesch et al. 2016; Gaysinskaya et al. 2018; Wang et al. 2020). Quantitation of expression of novel and known genes showed similar expression levels for both classes in these auxiliary data sets as in our primary data (Supplemental Fig. S2A). Indeed, PCA using only the transcript levels of novel genes was able to separate samples from multiple data sets based on their cell types, suggesting that the newly identified transcripts provide a fingerprint for cellular identity (Supplemental Fig. S2B).
Given the enrichment for cell type–specific gene expression among our novel genes, we hypothesized that this set of genes would be restricted in their expression to testicular male germ cells. To test this hypothesis, we quantified the levels of known and novel transcripts in a published RNA-seq data set covering several adult mouse somatic tissues (Wang et al. 2020). We observe that many previously annotated transcripts display substantial expression in somatic tissues, whereas our newly identified genes are much more lowly expressed in all adult tissues examined, with the exception of testes (Supplemental Fig. S2C).
As an additional confirmation of the validity of our transcriptome annotation, we performed chromatin immunoprecipitation (ChIP) sequencing for trimethylation of lysine 4 of histone H3 (H3K4me3) in cells isolated from adult testes. H3K4me3 is a well-known histone modification found around the transcription start sites of expressed genes in all eukaryotes (Santos-Rosa et al. 2002). We previously reported the pattern of H3K4me3 in RS (Erkek et al. 2013) and, here, extended this to include the LZ, PD, and EES+LES cell types. We observe similar patterns of enrichment for this mark around the start sites of both novel and known genes (Supplemental Fig. S3), strongly arguing that the novel expressed loci identified by our approach represent bona fide genes expressed in postreplicative male germ cells.
Differential features of novel genes
We next compared gene organizational features of novel versus known genes. We found that single-exon genes were overrepresented among novel genes compared with previously annotated genes (Fig. 2A). By dividing novel genes based on their expression profiles, we found that genes expressed in different cell types possess different proportions of mono- versus multiexonic genes (Fig. 2B). Genes expressed specifically in early meiotic prophase (LZ) display the most marked enrichment for monoexonic genes, whereas those expressed specifically in late spermatids (EES+LES) showed a fraction of spliced transcripts more similar to that seen in known genes (Fig. 2A,B).
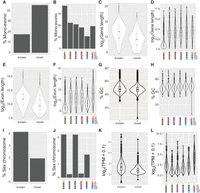
Characteristics of newly annotated spermatogenesis-expressed genes. (A,C,E,G,I,K) Comparisons between all novel to known genes. (B,D,F,H,J,L) Comparisons between classes of novel genes with different developmental expression patterns (indicated by colored ovals under the x-axes). (A,B) Bar plots showing percentage of identified genes containing only a single exon. (C–F) Violin plots showing distributions of log2 of gene length (C,D, including introns) or exon length (E,F). (G,H) Violin plots comparing distributions of GC% between different gene categories. (I,J) Bar plots showing fraction of genes mapping to the X or Y Chromosomes. (K,L) Violin plots comparing log2(transcripts per million[TPM] + 0.1) between different gene categories.
Consistent with the patterns of spliced and unspliced genes, the span of novel genes was shorter than that of GENCODE annotated genes (Fig. 2C), a feature also observed when considering just the length of exons (Fig. 2E). Comparing expression profiles to gene and exon lengths showed that this general feature obscures a more complex pattern. Novel genes expressed exclusively in LZ spermatocytes showed shorter gene and exon lengths (Fig. 2D,F), a notion consistent with the high percentage of monoexonic genes in this class (Fig. 2B). Novel genes expressed in all five cell types profiled show gene and exon lengths similar to those seen in previously annotated genes (Fig. 2C–F). Novel genes expressed exclusively in EES+LES show slightly decreased gene lengths compared with broadly expressed novel genes (Fig. 2D), whereas their exon lengths are substantially shorter (Fig. 2F). Coupled with the high fraction of multiexonic genes expressed specifically in late spermatids (Fig. 2B), this suggests that among the previously unannotated genes, those found exclusively during late spermiogenesis are enriched for spliced transcripts covering large genomic areas but producing relatively short mature RNAs.
In terms of sequence composition, novel and known genes show similar distributions of GC content (Fig. 2G). Large differences in GC percentages were also not observed in any particular expression class of novel genes (Fig. 2H).
The heteromorphic nature of mammalian sex chromosomes means that these chromosomes lack homologous partners for meiosis. This leads to a response known as meiotic sex chromosome inactivation (MSCI), causing genes on the X and Y Chromosomes to become transcriptionally repressed in pachytene (Turner 2007). Among our newly identified genes, fewer are located on the sex chromosomes compared with previously annotated genes (Fig. 2I). Consistent with the effects of MSCI, genes expressed in PD show a strong depletion in sex chromosome localization, whereas LZ- and RS-specific genes are more frequently localized to the X and Y (Fig. 2J).
We hypothesized that our newly identified genes may be expressed at lower levels and thus have escaped previous identification using less-sensitive methods. Given the difference in gene lengths between novel and known genes (and the differences between novel genes with different expression patterns), normalization by gene length is essential to allow a fair comparison. We compared TPM values for novel and known genes and found that, indeed, novel genes show a lower median value and a shift in distribution toward lower expression (Fig. 2K). Breaking down the levels of gene expression based on expression pattern showed that novel genes expressed in all cell types are expressed at nearly the same level as known genes, whereas cell type–specific genes show a generally lower expression level (Fig. 2L). Among genes expressed in specific cell types, those expressed in LZ spermatocytes show a higher expression level compared with those expressed later in spermatogenesis (Fig. 2L).
Newly identified genes are generally not evolutionarily conserved
We next examined the evolutionary origin of our novel genes. We compared conservation scores (calculated by comparing base pair evolutionary conservation in 60 vertebrate species) in exons for known and novel genes. Genes previously annotated in GENCODE showed a range of values from highly conserved to unconserved, whereas our novel annotated gene set shows a substantially lower level of conservation (Fig. 3A). This feature was true for both monoexonic and multiexonic genes (Fig. 3A). In fact, comparing the novel genes to a set of randomized regions (designed with matching lengths and splicing patterns) revealed only few genes with higher than random conservation levels (Fig. 3A). Restricting our conservation analysis only to six species more closely related to the mouse did not reveal a substantially increased level of conservation, suggesting a relatively recent evolutionary origin for the novel genes identified in this study (Fig. 3B).
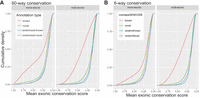
Conservation of novel genes. Cumulative density plots comparing mean 60-way (A) and six-way (B) exon conservation scores between novel and known single-exon and multiexon genes. Randomized known and novel refer to analyses in which annotations matching exon and intron length were assigned to random positions in the genome. Sixty-way conservation compares all vertebrates, and six-way conservation compares mouse, rat, guinea pig, kangaroo rat, naked mole rat, and squirrel.
Coding potential of newly annotated testicular genes
To assess whether the novel genes identified in our new annotation could be translated during spermatogenesis, we next assayed the coding potential of these genes. We used CPAT, a tool that estimates the probability that a given transcript is likely to encode a protein based on sequence features in open reading frames (ORFs) (Wang et al. 2013). Analysis of GENCODE annotated genes showed a bimodal distribution, with one group of genes showing a very high probability of coding for protein and another group showing very low probability (Fig. 4A). Known genes with either single or multiple exons fitted such distributions, although multiexonic genes display a much stronger density at high coding potential (Fig. 4A). In contrast, the distributions of coding probabilities of single-exon and multiexonic novel genes looked different from those of known genes, with most genes showing a low probability of coding for protein, as well as a tail of genes with higher coding potential (Fig. 4A). To look more closely at the set of putative protein-coding genes, we compared the length of predicted proteins to the CPAT-calculated coding probability score, as ORF length is known to be a major predictor of coding probability (Fig. 4B; Frith et al. 2006). We find, among known genes, that single-exon genes tend to encode shorter proteins and that proteins predicted in our novel gene set are also generally shorter than those found in previously annotated genes (Fig. 4B). To set cut-off values for likely protein-coding genes among our newly identified genes, we examined the subset of known protein-coding genes among the GENCODE annotation (Supplemental Fig. S5). There is a broad distribution of coding probabilities and protein lengths even among known coding genes; however, >75% of these genes had a coding probability of greater than 0.68 and a predicted protein length of 151 amino acids. Using these cut-offs on our set of novel genes identifies 209 putative protein-coding transcripts (or 2.11% of all newly identified genes). These unique loci produce 529 ORFs (accounting for splice variants and multiple ORF-containing transcripts) (Supplemental Table S1).

Coding potential of newly identified genes. (A) Violin plots showing CPAT-calculated coding probability for genes subdivided by overlap with GENCODE and number of exons (n indicates the number of genes in each class). (B) Plots showing predicted protein length versus CPAT-calculated coding probability for genes subdivided as in A. (C) Venn diagram showing fraction of ORFs overlapping with three databases: BLASTP, nonredundant BLASTP database; domain, conserved domain database; and Repbase, Repbase repeat elements. (D) Schematic cartoon of retrogene generation. (E) Table showing details for six identified retrogenes. The % identity refers to similarity between origin gene and retrogene in Mus musculus.
To further characterize these ORFs, we performed three comparisons of their sequences. First, we searched their predicted protein sequences against the nonredundant protein database using BLASTP (Altschul et al. 1990). Next, we searched these same predicted protein sequences against the conserved domain database (CDD) to identify putative protein domains in our novel genes (Lu et al. 2020). Finally, we compared the sequences of our ORFs to Repbase, a database annotating repetitive sequences throughout the genome (Jurka 2000; Bao et al. 2015). Of the 529 ORFs examined, 390 (73.7%) had a hit in at least of one of these searches, with 151 (28.5%) having a hit in all three databases (Fig. 4C). Of those ORFs possessing a BLASTP hit, 56.7% (183/323) overlap a Repbase element within their ORF. Domain searching identified 53 ORFs that lack a BLASTP hit but show similarity to a protein domain (of which 20 also do not overlap show any overlap with repetitive sequences) (Fig. 4C; Supplemental Table S1).
We also identified six so-called retrogenes (Brosius 1991), genes that arise through reverse transcription and subsequent genomic integration of an existing mRNA (Fig. 4D). These genes can be identified via the presence of a single-exon gene possessing a paralogous sequence found elsewhere in the mouse genome in a multiexonic form (Tubb-ps1, Hemgnl, Tekt5l, Nsa2-ps1, Ppp1r2-ps6, and Lonrf3l) (Fig. 4E). Three of these, Tubb3-ps1 (encoding a beta-tubulin isoform), Nsa2-ps1 (encoding a ribosome biogenesis factor), and Ppp1r2-ps6 (encoding a protein phosphatase inhibitor), were previously deposited in NCBI RefSeq but were subsequently removed during annotation updates. Two of these retrogenes (Hemgnl and Tekt5l) possess a best BLASTP hit not to a Mus musculus protein but rather to one from the related mouse species Mus caroli. The genes associated with these proteins in M. caroli also contain only a single exon, suggesting that the generation of these retrogenes occurred more than 3 million years ago (MYA) in the common ancestor of M. caroli and M. musculus (Thybert et al. 2018).
To further examine the evolutionary origin of our newly characterized retrogenes, we compared the nucleotide sequences of their ORFs to the genome sequences of M. musculus and four related rodent species: Mus spretus (diverged 1 MYA), M. caroli (3 MYA), Mus pahari (6 MYA), and Rattus norvegicus (12 MYA). As expected, all newly identified retrogenes showed two hits to the M. musculus genome (one with perfect identity and a second [from the origin gene] with lower identity). BLAST results showed that in addition to Hemgnl and Tekt5l, Tubb3-ps2 is also conserved in other mouse species (up to M. caroli), whereas the other three retrogenes do not appear to be present even in M. spretus (Fig. 4E). The conservation for Tekt5l extends through to rat (a homologous sequence is not found in rabbit). The origin gene of Tekt5l, Tekt5, is a filament protein enriched in the accessory structures of the sperm flagellum (Cao et al. 2011), so the identification of its expression during late spermatogenesis fits its protein localization. To date, no function for Tekt5 has been described.
We also observed, in addition to proteins encoded by repetitive sequences, several clusters of genes that appear to encode identical or nearly identical proteins. We found six copies of an ORF within an 892-kb region on mouse Chromosome 8 (mm10 coordinates Chr 8: 19,758,166–20,650,219). This region displays extensive internal direct and inverted duplication (Supplemental Fig. S4B). Three gene models are currently annotated within this region (Potefam3f, Potefam3a, and Potefam3b), all of which encode proteins containing ankyrin repeats and annotated as members of the POTE ankyrin domain 3 family. The expression pattern of one member of this family, named Potefam3, was previously characterized as postreplicative male germ cell specific (Bin et al. 2007), but function was not determined. Our newly identified genes do not show any homology with this gene family but instead show similarity to another gene model, Gm53405, ranging from 76%–90% over around one-third of the protein length (Supplemental Table S1). The protein encoded by Gm53405 has no identifiable domains and its function remains unknown.
Additionally, we found seven copies of a recently characterized X/Y Chromosome ampliconic region known as Laidx/y (Arlt et al. 2020) and nine copies of single-exon ORFs on the X Chromosome encoding proteins similar to proteoglycan 4–like from the African grass rat (Arvicanthus niloticus).
To examine whether the putative coding genes that we identified generate novel proteins, we examined published mass-spectrometry (MS) data generated from adult mouse testes, liver, and frontal lobes (Giansanti et al. 2022). We queried these MS data sets using a database composed of a union of the UniProt database and the predicted masses of proteins generated from our high CPAT scoring novel genes. We identified 19 proteins derived from novel genes that contained at least two uniquely identifiable peptides (Table 1). Most (15/19) of these proteins are only detectable in testis and not in the somatic tissues examined. Seventeen of 19 of these newly identified proteins were also found to have matches in database searches to nonredundant proteins or protein domains.
Confirmation of protein-coding genes by MS analysis
Among the proteins identified in this analysis, we observe two expressed from newly annotated expressed repetitive elements (one MERVK10c-int and one IAPEz-int), showing that protein-producing retrotransposons may be active in the mouse testis. We also observe that two of the previously mentioned retrogenes (Ppp1r2-ps6 and Tekt5l) both produce novel proteins, suggesting that they may play functional roles in the testis. We also observe database hits to nonmouse proteins for 10 of the newly identified proteins. This is in contrast to the generally low conservation observed for most of our novel genes (Fig. 3). This is consistent with evidence that loci encoding noncoding RNAs tend to evolve more rapidly than those encoding protein-coding genes (Necsulea et al. 2014). Analysis of our list of novel mouse genes producing protein identified six of 19 genes with a clear human homolog (Table 1). Of these five show homology with annotated genes in humans, all of which showed highly enriched expression in testis (compared with somatic tissues), with expression specifically observed in human postreplicative germ cells (Supplemental Fig. S5).
While analyzing putative protein-coding genes, we found that 217 of 530 (40.1%) of these transcripts showed transcription of the opposite strand within the same locus, often appearing as divergent transcription from the promoters. We further examined this in detail for all genes (not just those encoding proteins).
Many genes expressed in spermatogenesis overlap repetitive elements
Repetitive sequences comprise up to 45% of the mouse genome (Biémont 2010) and are generally transcriptionally repressed in most tissues. Low levels of repeat expression, however, have been observed during spermatogenesis in wild-type testes (Branciforte and Martin 1994; Soper et al. 2008; Brown et al. 2010), with enhanced expression observed upon removal of repressive mechanisms (Soper et al. 2008; Di Giacomo et al. 2013; Barau et al. 2016). We observed a substantial number of novel protein-coding genes whose ORFs overlapped with repetitive sequences (Fig. 4C). We thus chose to examine more globally the overlap between repetitive sequences and our new transcriptome annotation.
We first examined the developmental expression pattern of repetitive sequences through postreplicative spermatogenesis by quantifying expression of repetitive elements (found in Repbase) (Jurka 2000; Bao et al. 2015) in our RNA-seq data (Fig. 5A). We found that different families of repeats display distinct expression profiles, with many repeat families (particularly ERVK, ERV1, and ERVL) showing enrichment for cells in meiosis relative to later stages. Previous analysis of transposable elements in spermatogenic cells found members of these three families to be accessible and expressed in pachytene spermatocytes (Sakashita et al. 2020); however, the specific subclasses identified differ from those found in our analysis, possibly owing to different data sets or analysis pipelines.
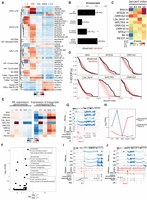
Overlap of spermatogenesis-expressed genes with repetitive sequences. (A) Heatmap showing relative expression of repeats during postreplicative spermatogenesis. Heatmap is displayed according to main repeat families (labeled on left axis) and subclustered by repeat subfamilies (labeled on right axis). (B) Plot showing number of multiexonic transcripts (and percentage) not overlapping with repetitive sequences compared to monoexonic transcripts and multiexonic transcripts where repeats overlap with the first exon or any other exon. (C) Heatmap showing Z-score of Jaccard indices comparing 5′ exons to repNames at defined levels compared with randomized distributions. repNames with 10 highest Z-score values are shown. (D) Reverse empirical cumulative density functions (ECDFs) of Jaccard indices for repNames with 10 highest Z-scores (black). Transcriptome annotation was randomized 100 times to generate null distributions (red). (E) Expression level of repetitive elements (REs) associated with repNames from C across five cell types examined in this study (left) versus expression level of multiexonic transcripts containing elements of those repNames in the same data set (right). (F) Enrichment of gffcompare class codes by repName when comparing our de novo annotation to GENCODE. x-axis = log2(fold enrichment); y-axis = significance [−log10(FDR)]. (G) Genome browser depiction of the Ceacam20 locus. Top panel shows RNA-seq in five cell types with spliced reads as curved lines. Middle panel shows H3K4me3 ChIP-seq enrichment. Bottom panel shows annotation from de novo annotation, GENCODE, and Repbase. (H) Quantification of transcript levels from the Ceacam20 locus. Trans 1 represents the reference GENCODE transcript, and Trans 2 initiates from the ORR1A2 element. (I,J) Examples of genomic loci where repetitive elements lead to new transcripts lacking part of the reference GENCODE transcript. Annotation is as in G.
To examine the role that repetitive sequences might play in shaping the spermatogenic transcriptome, we compared the genes found in our annotation to repetitive sequences found in Repbase (Jurka 2000; Bao et al. 2015). For comparison, we generated a randomized transcriptome annotation with features matching our novel annotation (with regards to transcript numbers and exon and intron length). We then compared the number of transcripts in four classes: (1) transcripts that do not overlap any Repbase elements, (2) monoexonic transcripts that overlap with Repbase elements, (3) transcripts in which an overlap with a Repbase element occurs within the first exon (and thus could putatively function as a novel promoter), and (4) transcripts in which an overlap with a Repbase element occurs within any other exon. In line with the fact that most repeats are transcriptionally silenced, we find that 34.4% of all genes identified in postreplicative male germ cells do not overlap a repeat, a percentage that is much greater than random (Fig. 5B). Multiexonic transcripts in which the first exon overlaps a repeat represent 18.1% of all transcripts identified, a value substantially less than that seen in our randomized transcriptome (Fig. 5B). These two findings suggest selection against a major role for repeat elements in testicular gene regulation and evolution. In contrast, monoexonic transcripts and multiexonic transcripts overlapping repeats in exons other than their first represent 8.4% and 39.0% of all identified genes, similar to what would be expected by chance (Fig. 5B). In total, 65% of all genes identified in our annotation overlap a repetitive element.
Focusing our analysis further on multiexonic transcripts whose first exon overlaps a repeat, we next examined whether any types of repetitive elements (repNames from Repbase) may potentially play a role as promoters of testicular gene expression. To examine transcripts in which the 5′ exon largely overlaps a repetitive element, we calculated the Jaccard index (JI; defined as the amount of intersection between the exon and repeat relative to the total merged length of exon and repeat) comparing the exons to the repeats. Next, for each repName, we compared observed distributions of JIs to distributions obtained by 100 rounds of randomization of the de novo transcriptome. To detect deviations of observed distributions of JIs from random, we plotted reverse empirical cumulative distributions (ECDFs) for observed and randomized JIs and calculated Z-scores of observed reverse ECDFs from distributions generated from 100 rounds of transcriptome randomization at four values of JI (0.2, 0.4, 0.6, and 0.8). Figure 5C shows the top 10 repNames ordered by maximum Z-score. Figure 5D illustrates observed and randomized reverse ECDFs of JIs for repNames with the highest Z-scores. In general, most ECDFs comparing 5′ exons to the repeats they overlap show very little enrichment relative to the random distributions (Supplemental Fig. S6). However, for some values of JI, we observed enriched Z-scores, suggesting a greater than expected overlap between 5′ exons and some repNames (Fig. 5C).
We next examined whether the differential expression patterns seen in repetitive elements (Fig. 5A) would drive similar cell type expression patterns for overlapping genes. Focusing on the top 10 repNames identified as having a higher than random JI in 5′ exons (Fig. 5C), we plotted expression for repetitive elements and for multiexonic transcripts harboring repeats of that repName across the five cell types examined in this study (Fig. 5E). We generally found poor correlations in gene expression profiles between repeats and genes with the exception of IAPLTR3 elements, which show peak expression of both the repetitive element and the overlapping transcripts in PD spermatocytes and a sharp down-regulation of expression in elongating spermatids (Fig. 5E).
Although our global data suggested that repeats do not serve as the major determinants of gene expression profiles in male germ cells, we wondered whether they may serve as alternative promoters driving additional expression of reference transcripts in specific cell types. We thus further compared our de novo transcriptome annotation with GENCODE using gffcompare (Pertea and Pertea 2020). This analysis returns a series of class codes ranging from completely matching (“=”) to completely unknown (“u”). We were particularly interested in transcripts overlapping repeats whose comparison to GENCODE suggested a partial overlap consistent with an alternative 5′ exon serving as a new site for transcription initiation (gffcompare class code “k”). We calculated the enrichment for class codes in transcripts corresponding to specific repNames (Fig. 5F). We observed that some repNames are enriched for particular variation with respect to the reference annotation. In particular, we observed nine transcripts overlapping Charlie7 elements present in the introns of reference transcripts and 29 transcripts with ORR1E elements in their 5′ exons that are present on the opposite strand of exons of reference transcripts. We also observed 39 transcripts whose 5′ exons overlap MMERVK10C (13) or RLTR10C (26) and whose position in the genome contains no reference annotation. We did observe seven transcripts overlapping with reference transcripts that possess 5′ MERVL-containing exons; however, these elements were not substantially enriched in their JI relative to the full 5′ exon length. In total, we find no strong evidence for a general phenomenon of repetitive elements functioning as alternative promoters during spermatogenesis.
Although repetitive elements may not function generally as an alternative regulatory mechanism for cell type–specific expression in the testis, we did observe specific examples in which repeats seem to play such roles. For example, our de novo annotation of the Ceacam20 locus suggested a 5′ ORR1A2 element could splice into the reference annotated transcript (Fig. 5G). This alternative 5′ exon appears most abundantly in the transcript during spermatid elongation, and we also observed enrichment of H3K4me3 at the repetitive sequence in elongating spermatids. Quantification of transcript levels for these two isoforms shows that, indeed, the canonical transcript (Trans 1) is most expressed in LZ & PD and is not detectable in EES and LES, whereas the ORR1A2-initiating transcript (Trans 2) shows high expression specifically in EES and LES (Fig. 5H). Trans 2 of Ceacam20 encodes a protein with a 43-amino-acid truncation at its N terminus, relative to the protein encoded by canonical Trans 1, suggesting the possibility of modified protein function in elongating spermatids. We also observe transcripts in which a repeat found within an intron of a reference transcript splices into the reference transcript, generating truncations or out of frame transcripts (Fig. 5I,J). Thus, although we did not find evidence for a general function of repetitive elements in shaping transcriptional patterns across postreplicative spermatogenesis, we did observe clear cases in which repeats can impact transcriptional activity (and coding potential) during spermatogenesis.
Bidirectional promoter activity in spermatogenic cells
Based on the observation that many putative novel protein-coding genes displayed transcription on the opposite strand from known genes, we proceeded to further characterize antisense promoter activity throughout the genome. Examination of many known genes in the genome browser revealed that spermatogenic cell types showed bidirectional transcription across multiple cell types. For example, the Actb locus, encoding the beta actin protein, is expressed in all of the spermatogenic cell types examined and shows a clear long transcript expressed from the opposite strand in all cells as well (Fig. 6A). To generate an unbiased set of promoters to examine bidirectional promoter activity at a genome-wide scale, we used our H3K4me3 ChIP-seq data (Supplemental Fig. S3) to define active promoter regions. We identified peaks of H3K4me3 throughout the genome in each of four spermatogenic cell types (LZ, LD, RS, and EES + LES) using MACS. We then defined a merged set of 11,140 anchor sites, in which an H3K4me3 peak was within 2 kb of a defined transcript and was 5 kb distal from other peaks, such that transcription from neighboring genes would not interfere with interpretation of RNA-seq signal at these sites. We then assigned transcripts associated with these anchors to be either sense or antisense based on the following criteria: (1) if only one transcript was found, it is considered sense; (2) if two multiexonic transcripts were found, the one with a TSS closer to the H3K4me3 anchor was considered sense; (3) if one multiexonic transcript and one monoexonic transcript were found, the multiexonic one was considered sense; and (4) if two monoexonic transcripts were found, the one with highest expression was considered sense. We quantified RNA-seq signal in exons of the sense transcript and for 2 kb upstream of the H3K4me3 anchor for antisense.
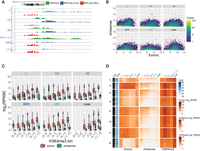
Antisense transcription in the testis. (A) Genome browser screenshot of Actb locus showing strong antisense transcription in all cell types profiled. Green indicates H3K4me3 ChIP-seq; blue, (+) strand RNA; and red, (−) strand RNA. (B) Scatter plot showing exonic log2(RPKM) versus antisense log2(RPKM) in testicular cell types (this study) and the liver (from Wang et al. 2020). (C) Boxplot showing log2(RPKM) of exonic (red) and antisense (blue) sequences binned by levels of H3K4me3 in testicular cells and the liver. (D) Heatmap showing levels of exonic and antisense RNA and ChIP-seq enrichment for H3K4me3 at TSS ±3 kb in testicular cells types and the liver.
We then plotted sense versus antisense RNA levels for each anchor in each cell type (Fig. 6B). A strong antisense signal is most clearly observed in LZ, with substantial signals also seen in PD and RS. In these early populations, two groups of transcripts show substantial antisense RNA level: one with high levels of sense strand transcripts and a second in a group of more lowly expressed genes (Fig. 6B,D). These trends are much less visible in EES and LES, in which lowly expressed genes show very little antisense RNA and more highly expressed genes show less correlation between sense and antisense levels (Fig. 6B,D). This may reflect the fact that EES and LES undergo global transcriptional silencing (Kierszenbaum and Tres 1975), and therefore, unstable antisense RNA produced during transcription may not persist in these cells.
To examine whether the features we observe in our spermatogenic cells are particular to these cell types, we examined publicly available RNA-seq and H3K4me3 ChIP-seq data for the mouse liver (Stamatoyannopoulos et al. 2012; Wang et al. 2020). We identified H3K4me3 anchors identically as for the testicular cell types and found 9554 unique H3K4me3 anchors in this tissue. We then quantified transcripts surrounding these anchors using known exons. We did not observe, in contrast to early postreplicative spermatogenic cell types, the same cloud of genes with high antisense and low sense strand RNA in the liver (Fig. 6B).
We next examined whether the level of H3K4me3 found in the promoters of these genes could predict the sense and antisense RNA levels. We thus divided our promoters into six bins based on their H3K4me3 enrichment levels and then plotted sense and antisense RNA levels in these groups (Fig. 6C). We see a clear correlation between sense strand RNA levels and H3K4me3 levels in all cell types examined. Antisense transcript levels also correlate with H3K4me3 levels for promoters with higher levels of H3K4me3 (the upper three of six bins); at lower levels of H3K4me3, antisense transcript levels are very low (Fig. 6C).
When we compared levels of sense and antisense RNA driven from the same promoters in spermatogenic cells and liver, we found patterns that differ between tissues (Fig. 6D). As expected (given the nature of anchor point selection), H3K4me3 levels are high in all promoters examined. As was seen looking only at spermatogenic promoters (Fig. 6B), antisense RNA levels were most clearly detectable in early meiotic prophase (LZ) across all promoters examined. We next divided promoters into eight groups using k-means clustering. Cluster 2 shows genes expressed at high levels in all cell types examined (with promoters marked with high levels of H3K4me3). Antisense transcript levels are high in LZ, PD, and RS but much less so in EES, LES, and liver (Fig. 6D). Cluster 8, which contains genes highly expressed in LZ and liver (with lower expression in later spermatogenic cell types), shows much clearer antisense transcript levels in LZ compared with liver despite expression of sense strand transcripts in both cell types, suggesting that transcriptional mechanisms, as opposed to intrinsic sequence features, may be responsible for the differences in antisense transcription seen in postreplicative male germ cells.
Discussion
The completion of the initial sequencing of the mouse genome in 2002 (Mouse Genome Sequencing Consortium et al. 2002) was a major milestone in enabling our understanding of gene regulation in this model organism. Improvements to both sequence assembly and annotations of known genes have continued to refine these analyses (Frankish et al. 2021). Nonetheless, the existing genome annotation is a massive, combined effort spanning decades and, as such, may harbor biases based on historically used pipelines. These biases include a strong focus on the identification of conserved, spliced, and nonrepetitive genes. Additionally, genome annotations are dependent on transcriptome data, which for rarer cell types may not be exhaustive. We have used a deep-coverage expression analysis combined with an unbiased annotation approach to expand the known repertoire of loci expressed in postreplicative spermatogenesis, including cell types in five distinct phases of development. Our results, indeed, show a large number of nonconserved, monoexonic, and repeat-overlapping genes previously missing from the genome annotation (Fig. 7).
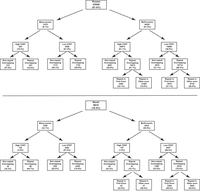
Summary of genes identified as expressed in postreplicative spermatogenic cells. Flow chart showing number of genes in each category: known versus novel, monoexonic versus multiexonic, high versus low coding potential, and repeat-overlapping versus nonoverlapping. The percentage in each box refers to percentage of the parent category.
Gamble et al. (2020) previously reported a new transcriptome annotation generated from RNA-seq data from 25 dpp total testes (a time point when RS are abundant and EES are just forming and LES have not yet developed) (Bellve et al. 1977). Comparing the 2507 novel genes identified in their study to the 7653 novel loci identified in this study, we found 1447 genes in common. This represents 18.9% of the novel genes identified in our study and 57.7% of the novel genes identified by Gamble et al. (2020). Differences in assembly protocols, read length (PE 150 bp for this study vs. PE 100 bp for Gamble et al. 2020), and cell populations (the inclusion of more late spermatids in this study) may account for the greater number of novel genes reported in this study. The presence of substantial overlap between the two studies provides confidence in the validity of both studies, whereas the presence of novel genes in both individual studies suggests that both novel annotations generated may remain incomplete, and further new transcripts may remain to be discovered in spermatogenic cells.
We find that cells in the early parts of meiotic prophase (leptonema and zygonema [LZ]) show distinct transcriptional properties. These cells express many short monoexonic transcripts (Fig. 2B,D), as well as substantial numbers of distinct repeat subfamilies (Fig. 5A). We did not, however, observe these repetitive elements functioning as novel promoters to drive expression of other transcripts in an LZ-specific pattern. In general, we do not find that repetitive sequences overlapping 5′ exons of transcripts lead to clear cell type–specific expression patterns mirroring the expression of isolated repetitive elements. We do observe that IAPLTR3 elements peak in expression in mid-to-late meiotic prophase (PD), a pattern of expression that is also found in the 56 genes in which these elements serve as 5′ exons. Specific IAPLTR elements show different activities in different tissues, and these elements have been suggested to sensitize gene expression to environmental cues (Sharif et al. 2013). Whether these elements function in PD spermatocytes to coordinate the regulation of transcripts required for meiotic progression is an interesting potential future research direction.
Our analysis of spermatogenesis expressed genes found that the vast majority of our 9919 novel transcripts (98.2%) are likely to generate long noncoding RNAs (lncRNAs). This is not surprising given the extensive historical focus of research on coding genes. Additionally, we found that most of our novel genes are not well conserved, a feature found much more frequently in lncRNAs than in coding genes (Necsulea et al. 2014). Despite this, we identified over 50 new putative protein-coding genes in the mouse genome, 19 of which show clear evidence for protein production (Fig. 4; Table 1). We found six unannotated retrogenes, of which three were previously annotated but subsequently removed from the annotation (Fig. 4D). Some of these were previously annotated as “processed pseudogenes” (Cheetham et al. 2020), an ambiguous term because all these loci contain ORFs that could generate proteins (and, indeed, we found evidence of testicular protein expression for two of these retrogenes). Many retrogenes have been found to be expressed in testes of many species (Carelli et al. 2016). Such genes may provide supplemental expression support to their spliced counterparts or have begun evolving toward novel functions. We also observed the presence of testis expressed protein-coding genes in a repetitive region of Chromosome 8 (Fig. 4F). Expression of testis genes in so-called ampliconic regions has been previously shown on both the X and Y Chromosomes (Skaletsky et al. 2003; Mueller et al. 2008). In sex chromosomes, the amplification of these genes is thought to provide an alternative mechanism for recombination, allowing eventual removal of mutations deleterious to fertility (Rozen et al. 2003). The function of such a region in spermatogenesis on an autosome remains less obvious. These ampliconic genes, as well as the retrogenes and additional novel protein-coding genes, are all interesting targets for further analysis of both their regulation and function.
We also found an interesting feature of promoters expressed in postreplicative spermatogenesis: their bidirectional activity. Previous studies have shown that many mammalian promoters produce RNA from both strands during transcription initiation, but the antisense transcripts are not subject to further transcription elongation (Seila et al. 2008). To observe these initiating RNAs, Seila et al. (2008) sequenced short RNAs that were found proximal to transcription start sites. In contrast to these RNAs, we observed much longer RNAs in spermatogenic cells (Fig. 6A). We find that promoters active both in the liver and during spermatogenesis display different patterns of antisense transcript levels (Fig. 6D). This observation suggests either that promoters fire in a very different way during postreplicative spermatogenesis or that antisense transcripts are more strongly stabilized in these cells. The fact that elongating spermatids (EES & LES), which are in the process of undergoing genome-wide transcriptional silencing (Kierszenbaum and Tres 1975), show lower antisense transcript levels compared with transcriptionally active (round) spermatids (Fig. 6B,D) suggests that antisense transcripts may be short lived and not stabilized during late spermiogenesis. Variant subunits of the core transcription machinery have been identified during germ cell development in mice (Freiman 2009). Work in human cells has also shown that the dynamics of RNA Polymerase II occupancy (and its phosphorylation) regulate the directionality of transcription, with higher RNA Polymerase II–Ser2P leading to increased bidirectional promoter activity (Fong et al. 2017). Whether differences in transcriptional dynamics or transcription complex variation could be responsible for differences in promoter activity in these germ cells is an intriguing possibility.
Altogether, our analysis has extended the number of known transcription units in late mouse spermatogenic cells extensively. Using an annotation approach that does not depend on conservation, splicing, or repeat masking allowed us to fill in gaps in the existing testicular transcriptome (Fig. 7). These novel coding and noncoding transcripts now represent interesting targets for further analysis as potential regulators of spermatogenesis and male fertility. Future work is needed to functionally validate the role of these transcripts via targeted mutagenesis. It will also be interesting to expand this analysis to other mammalian species to determine if novel transcripts (not likely homologous to those found here) may be found there as well. In the future, this annotation can be combined with additional transcriptomic and epigenomic data to further understanding of mammalian spermatogenesis.
Methods
Cellular population isolation
Cells were purified from the testes of 3-mo-old C57BL/6JRj/6 mice purchased from Janvier Labs. All experiments were performed in accordance with Swiss animal protection laws (license 2670, Kantonales Veterinäramt) and institutional guidelines.
For all cell populations except LESs, biological replicates were derived from cells isolated from a single individual. Owing to the low overall RNA content of LESs, for this population, cells for each biological replicate were derived from five individuals sorted separately.
A detailed description of the cell purification is provided elsewhere (Gill et al. 2022). Essentially, single-cell preparations from total testes were generated by sequential digestion with collagenase followed by trypsin dissolved in Gey's balanced salt solution (GBSS). Trypsin was then inactivated using 5% fetal bovine serum (FBS). These cells were then stained simultaneously with 20 μg/mL Hoechst 33342 (Thermo Fisher Scientific) and 1 μM SYTO 16 (Thermo Fisher Scientific) for 45 min at room temperature protected from light. Cells were then centrifuged, resuspended in GBSS, and stained with the cell viability dye DRAQ7 (Biostatus) before being sorted on a BD FACS Aria III. Cells were gated for size and viability and then analyzed for DNA content (via Hoechst blue fluorescence). Cells with 4C DNA content were separated based on their Hoechst red fluorescence (as previously described) (Bastos et al. 2005) to discriminate LZ spermatocytes and PD spermatocytes. Cells with 1C DNA content were separated based on their SYTO 16 fluorescence levels (elongating spermatids show much higher fluorescence with SYTO 16 than RSs). Elongating spermatids were then further separated based on their size (as measured by FSC).
A portion of each cell population was used to determine the purity of the fractions (reported by Gill et al. 2022). For the two spermatocyte populations (LZ & PD), purity was ∼90% (with 5%–7% contamination for the other spermatocyte populations). RSs were 95% pure (with 1%–2% contamination with elongating spermatids). EESs and LESs were ∼86% and ∼77% pure (with essentially all remaining cells coming from the other elongating spermatid population) (Gill et al. 2022).
Additional RS samples were collected from testes of juvenile C57BL/6J mice (at 23 dpp, when the first elongating spermatids have not yet formed) in a similar manner excluding the SYTO 16. Additional elongating spermatids were collected from mice overexpressing a PRM1-EGFP fusion protein (Haueter et al. 2010). Single-cell suspensions were isolated from the testes of these mice as described above. These cell suspensions were then stained with Hoechst 33342 and subjected to FACS and 1C; PRM1-EGFP-positive cells were isolated as elongating spermatids. For H3K4me3 ChIP-seq in elongating spermatids, a mixed pool of EES and LES was isolated using centrifugal elutriation as previously reported (Grabske et al. 1975; Wang et al. 2020).
RNA extraction
Isolated cells were pelleted at 2000g for 10 min at 4°C. Supernatant was removed, and cells were resuspended in 300 μL TRI Reagent (Zymo Research). Tubes were then flash-frozen in liquid nitrogen and stored at −80°C until RNA isolation. RNA was isolated using the Direct-zol micro kit (Zymo Research) including the recommended DNase treatment. RNA quantity was determined using the Qubit RNA high-sensitivity assay (Thermo Fisher Scientific). The quality of total RNA was analyzed using an Agilent Bioanalyzer RNA 6000 pico chip.
RNA sequencing
High-throughput sequencing libraries were prepared using the Illumina TruSeq protocol. Libraries were sequenced with an Illumina NextSeq 500 sequencer in paired-end mode, with each read being 150 bases long.
H3K4me3 ChIP-seq
Chromatin was isolated from FACS isolated LZ and PD spermatocytes and elutriated EES/LES samples as previously described (Erkek et al. 2013). Briefly chromatin was isolated from cells by incubating with 15 mM Tris-HCl (pH 7.5), 60 mM KCl, 5 mM MgCl2, 0.1 mM EGTA with 0.5% NP-40, and 1% sodium deoxycholate for 30 min on ice. Chromatin was digested using 5 U MNase (Roche Nuclease S7 10107921001) for 30 min per 1 million cells at 37°C. H3K4me3-containing fragments were isolated using 2 µL of anti-H3K4me3 antibody (Millipore 17-614) per million cells. Library preparation for ChIP-seq was performed with the Illumina ChIP-seq DNA sample prep kit (IP-102-1001).
NGS data processing
Adaptors were trimmed from raw reads using cutadapt (Martin 2011), and quality was assessed with FastQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc). Reads were aligned to the mouse reference genome (build mm10) using STAR (Dobin et al. 2013); duplicated alignments were removed using SAMtools (Li et al. 2009), with only uniquely aligned reads from proper pairs retained. Read quantification (TPM) was performed at the transcript level with Salmon v0.12 in quasi-mapping mode (Patro et al. 2017), and quantification of alignments (RPKM) was performed at the gene level in R (R Core Team 2021) using the QuasR package (Gaidatzis et al. 2015), the latter for both RNA-seq, Ribo-seq, and ChIP-seq data.
De novo transcriptome assembly
Transcriptomes were first built individually for each of the five cell types by providing Scallop (Shao and Kingsford 2017) with pooled alignments from the three biological replicates from each cell type and using the following parameters: ‐‐min_transcript_coverage 10 min_splice_boundary_hits 10 ‐‐min_flank_length 10 -. Transcripts overlapping ENCODE blacklisted regions (mm10 v2) were then removed (Amemiya et al. 2019). Salmon (Patro et al. 2017) was used to quantify expression of transcripts from the five cell type–specific transcriptomes in each of the associated samples. Transcripts with expression <0.5 TPM in all three replicates were discarded. Finally, the five individual cell type–specific transcriptomes were merged using StringTie (Pertea et al. 2015) to generate a final single postreplicative male germ cell transcriptome.
Categorization of predicted transcripts into known or novel classes
Predicted transcripts were compared to transcripts from GENCODE v19 using gffcompare v0.10.4 (Pertea and Pertea 2020): Transcripts with output class codes {=,c,k,m,n,j,e,o,i,y,p} were considered known, whereas transcripts with output class codes {s,x,u} were considered novel.
Analysis of published RNA-seq data
RNA-seq data from published studies examining similar cell types to those examined in this study were downloaded from the NCBI Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/) and processed identically to in-house generated data. These studies are Gan et al. (2013) (poly(A) RNA-seq from spermatocytes and spermatids isolated by STA-PUT gradient purification) (Bryant et al. 2013), Lesch et al. (2016) (total RNA-seq from pachytene spermatocytes and RSs isolated by STA-PUT gradient purification), da Cruz et al. (2016) (total RNA-seq from LZ and pachytene spermatocytes and RSs isolated by Vybrant DyeCycle Green FACS) (Rodríguez-Casuriaga et al. 2014), Gaysinskaya et al. (2018) (total RNA-seq from leptotene, zygotene, pachytene, and diplotene spermatocytes isolated by Hoechst 33342 FACS) (Gaysinskaya et al. 2014), and Wang et al. (2020) (total RNA-seq and Ribo-seq from pooled spermatocytes, RSs, and elongating spermatids isolated by centrifugal elutriation) (Grabske et al. 1975).
Conservation of novel transcripts
Conservation scores from UCSC phastCons60way (probability of a base to belong to a conserved element computed from multiple alignments of 60 vertebrate genomes) (Siepel et al. 2005; Pollard et al. 2010), as well as those computed from a subset of six genomic alignments from mm10 (mouse), rn5 (rat), dipOrd1 (kangaroo rat), cavPor3 (guinea pig), hetGla2 (naked mole rat), and speTri2 (squirrel), were used to analyze conservation. For multiexonic transcripts, the mean conservation score across exonic bases versus intronic bases was calculated (10% of the bases at the beginning of the first exon [10% of first exon length] and the last 10% bases from the last exon were removed). For monoexonic transcripts, the mean conservation score across exonic bases was compared with the conservation score for the same number of bases downstream from the transcript after a 50-bp gap (excluding any overlap with other predicted transcripts). As a baseline for comparison, we assigned a set of random start sites in the genome to generate a set of transcripts with the same exon structure as our novel and known transcripts, excluding any regions overlapping predicted transcripts.
Coding potential of novel transcripts
To assess the coding potential of transcripts, we used CPAT v2.0, which outputs for each transcript a coding probability based on sequence (Wang et al. 2013). We used the CPAT-predicted coding probabilities (and ORF length) from known protein-coding genes to set cut-offs for identification of putative protein-coding genes in our novel gene set.
For putative novel protein-coding genes, we identified ORFs encoding proteins of greater than 151 amino acids using the ORFik package in R (Tjeldnes et al. 2021). Predicted protein sequences were then compared with the database of nonredundant protein sequences from all organisms via BLASTP (Altschul et al. 1990) or the CDD (Lu et al. 2020). To identify repeat-overlapping ORFs, the genomic position of the predicted ORF was compared with Repbase elements (Jurka 2000; Bao et al. 2015).
Self-similarity of a region on Chromosome 8 was determined using YASS to identify local alignments, with default parameters (Noe and Kucherov 2005) loading the region.
MS analysis
RAW MS files were downloaded from the PRIDE database (https://www.ebi.ac.uk/pride/; project PXD030983) (Giansanti et al. 2022). Data were compared with a combined database consisting of the mouse UniProt database and predicted protein sequences for genes with high CPAT scores, and LFQ quantification was performed. A database search was performed using both MaxQuant (v.2.2.0.0) (Cox and Mann 2008) and FragPipe (v.19.1) (Kong et al. 2017), and only predicted proteins with unique peptides identified with both algorithms are reported.
Analysis of human homologs of novel protein-coding genes
DNA sequences for the ORFs of novel genes with evidence for protein expression were compared with the human genome (build hg38) using BLAT (Kent 2002) via the UCSC Genome Browser (Kent et al. 2002). Genes with one high-scoring BLAST hit were identified. For these genes, tissue panel RNA-seq data from the GTEx project were examined in the UCSC Genome Browser (The GTEx Consortium 2013). Preprocessed single-cell RNA-seq data for human testicular cells (Guo et al. 2018; Karlsson et al. 2021) were queried for each of these genes via the Human Protein Atlas website (http://proteinatlas.org).
Overlap of transcripts with repetitive sequences
RepeatMasker alignments for mm10 genome were downloaded from the UCSC GenomeBrowser (file rmskAlignBaseline.txt.gz, version from 2015-03-22) and processed using custom R scripts (Smit et al. 2015). Only repeat elements belonging to classes “LTR,” “LINE,” “SINE,” “DNA,” and “Satellite” and not <10 bp were selected for further analysis. Among overlapping repeat elements with same ID (with minimum overlap of 15 bp), the element with the highest Smith–Waterman alignment score was chosen. To remove overlaps between joined elements, we kept only the longest one (not <30 bp) for further analysis.
Overlaps of GRanges objects for exons and repeats were found using the findOverlaps function (from the R package GenomicRanges) (Lawrence et al. 2013), ignoring strands and setting a minimum overlap of 5 bp. For each overlap, we calculated lengths of exon–repeat intersection and exon–repeat union. Based on these numbers, we calculated a JI for each exon–repeat overlap as a ratio intersection length/union length. If an exon overlapped several repeat elements, we chose the repeat element with the highest JI. Based on resulting set of exon–repeat overlaps, we annotated each transcript as containing any exon overlap, a 5′ exon overlap, or no repeat overlap.
To understand how much intersection between repeats and exons would be expected by chance, we randomized the de novo transcriptome in the following way. We picked each gene and placed it randomly in the genome, preserving all transcripts and their exon–intron structure. We performed 100 rounds of such randomization and performed the same analysis as for the real de novo transcriptome.
To identify repNames with distribution of JIs different from random, we calculated reverse ECDFs of JIs for observed and each
round of randomized transcriptomes. Next, we chose four values of JIs (0.2, 0.4, 0.6, and 0.8), estimated values of reverse
ECDFs at these values for observed and randomized transcriptomes, and calculated Z-scores as 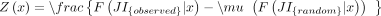
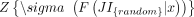 , where F(JI|x) is an estimate of reverse cumulative distribution of JIs at one of the chosen values x ∈ {0.2, 0.4, 0.6,0.8},
and
, where F(JI|x) is an estimate of reverse cumulative distribution of JIs at one of the chosen values x ∈ {0.2, 0.4, 0.6,0.8},
and 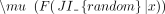 and
and 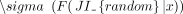 are the mean and standard deviation of reverse cumulative estimates across randomized transcriptomes (Fig. 5C,D).
are the mean and standard deviation of reverse cumulative estimates across randomized transcriptomes (Fig. 5C,D).
To investigate correlation between genome-wide expression of repeats and transcripts with 5′ exon overlapping repeats, we first calculated aggregated expression of all repeat elements across the genome having corresponding repName annotations and calculated relative expression by centering log2(CPM) values around mean across all stages. Next, we quantified expression for each transcript with Salmon, found all transcripts with 5′ exon overlapping corresponding repNames, and calculated relative expression by centering log2(TPM) values around mean across stages. Finally, we averaged relative expression across all transcripts with 5′ exon overlapping corresponding repName (Fig. 5E).
To investigate relationship of repeat-overlapping transcripts with reference transcriptome in GENCODE M23 (basic) annotation, we used gffcompare (v0.12.6) (Pertea and Pertea 2020) with the default parameters and obtained class codes for each transcript in de novo transcriptome. Next, for all transcripts overlapping with their 5′ exon a certain repName, we created contingency tables and performed Fisher's exact tests for each class code separately. For multiple testing correction of P-values, we used the p.adjust function in R with the default parameters.
Quantification of bidirectional promoter activity
Anchors were defined from spermatogenic H3K4me3 ChIP-seq data as follows:
-
Definition of a unique set of H3K4me3 summits for all cell types:
-
- Peak calling with MACS2 in each of the four cell types;
-
- Selection of the most statistically significant summit per peak for all cell types;
-
- Take union of all these summits;
-
- “Merge” adjacent summits—all summits that were <100 bp apart were grouped, and within each group, the summit with the lowest q-value was considered as representative one;
-
- “Merge” even further to end up with ideally one H3K4me3 summit per promoter region—H3K4me3 summits that are located <1 kb apart are grouped, and within each group, the one with the lowest qvalue was kept.
-
-
Association of the H3K4me3 summits to transcriptome TSS:
-
- Main transcript from each gene in the transcriptome was considered (the one with the highest mean expression across all five stages);
-
- H3K4me3 summits located <2 kb away from a TSS were kept;
-
- H3K4me3 summits that have at least two associated multiexonic TSS on opposite strands were discarded.
-
-
Definition of “gene” strand for each anchor:
-
- When we have only one transcript associated with a H3K4me3 summit, its strand will be the gene strand;
-
- When we have at least two transcripts—
-
- If there is only one multiexonic transcript, its strand will be the gene strand;
-
- If there is more than one multiexonic gene, the strand of the closest to the H3K4me3 summit will be the gene strand;
-
- If there is no multiexonic gene, the strand of the most highly expressed transcript will be the gene strand.
-
-
-
Discard the H3K4me3 summits for which the associated transcript is fully located upstream.
-
In cases in which there were two anchors separated by <5 kb and each associated with a monoexonic or multiexonic transcript, the first one was discarded, and when there were two anchors separated by <5 kb and one associated with a monoexonic transcript and the other with a multiexonic transcript, the anchor associated with the monoexonic transcript was discarded.
Total number of anchors identified was 11,140. For each anchor, QuasR (Gaidatzis et al. 2015) was used to count the exonic signal within the exons of the associated gene, the antisense signal in a 2-kb region located on the opposite strand and starting from the anchor, and the H3K4me3 signal in a 2-kb region centered around the anchor (both strands considered). For liver ChIP-seq data, the same procedure was applied but in this case using the set of UCSC known genes (R package TxDb.Mmusculus.UCSC.mm10.knownGene), as well as mouse liver RNA-seq (European Nucleotide Archive [ENA; https://www.ebi.ac.uk/ena/browser/home] accession PRJEB28810) and H3K4me3 ChIP-seq data (ENCODE, accession GSM769014), to get a set of 9554 anchors.
Data access
All raw and processed sequencing data generated in this study have been submitted to the NCBI Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/) under accession number GSE214316. The novel annotation is available in GTF form in our GEO submission and as a browser track session from the UCSC Genome Browser at https://genome-euro.ucsc.edu/s/mgill80/Gill_Transcriptome_2023. R code used in this analysis is available as Supplemental Code.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
We thank members of the FMI animal facility for their support. We thank Hubertus Kohler (FMI) for isolation of cells via FACS and the FMI Functional Genomics platform, particularly Sebastien Smallwood and Stephane Thiry for library generation and sequencing. We thank Jan Seebacher (FMI) for advice and processing of mass-spectrometry data. We thank the FMI Computational Biology group, particularly Michael Stadler and Charlotte Soneson, and members of the Peters’ laboratory for helpful discussions and critical feedback. We thank Elizabeth Snyder (Rutgers University) for providing the GTF files for the annotation from Gamble et al. (2020). This research was supported by the Novartis Research Foundation and the Swiss National Science Foundation (31003A-146293; 31003A-172873). This work also received funding from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation programme (grant agreement ERC-AdG 695288–Totipotency).
Author contributions: M.E.G. and A.H.F.M.P. conceived the study. M.E.G. isolated cells and total RNA for primary RNA-seq experiments. S.E. isolated cells and total RNA for secondary (round and elongating spermatid) RNA-seq experiments. C.-Y.L., S.E., and S.C. isolated cells and performed H3K4me3 ChIP-seq. M.E.G., A.R., and E.A.O. performed bioinformatic analyses. E.A.O. supervised bioinformatic analysis. M.E.G. and A.H.F.M.P. supervised the project. M.E.G. wrote the manuscript. M.E.G. and A.H.F.M.P. edited the manuscript, with input from all authors.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.278060.123.
-
Freely available online through the Genome Research Open Access option.
- Received May 2, 2023.
- Accepted September 29, 2023.
This article, published in Genome Research, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








