
Proteome-wide structural analysis quantifies structural conservation across distant species
Abstract
Traditional evolutionary biology research mainly relies on sequence information to infer evolutionary relationships between genes or proteins. In contrast, protein structural information has long been overlooked, although structures are more conserved and closely linked to the functions than the sequences. To address this gap, we conducted a proteome-wide structural analysis using experimental and computed protein structures for organisms from the three distinct domains, including Homo sapiens (eukarya), Escherichia coli (bacteria), and Methanocaldococcus jannaschii (archaea). We reveal the distribution of structural similarity and sequence identity at the genomic level and characterize the twilight zone, where signals obtained from sequence alignment are blurred and evolutionary relationships cannot be inferred unambiguously. We find that structurally similar homologous protein pairs in the twilight zone account for ∼0.004%–0.021% of all possible protein pair combinations, which translates to ∼8%–32% of the protein-coding genes, depending on the species under comparison. In addition, by comparing the structural homologs, we show that human proteins involved in the energy supply are more similar to their E. coli homologs, whereas proteins relating to the central dogma are more similar to their M. jannaschii homologs. We also identify a bacterial GPCR homolog in the E. coli proteome that displays distinctive domain architecture. Our results shed light on the characteristics of the twilight zone and the origin of different pathways from a protein structure perspective, highlighting an exciting new frontier in evolutionary biology.
The rapid development of DNA sequencing technologies has delivered high volumes of genome information for many species (Ng and Kirkness 2010). The availability of the entire genome for many model organisms has enabled researchers to perform a significant number of large-scale comparative genomic, phylogenetic, and evolutionary studies. In contrast, information on protein structures has accumulated slower than genome sequence. So far, Homo sapiens is the best represented species in the Protein Data Bank (PDB) (Berman et al. 2000; Burley et al. 2021), yet the experimental structures in the database still cover no more than half of the protein-coding genes in the human genome.
It is widely accepted that tertiary structures of proteins are more conserved than their amino acid sequences (Chothia and Lesk 1986; Rost 1999; Wood and Pearson 1999; Illergård et al. 2009) and that the structures determine the biological functions of proteins. As Rossman and Liljas noted half a century ago, “the preservation of structure may originate in the three-dimensional requirement for the conservation of essential functions” (Rossman and Liljas 1974). Proteins related by evolution can have similar tertiary structures, even when the sequence similarities are statistically marginal or seemingly nonexistent (Eventoff and Rossmann 1975; Pastore and Lesk 1990). For example, the globin family of proteins shows how two proteins from distant species can have nearly identical tertiary structures, despite having sequence identities as low as 16% (Lesk and Chothia 1980). Another quintessential demonstration is the Rossman fold, which is a nucleotide binding motif discovered in the 1970s (Rao and Rossmann 1973). Two distantly related proteins with a Rossman fold can have strictly conserved structures with no detectable sequence identities (Toledo-Patiño et al. 2022). In this regard, structural similarity could be a better indicator than sequence similarity for determining the evolutionary relationship in which there is a huge evolutionary gap.
Currently, we have limited knowledge about the conservation of protein structures at the proteomic level. One key factor contributing to this undesirable situation is the limited number of experimentally determined protein structures. Fortunately, the development of AlphaFold2 (Tunyasuvunakool et al. 2021), a machine-learning algorithm, has enabled us to model millions of proteins at proteomic scales. AlphaFold2 can provide reliable in silico modeled structures roughly as good as experimentally determined structures. Combining experimental structures in PDB and computationally modeled structures by AlphaFold2 should allow us to analyze protein structures on a large scale.
The term “twilight zone” in the field of evolutionary biology refers to the evolutionary relationships between proteins that are not easily detectable by sequence similarity alone (Doolittle 1986). Typically, the twilight zone is defined as having a sequence identity of 20%–25% (Doolittle 1986; Sander and Schneider 1991). Proteins in the twilight zone have diverged so much over evolutionary time that their sequences no longer show significant similarity, making it difficult to infer evolutionary relatedness based on sequence alone. Protein structure, on the other hand, may hold the key to unlocking the evolutionary relationships between these proteins. For many years, we have been limited by the availability of structural information at the genomic level, making it difficult to determine the characteristics and size of the twilight zone, but with the advent of computational tools such as AlphaFold2, we now have the opportunity to gain insight into the evolutionary relationship between highly diverged proteins in the “twilight zone” and beyond.
Having access to the structural information for the whole proteome should also enable us to compare distantly related species at a genome scale. This information can be particularly valuable when investigating species separated by significant evolutionary gap, such as eukaryotes and prokaryotes, including bacteria and archaea. H. sapiens is well studied at the protein structure level. Escherichia coli is the most frequently used model organism for bacteria and the best-represented bacteria in the PDB (Berman et al. 2000; Burley et al. 2021). Methanocaldococcus jannaschii was the first archaeon to have its genome sequenced (Garrett 1996). It has been suggested that the first eukaryote originated from the endosymbiosis of ancestral bacteria and archaea (Martin and Müller 1998; Koonin 2006, 2015; Martin and Koonin 2006; Archibald 2015; Martin et al. 2015; Eme et al. 2017). However, links between eukaryotes and bacteria or archaea have become obscured as protein sequences diverge over evolutionary time. Gene loss and gain along the eukaryote tree of life has further accelerated the change in the distribution of bacterial or archaeal descent genes since the last common ancestor of eukaryotes. Consequently, how many protein-coding genes in the human genome are structurally conserved to their prokaryotic homologs and how these proteins were distributed remain an open issue. The uncertainty in sequence alignment at this phylogenetic distance highlights the importance of using structural comparison approaches to explore the relationship between organisms across different domains of life.
Here, we present the first attempt to make a global structural-based comparison of the protein-coding genes from the three model organisms at the whole proteomic level. We used a sequence-independent protein structural comparison algorithm to compare the structures in the PDB and AlphaFold2 database in a pairwise manner. Using E. coli and M. jannaschii as representative bacterial and archaeal proteomes for the comparative analysis, we mapped the distribution of structural similarity with sequence identity at the genomic level and characterized the twilight zone. We also investigated enrichment patterns in human proteins that are structurally conserved with archaeal and bacterial orthologs. These results presented here showed an exciting new frontier in the field of evolutionary biology and provided insight into the chimeric nature of eukaryotic genomes.
Results
Global analysis of the AlphaFold2-predicted structures reveals distinct statistical characteristics between H. sapiens and E. coli or M. jannaschii proteomes
We obtained a total of 36,764 structures from the PDB (Berman et al. 2000; Burley et al. 2021) and 29,527 modeled structures for H. sapiens, E. coli, and M. jannaschii from the AlphaFold2 database (Tunyasuvunakool et al. 2021) (for a schematic representation for the workflow and methods used in this study, see Supplemental Fig. S1). Although the number of the structures from the PDB is greater than that from the AlphaFold2 database, the experimental structures cover no more than one-third of the proteomes for any of the three species because there are a lot of redundant structures obtained under various experimental conditions for a same protein. By comparison, the AlphaFold2 database almost covers the entire proteome, except for the noncanonical isoforms for some protein-coding genes.
Before the global analysis of all the structures, we used a graph-based community clustering approach to trim the unfolded regions and separate multidomain proteins into single domains based on the predicted alignment error matrix generated by AlphaFold2. An example of the structure trimming is depicted in Figure 1, A and B, in which human HMGB1, an abundant architectural chromosomal protein, was separated into two HMG box domains. The predicted aligned error (PAE) matrix is a heatmap image generated by AlphaFold2 that displays the distance error for every pair of residues (Fig. 1A). The PAE value for any of the two residues in a given protein indicates how confident the relative three-dimensional position between the two residues can be determined. Therefore, if two individual domains are connected by a flexible linker, as in the case of the two HMG box domains in HMGB1 (Fig. 1A), then the relative position between inter-domain residues cannot be confidently determined, but the relative position between two residues within a single domain can be confidently determined with a high PAE value. In addition, the PAE matrix can suggest nonstructured regions in a protein. In the case of HMGB1, the nonstructured C-terminal tail shows low PAE values between most intra-domain residues (Fig. 1B). To define individual domains and trim nonstructured regions without human intervention for hundreds of thousands of protein structures, we used a graph-based clustering algorithm (Leiden) to detect clusters of residues based on the graph connectivity in the heatmap image (Fig. 1B).
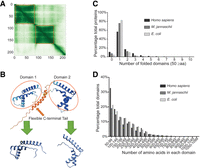
Analysis of the AlphaFold2-predicted structures reveals distinct statistical characteristics between the proteomes of the three model organisms. (A,B) An example illustrating how AlphaFold modeled structures were trimmed into single domains. The orange box indicates the range for each domain. The boundaries of the orange boxes were determined using the Leiden algorithm. (A) The alignment error matrix of the protein HMGB1 (UniProt ID P09429). The axes represent the positions of residues along the sequence of the protein. The x-axis corresponds to the reference position, and the y-axis corresponds to the predicted position. The color at the coordinate (x,y) indicates the AlphaFold2's expected position error of residue y if residue x serves as the reference and the predicted and true positions of residue y are compared. Darker color indicates lower errors or higher accuracy, whereas lighter color indicates higher error or lower accuracy. Thus, the matrix is an indication of the confidence of the relative position between any of the two residues. (B) The modeled tertiary structure of HMGB1 and its relationship with the two individual domains. Blue color indicates high model confidence, and orange color indicates low model confidence. (C) Distribution of the number of domains in each protein structure for all the modeled protein structures from each of the three species in the AlphaFold2 database. Unfolded domains and domains smaller than 50 aa were excluded from the analysis. (D) Distribution of the number of amino acids in each domain for all the modeled protein structures in the AlphaFold2 database. Unfolded domains and domains smaller than 50 aa were excluded from the analysis
Using the trimmed single-domain structures from the AlphaFold2 database, we examined the statistical characteristics of the H. sapiens, E. coli, and M. jannaschii proteomes (Fig. 1C,D). The results show that most proteins in the proteomes of the three species contain only one single domain. A small fraction of genes cannot encode any folded domain >50 aa (11.5% in H. sapiens, 7.6% in E. coli, and 4.2% in M. jannaschii). In general, multidomain proteins are more frequently observed in the human proteome than the other two prokaryote proteomes (average number: 1.15 for M. jannaschii, 1.16 for E. coli, and 1.58 for H. sapiens) (Fig. 1C). This result agrees with the previous finding that the fraction of proteins with two or more domains is greater in eukaryote and multicellular organisms (Björklund et al. 2005; Han et al. 2007). Although the average number of domains (excluding unfolded domain and domain size <50 aa) is greater, the average size of the domains is smaller in the human proteome than in the other two prokaryote proteomes (average size: 225 aa for M.jannaschii, 246 aa for E. coli, and 204 aa for H. sapiens, excluding domains <50 aa) (Fig. 1D). We reckoned that these observations are reasonable, as it has been established that metazoans tend to have more multidomain proteins than fungi and protozoa, which, in turn, have more than bacteria and archaea (Tordai et al. 2005). Additionally, domain size determines the versatility and mobility of the domain building blocks, and smaller domains are more likely to be used in multidomain proteins (Altschul et al. 2005; Pozzoli et al. 2008). Thus, as the complexity of the organism increases, the average size of the domains decreases.
Protein structure comparison can reveal evolutionary relationships between distantly related species
In most cases, apart from the functional sites on the protein surface and the hydrophobic core maintaining the structural integrity, a large portion of amino acid residues in a given structure is not evolutionarily important. Therefore, tertiary structures of proteins are often more conserved than their amino acid sequences.
An example to illustrate this point was shown in Figure 2, A and B, in which structural and sequence comparisons of H. sapiens glutathione S-transferase mu 4 and E. coli glutathione S-transferase yfcF were made. Here, we used a sequence-independent algorithm, TM-align, to measure the similarity between the structures. We used EMBOSS Stretcher (Myers and Miller 1988) to calculate the similarity between the amino acid sequences. As is shown, the structures of H. sapiens and E. coli glutathione S-transferase are nearly superimposable, whereas the amino acid sequences of the two proteins only share 35% similarity and 19% identity.
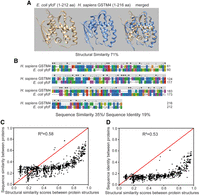
Comparison between structural similarity and sequence similarity or identity. (A,B) An example showing that proteins with nearly superimposable tertiary structures may have a low level of sequence similarity or identity. (A) AlphaFold2 modeled protein structures for E. coli and human glutathione S-transferase and alignments of the two structures. (B) Sequence alignment of E. coli and human glutathione S-transferase. The color indicates residue groups (blue, hydrophobic; red, positive charge; magenta, negative charge; green, polar). Sequence identity and similarity are indicated on top of the amino acid letters. An asterisk suggests a fully conserved position; a colon suggests strong similarity, and a period suggests weak similarity. (C,D) Comparison between structural similarity (TM-score) and sequence similarity (C) or identity (D) for 1000 pairs of selected experimental structures. The samples were obtained through stratified sampling, in which random sampling was performed at each stratum level (structural similarity ranging from x to x + 0.1, where x counts from 0.0 to 0.9). In each structure pair, one structure is from H. sapiens, and the other is from E. coli. Note that these two figures illustrate the correlation between structural similarity and sequence similarity or identity. The stratified sampling approach used here means that the figures do not represent the actual distribution of the structural similarity and sequence similarity or identity. For the list of selected experimental structures, see Supplemental Table S1. For the actual distribution at a genomic level, see Figure 3, D through F.
To further elucidate how structural similarity is related to sequence similarity in distantly related species, we randomly selected 1000 unique pairs of experimental structures with similarity scores roughly equally distributed in the range from zero to one (we randomly picked 100 pairs of experimental structures with structural similarity ranging from x to x + 0.1, where x counted from zero to 0.9 with an increment of 0.1). The comparison between the structural and sequence similarities in the selected structure pairs suggests that high sequence similarity is always accompanied by high structural similarity. However, the conclusion is not necessarily vice versa because when the sequence similarity is low, the proteins may still share a high structural similarity (Fig. 2C; Supplemental Table S1). This trend persists when sequence identity, rather than sequence similarity, is examined (Fig. 2D; Supplemental Table S1). Thus, sequence similarity and identity are not good indications of the relationship between two protein structures when the two are only moderately alike.
An enormous number of structural homologs can be discovered through global structural alignment, amounting to hundreds of thousands of pairs
When studying the evolutionary relationships between distantly related species, relying solely on sequence similarity and identity can be challenging due to the limited and ambiguous information they provide. Proteins often diverge to the extent that sequence-based methods alone cannot detect the similarities between them. Therefore, we wanted to investigate how much we could discover by using the structural information of the entire proteome. To accomplish this, we compared all possible combinations of individual domain structures between three species in a pairwise manner. To gain an estimation of the distribution of structural similarities between protein pairs and prevent duplicated counting of numbers, we exclusively used the AlphaFold2 modeled structures.
The global pairwise structural alignment revealed hundreds of thousands of between-species protein pairs with good structural similarity (Fig. 3A–C). For instance, when we compared the proteomes of E. coli and H. sapiens, we found 634,284 protein pairs with a structural similarity above 0.4, 122,102 pairs with a similarity above 0.5, and 33,323 pairs with a similarity above 0.6. Similarly, when we compared the proteomes of M. jannaschii and H. sapiens, we found 296,285 protein pairs with a structural similarity above 0.4, 49,986 pairs with a similarity above 0.5, and 8736 pairs with a similarity above 0.6. As the protein similarities cutoff increases, the number of protein pairs decreases sharply. It should be noted that the proteome sizes of M. jannaschii, E. coli, and H. sapiens are different, with H. sapiens being the largest, E. coli being the next largest, and M. jannaschii being the smallest. Although the absolute number of pairs at each similarity level differs, the percentage numbers are actually similar. Using a cut-off score of 0.5 (this is a widely cited and empirically determined cutoff for the TM-score [Zhang and Skolnick 2005; Xu and Zhang 2010]), we found that 0.09% of protein pairs had similar structures when comparing H. sapiens and E. coli, 0.09% when comparing H. sapiens and M. jannaschii, and 0.19% when comparing E. coli and M. jannaschii. Around 9500 protein-coding genes from human proteomes are involved in the protein pairs between E. coli and H. sapiens with structural similarity above the cutoff, and around 7200 human protein-coding genes from human proteome are involved in protein pairs between M. jannaschii with structural similarity above the cutoff. (Note that one protein may share a similar structure with multiple proteins from another proteome, so the number of protein pairs is greater than the number of protein-coding genes.)
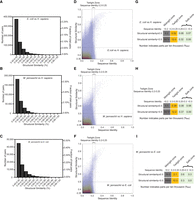
Distribution of structural similarity and sequence identity when comparing proteins at the proteomic scale. (A–C) Distribution of the numbers of protein pairs with structural similarity above 0.4 when comparing AlphaFold2 modeled proteins from one species with those from another. The subfigures compare the proteomes of E. coli versus H. sapiens (A), M. jannaschii versus H. sapiens (B), and M. jannaschii versus E. coli (C). Note that the number of protein-coding genes in each species differs, so a greater absolute number does not necessarily indicate a greater proportion. The left y-axis indicates the absolute number, and the right y-axis indicates the proportion in all possible protein combinations. (D–F) Overlay of 2D scatter plot and 2D contour plot depicting the distribution of structural similarity and sequence identity of the proteins. The subfigures compare the proteomes of E. coli versus H. sapiens (D), M. jannaschii versus H. sapiens (E), and M. jannaschii versus E. coli (F). The vertical dashed lines indicate the sequence identity of 0.2 and 0.25, and the horizontal dashed line indicates the structural similarity of 0.5. (G–I) The proportion of protein pairs of all possible combinations in each zone. The subfigures compare the proteomes of E. coli versus H. sapiens (G), M. jannaschii versus H. sapiens (H), and M. jannaschii versus E. coli (I). The twilight zone is defined as a sequence identity between 0.2 and 0.25. The safe zone is defined as sequence identity greater than 0.25, and the midnight zone is defined as sequence identity less than 0.2.
The AlphaFold2 modeled structures enabled traversal of the “twilight zone” at the boundary of the limitation of sequence alignment
The limit for confidently detecting evolutionary relationships among proteins in the sequence-alignment analysis is often referred to as the “twilight zone,” with a threshold of ∼20%–25% or 20%–30% sequence identity (Doolittle 1986; Chung and Subbiah 1996; Rost 1999). Esser and coworkers determined that there are only 850 proteins that share sequence identity >25% between yeast and 60 prokaryotic genomes using a local-alignment approach (Esser et al. 2004). Apparently, our findings reveal a larger number of structurally similar protein-coding genes compared with the number of homologs determined by sequence identity a decade ago, despite the greater evolutionary distance between human and prokaryotes than between yeast and prokaryotes.
So where are these structurally similar proteins located? How many of them are in the twilight zone (sequence identity 20%–25%), and how many are hiding in the “midnight zone” (sequence identity <20%)? What is the size of these zones? To answer these questions, we performed global sequence alignments between the individual domain structures and plotted the distributions of the structural similarity and sequence identity for all protein pairs with a structural similarity above 0.35 (Fig. 3D–F). As we were performing pairwise alignment of all possible combinations, most data points were concentrated in the region below the structural similarity and sequence identity thresholds, which is expected as they represent irrelevant proteins. Most protein pairs with structural similarity above 0.5 have a sequence identity below the 25% threshold. A significant number of proteins with high structural similarity are found in the midnight zone, where sequence identity is almost undetectable.
The global alignment algorithm is typically slow. Because of the limitation in computational resources, we only calculated the sequence identity for the protein pairs with a structural similarity above 0.35. Nevertheless, we used random sampling to pick a fraction of the total and obtain a representative subset to approximate the distribution of the structural similarity and sequence identity below the structural similarity 0.35 (for the distribution of random sampling, see Supplemental Fig. S2). Combining the distribution derived from the random sampling (Supplemental Fig. S2) and the complete data for proteins with structural similarity above 0.35 (Fig. 3D–F), we estimated the sizes of the safe zone (sequence identity > 25%), twilight zone, and midnight zone (Fig. 3G–I). As expected, the midnight zone is much larger than the twilight, which is in turn larger than the safe zone. The size of the safe zone (relative to the number of total possible combinations) is larger in the comparison of E. coli and M. jannaschii proteomes than in the comparisons of H. sapiens with either E. coli or M. jannaschii proteomes. For proteins with evident structural homology (>0.5), the twilight zone (with sequence identities 20%–25%) is two to five times larger than the safe zone, and the midnight zone is even larger, spanning 10–20 times the size of the twilight zone. These results indicate that in large-scale phylogenetic surveys encompassing all alignments with sequence identities >25%, we are merely sampling a tiny fraction of structurally homologous proteins. Furthermore, the proportions of numbers with structural similarity above and below 0.5 suggest that even the safe zone is not entirely safe. However, this largely depends on how we set the boundary between the twilight zone and the safe zone. If the boundary was set to a sequence identity of 0.3, then >98% of the proteins in the safe zone should share more than 0.5 structural similarities (Fig. 4A–C). In fact, the distribution histogram for structural similarities within the twilight zone shows two clear clusters: one centered at around 0.7 structural similarity, representing the true homologous protein pairs; and the other right-skewed cluster represents groups of data points at the extreme tail of the population, corresponding to nonhomologous protein pairs (Fig. 4D–F). Given a sufficiently large number of nonhomologous protein pairs, it is inevitable that the sequence identity between some irrelevant proteins will exceed the cutoff of 0.2 by chance alone, leading to uncertainty in the determination of the homologous relationship solely based on the sequence.
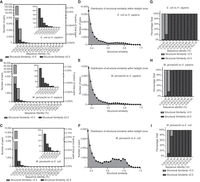
Examination of the distribution of structural similarity across different ranges of sequence identity. (A–C) Numbers of protein pairs (left, y-axis) and proportion in all possible combinations (right, y-axis) across different sequence identity ranges. The inserted pictures at the top right corner depict zoomed-in figures for protein pairs with sequence identity above 0.3. The subfigures compare the proteomes of E. coli versus H. sapiens (A), M. jannaschii versus H. sapiens (B), and M. jannaschii versus E. coli (C). The light gray color bar indicates those with structural similarity less than 0.5, and the dark gray color bar indicates those with structural similarity equal to or greater than 0.5. The light gray bar is invisible in the zoomed-in pictures at the corners, because very few protein pairs are identified with structural similarity less than 0.5 within the indicated ranges. (D–F) Distribution of structural similarity within the twilight zone, defined as sequence identity between 0.2 and 0.25. The subfigures compare the proteomes of E. coli versus H. sapiens (D), M. jannaschii versus H. sapiens (E), and M. jannaschii versus E. coli (F). The bin size for the distribution plot is 0.25. (G–I) The percentage fraction of protein pairs with structural similarity greater or less than 0.5 across different sequence identity ranges. The subfigures compare the proteomes of E. coli versus H. sapiens (G), M. jannaschii versus H. sapiens (H), and M. jannaschii versus E. coli (I). The x-axis ends at a sequence identity range of 0.55–0.70 because no protein pairs are identified above this value range.
Figure 3, G through I, shows that if the boundary between the safe zone and twilight zone is set at a sequence identity of 0.25, then the safe zone would make up 0.0014% of the total possible protein combinations, whereas the twilight zone would constitute 0.0052% of the total possible protein combinations for E. coli versus H. sapiens. Similar numbers were obtained when we compared the H. sapiens proteome with the M. jannaschii proteome. The fraction numbers increased to 0.009% and 0.021% when we compared the M. jannaschii proteome with the E. coli proteome, suggesting that the selection of species may have a significant impact on the size of the twilight zone. Above the structural similarity of 0.5, the twilight zone for comparing the H. sapiens and E. coli proteomes involves around 2854 (16%) human genes and 1279 (32%) E. coli genes, and the twilight zone for comparing the H. sapiens and M. jannaschii proteomes involves around 1359 (7.5%) human genes and 497 (29%) M. jannaschii genes. In contrast, 9172 (51%) and 6846 (38%) human genes are involved in the midnight zone above a structural similarity of 0.5 when comparing the H. sapiens proteome with the E. coli and M. jannaschii proteomes, respectively. If a structural similarity above 0.5 is used as the criterion for evaluating the false-positive rates of global sequence alignment, then most of the aligned protein detected in the range of sequence identity of 0.2–0.3 should be considered as false-positive hits. The false-positive rate drops sharply when the sequence identity increases to above 0.3, which agrees with the empirical determination for the range of the twilight zone (Fig. 4G–I).
Next, we inspected the identities of the proteins found within the twilight zone. Tables 1 and 2 display the lists of the most structurally similar protein pairs in the twilight zones when comparing the H. sapiens proteomes with either the E. coli proteome or M. jannaschii proteome (Tables 1, 2; Supplemental Tables S2, S3). Membrane transporters are prominent in the most structurally conserved protein list of the twilight zone when comparing H. sapiens to E. coli (Table 2; Supplemental Table S2). Incidentally, it is known that membrane proteins are less conserved than water-soluble proteins across the tree of life in terms of sequence, presumably due to a stronger adaptive selection in changing environments (Sojo et al. 2016). Apart from the membrane proteins, other proteins in the two tables participate in various biological processes, such as small-molecule metabolism, protein synthesis, and protein degradation. No other generalizable features are readily apparent from direct observation of the tables.
List of the most structurally conserved proteins between H. sapiens and E. coli in the twilight zone
List of the most structurally conserved proteins between H. sapiens and M. jannaschii in the twilight zone
Global comparison of the structures between H. sapiens and E. coli or between H. sapiens and M. jannaschii indicates that structurally conserved genes are enriched in specific pathways
Next, we asked which protein is the most structurally conserved when comparing all human proteins with those from E. coli or M. jannaschii. Because we are not examining the distribution of structural similarity and size of the twilight zone, duplicate counting is no longer an issue. We included all the experimental structures along with the computed structures in the analysis. The best aligned score among all the structures was used for the evaluation of the level of structural conservation. Shown in Tables 3 and 4 are lists of proteins (domains) sharing the greatest structural similarities upon comparing the structures of the two species. As is evident from the tables, all the structurally conserved proteins between H. sapiens and E. coli are metabolic enzymes involved in carbohydrate, amino acid, and nucleotide metabolism (Table 3), whereas almost all the most conserved proteins between H. sapiens and M. jannaschii are involved in protein synthesis (Table 4). These results imply that eukaryotic proteins that evolved from bacteria and archaea are enriched in different biological processes, consistent with the findings of Esser and colleagues (Esser et al. 2004).
List of the most structurally similar proteins between H. sapiens and E. coli
List of the most structurally similar proteins between H. sapiens and M. jannaschii
It is important to note that single gene comparison may not always provide reliable information on the evolutionary history of an organism. For instance, as shown in Table 3, the most structurally conserved gene MTR is associated with methionine synthesis. However, humans have just one enzyme related to methionine synthesis, although E. coli has all the genes required for this pathway. Humans may have lost the other genes in this pathway during evolution, but horizontal gene transfer could also be a reasonable explanation for the high similarity. The use of multiple genes is necessary to gain a more comprehensive understanding of an organism's evolutionary history.
Therefore, to further investigate the enrichment preference of the structurally conserved genes, we ranked all the human protein-coding genes based on the structural conservation scores for each gene (Supplemental Table S4) and performed gene set enrichment analysis (Mootha et al. 2003; Subramanian et al. 2005) with KEGG pathway or module gene sets (Supplemental Table S5). In line with the observation of the most conserved proteins, human proteins bearing a strong resemblance to E. coli proteins were selectively enriched in various small-molecule metabolic pathways (Fig. 5A). The glycolytic pathways and the subsequent catabolism of pyruvate were ranked at the top of the enriched pathway list, which is in line with the observation that the rate of evolution of the glycolytic enzymes is exceptionally slow (Fothergill-Gilmore 1986). Fatty acid metabolism was ranked immediately after the glycolytic pathway, consistent with the fact that eukaryotes harbor the same lipid type as bacteria (Lykidis 2007). Sugar and fat are the primary energy sources for cells. Thus, all three top enriched pathways were linked to the energy supply. In contrast, human proteins resembling those from M. jannaschii were preferentially shown as subunit compositions of cytosolic ribosome and proteasome, two complexes in charge of protein synthesis and degradation, respectively (Fig. 5B). Concordantly, proteasome and cytosolic ribosome proteins are also prominent among the 50 most conserved yeast proteins that are specific to archaea in a previous study, which identifies 850 yeast proteins with homologs in prokaryotes (Esser et al. 2004). Unlike the top enriched pathways for conserved genes to E. coli proteins, these two complexes deal with macromolecule metabolism instead of small-molecule metabolism. To confirm the differential enrichment pattern, we directly compared the structural conservation scores to E. coli and M. jannaschii. Indeed, human proteins related to energy supply are more likely to resemble E. coli structures than M. jannaschii structures (Fig. 5C), whereas the human proteins constituting cytosolic ribosome or proteasome are more similar to M. jannaschii homologs than E. coli homologs (if any) (Fig. 5D).
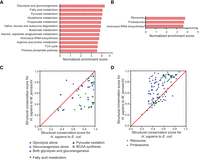
Global comparison of the structures between distantly related species indicates that structurally conserved genes are enriched in specific pathways. (A,B) Gene set enrichment analysis for all protein-coding genes in the human genome with structural conservation scores for H. sapiens to E. coli (A) and H. sapiens to M. jannaschii (B) used as ranking metrics. The built-in KEGG pathway gene sets were used to evaluate the enriched gene set. The bar charts show significantly enriched gene sets with normalized enrichment scores over 3.5. The enrichment analysis reveals the likelihood of the genes in the indicated gene set being ranked at the top of the ranked list based on structural similarity. (C,D) Comparison of the structural conservation scores for H. sapiens to E. coli and H. sapiens to M. jannaschii of protein-coding genes in selected gene sets. Points above the diagonal indicate that the conservation score on the y-axis is better than that on the x-axis. Part C corresponds to the gene sets enriched in the conserved genes for H. sapiens to E. coli. Part D corresponds to gene sets enriched in the conserved genes for H. sapiens to M. jannaschii. For the lists of genes in the gene sets, see Supplemental Table S5.
To further investigate the structural similarity between the three species, we compared the similarity scores for all the ribosome subunits from M. jannaschii to their annotated orthologs in the E. coli ribosome and human ribosome (Supplemental Table S6). It is worth noting that humans have two types of ribosomes, those in the cytosol, which synthesize all the protein-coding genes in the nuclear genome, and those in mitochondria, which are responsible for synthesizing 13 protein-coding genes in mitochondrial DNA. Consistent with the analysis results above, the paired comparison revealed the H. sapiens cytosolic ribosome resembles the M. jannaschii ribosome more than the H. sapiens mitochondrial ribosome and E. coli ribosome do (Supplemental Fig. S3). The human mitochondrial ribosome subunits bear the least structural similarity to the M. jannaschii ribosome subunits.
Global structural comparison provides insight into the origin of G protein-coupled receptor (GPCR) signaling pathway
Not every human protein can be assigned with a structural homolog from the E. coli or M. jannaschii proteomes. Some proteins may have homolog(s) in only one of the two prokaryotic organisms, whereas others may not have a homolog in any of the two organisms. Then what is the distribution of these proteins? If the cut-off structural similarity score is set at 0.5, then 42% of the human proteins cannot be assigned with a structural homolog from either the E. coli or M. jannaschii proteomes, 18% of the proteins can be matched to structural homolog(s) in the E. coli proteome only, and 4% of the proteins can be matched to structural homolog(s) in the M. jannaschii proteome only. The remaining 35% of the proteins have homologs in both prokaryotic organisms (Fig. 6A; Supplemental Table S7). In general, the proteins with structural homologs in E. coli outnumbered those with homologs in M. jannaschii.
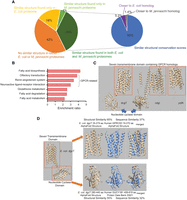
Global structural comparison sheds insight into the origin of the GPCR signaling pathway. (A) Distribution of structurally conserved and nonconserved proteins in the human proteome. Fractions of human proteins with structural homologs in the E. coli or M. jannaschii proteomes are shown. A sub-pie chart is presented for proteins with structural homologs found in both the E. coli and M. jannaschii proteomes. “Similar structural conservation scores” indicates a difference smaller than 0.1 between the scores, whereas “closer to E. coli homolog” or “closer to M. jannaschii homolog” indicates that the difference in the structural conservation score is greater than 0.1. (B) Gene overrepresentation analysis for proteins with a structural homolog found in E. coli only. For the list of proteins used for the overrepresentation analysis, see Supplemental Table S7. (C) The tertiary structures of the three GPCR-like proteins found in the E. coli proteome. The orange box indicates the seven-transmembrane domain. (D) Structural analysis for E. coli dgcT. The protein consists of two domains, one seven-transmembrane domain, and one nucleotide cyclase. The most structurally similar homolog for each domain is shown.
Consistent with the gene set enrichment analysis (Fig. 5A), overrepresentation analysis showed that genes involved in fatty acid metabolism are overrepresented in the subset that has homolog(s) in the E. coli proteome only (Fig. 6B). Three seemingly irrelevant pathways (olfactory transduction, renin–angiotensin system, and neuroactive ligand–receptor interaction) are also overrepresented in the subset. In fact, a common feature between the three pathways is that all three pertain to the communication between the extracellular environment and the GPCR-mediated signaling cascade. In particular, the olfactory transduction gene set contains more than 300 GPCR proteins, and all these proteins were present in the subset with structural homologs in the E. coli proteome only.
The GPCR superfamily is the largest superfamily in the human genome, with the seven-transmembrane (7TM) domain being its most prominent feature. Although proteins with the 7TM topology have been found in prokaryotes such as rhodopsin, the phylogenetic relationships between the prokaryotic 7TM proteins and eukaryotic GPCR proteins cannot be inferred unambiguously from sequence similarity alone (Strotmann et al. 2011). However, by referencing protein structures, a previous study has provided evidence that eukaryotic GPCR may share common ancestors with prokaryote rhodopsins (Shalaeva et al. 2015).
Herein, upon close inspection of the overrepresented gene sets, we found that all the overrepresented human GPCR proteins (Fig. 6B) share a moderate structural resemblance to three E. coli proteins: dgcT, cdgI, and trhA (also known as yqfA) (Fig. 6C). What is interesting here is that both dgcT and cdgI contain two domains: one 7TM domain and one nucleotide cyclase domain (Fig. 6D). The cyclase domains from dgcT and cdgI share similar structures with an experimental protein structure for human guanylate cyclase (PDB 2WZ1). As its name indicates, GPCR interacts with G-protein, and G-protein binds to adenylate cyclase to signal the downstream events. The core tertiary structures for adenylate cyclase and guanylate cyclase are essentially the same (Roelofs et al. 2001; Winger et al. 2008). Additionally, structure-guided sequence alignment showed that the 7TM domains of dgcT and cdgI share some sequence similarity with known human GPCR proteins (Supplemental Fig. S4). Therefore, the fusion protein composed of a 7TM domain and a cyclase domain can be considered as a primitive template for the GPCR signaling pathway, in which G-protein is omitted and GPCR directly interacts with the nucleotide cyclase. Protein with such a domain architecture is absent in the M. jannaschii proteome. These results suggest that the link between GPCR-like transmembrane protein and cyclic nucleotide secondary messenger may have been established before the emergence of eukaryotes.
The global structural comparison suggests an archaeal origin of core proteins relating to information storage and processing
Next, we investigated the overrepresented protein groups in the protein subset, of which structurally similar homologs can be found in the M. jannaschii but not the E. coli proteome. The overrepresentation analysis revealed that proteins relating to systemic lupus erythematosus and alcoholism were overrepresented in this protein subset (Fig. 7A). However, further evaluation of the enriched genes suggests that the computational algorithm determined that these two pathways were overrepresented because a good number of histones were included in the gene sets. (Somehow, the KEGG database does not include a gene set dedicated to histones.) Histones have multiple copies in the human genome, and each copy was assigned a unique gene entry, leading to a disproportionated number in the two gene sets. Thus, the overrepresentation analysis actually indicates that histones are among the overrepresented proteins with homologs in M. jannaschii only. In addition to histones, the analysis reveals that proteins involved in RNA transcription (general transcription factors and RNA polymerase) and DNA replication are overrepresented in the protein subset. Given that ribosomal proteins are more conserved to M. jannaschii than E. coli (Fig. 3D), we wondered if most of the proteins relating to the central dogma of molecular biology are more similar to their archaeal homologs.
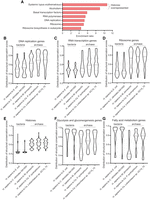
The global structural comparison suggests an archaeal origin of core proteins relating to information storage and processing. (A) Gene overrepresentation analysis for proteins with a structural homolog found in M. jannaschii only. For the list of proteins used for the overrepresentation analysis, see Supplemental Table S7. (B–G) Distribution of the structural conservation scores for protein-coding genes in the selected gene sets upon comparing H. sapiens to E. coli, Caulobacter vibrioides NA1000, M. jannaschii, Candidatus Prometheoarchaeum syntrophicum, or Candidatus Lokiarchaeum sp. GC14_75. Violin plots were used to represent the distribution of the structural similarities, with the name of the gene set marked at the top of each panel. The three dashed lines represent 25% quartile, median value, and 75% quartile. For the lists of genes in the gene sets, see Supplemental Table S5.
Herein, we expanded our analysis to include two additional archaeal species, Lokiarchaeum sp. GC14_75 and Candidatus Prometheoarchaeum syntrophicum, and one bacterial species Caulobacter vibrioides. We compared the distribution of structural similarities for genes involved in gene sets relating to DNA storage (histones), DNA replication, RNA transcription, and protein translation (ribosome), as well as the distribution of structural similarities for genes relating to fatty acid metabolism and sugar metabolism (Fig. 7B–F). Lokiarchaeum sp. GC14_75 and C. Prometheoarchaeum syntrophicum are two recently discovered archaeal species that are closely related to eukaryotes (Zaremba-Niedzwiedzka et al. 2017; Imachi et al. 2020). These species possess genes involved in functions typically associated with eukaryotes, such as membrane trafficking and cytoskeletal organization. These features suggest that the two species may be closely related to the ancestral archaea involved in the symbiotic interactions that led to the emergence of the first eukaryotic cells. In contrast, although it has been established that mitochondria originated from an alphaproteobacterial-like ancestor (Martijn et al. 2018), identifying the closest species to mitochondria poses a challenging task. This difficulty arises because mitochondria have lost most of their genes over the course of evolution, transferring them to the nucleus. Although endosymbiotic alphaproteobacteria (e.g., Rickettsia prowazekii) may resemble mitochondria, they have lost many modules and pathways, making them less suitable for evaluating the level of structural similarities of pathways. Therefore, we opted to include the free-living alphaproteobacterium, C. vibrioides, in our analysis to complement the comparison between E. coli and H. sapiens. By including these three additional species, we hoped to gain further insight into the evolution of the key cellular functions.
Indeed, proteins relating to central dogma are more likely to show stronger structural resemblance to homologs in archaeal species than in bacterial species (Fig. 7B–E). Conversely, proteins relating to energy supply are more likely to show a stronger resemblance to homologs in bacterial species than in archaeal species (Fig. 7F,G). Overall, the proteins relating to energy supply tend to be more structurally conserved than those involved in central dogma machineries in the species examined here. Human genes involved in fatty acid metabolism display significantly higher structural similarities to their homologous proteins in the two Asgard archaeal species compared with M. jannaschii, although bacterial proteins still show greater overall structural resemblance for these human genes. In summary, our analysis of structural similarities supports the idea that the central dogma machineries in eukaryotes evolved from those in an ancestral archaeal species, which is consistent with the findings of a recent study that showed the genetic information processing genes of eukaryotes have evolved from archaeal species (Brueckner and Martin 2020).
SHF: an online resource for structural homolog identification
As shown above, we have gained a lot of interesting information on the molecular evolution of proteins by comparing the protein structures and identifying structural homologs at the proteomic scale. We believe what has been discovered here is only the tip of the iceberg. Some more helpful information could be extracted if we analyze the data from other perspectives. Therefore, we have made these data available to the public as a searchable online resource (the Structural Homolog Finder [SHF]). We have additionally included some structural analog information for Saccharomyces cerevisiae, the unicellular eukaryotic model organism, in the online resource. The webpage can be accessed at http://micc.tmu.edu.cn/shf. On the web interface, the user may enter either the gene symbol or UniProt ID of the gene of interest for querying structural homologs in another species. Then, the user should specify the species names for the source and target organisms (Supplemental Fig. S5A). The backend program will search for structural homologs in the collection of protein structures from the target organism for the specified protein from the source organism. A typical run of the query takes <10 sec. An example of the output page is shown in Supplemental Figure S5, B and C. A ranked list based on structural similarity and a visualization of the structure overlay are provided to the user. For multidomain proteins, homologs to all the individual domains were listed and ranked together. If no structurally similar homologs were found, the query will return “No similar structure found” but will still return one most similar protein structure (of course, with a poor similarity score) from the target organism.
Discussion
In this study, we conducted pairwise tertiary structural comparisons at a proteomic scale and used the structural information to characterize the twilight zone and investigate the evolutionary relationship of genes from distantly related species. The use of protein structures enabled us to surpass the boundary of the twilight zone and readily evaluate the phylogenetic relationship between highly divergent sequences. A simple explanation for why the investigation of the twilight zone can be realized with structural information would be that “protein structures are generally more conserved than protein sequence over evolutionary time.” However, the underlying reason determining why we could obtain more information from structures is that only a small number of key conserved residues (∼8%) coordinate the topology of proteins and shape their functions (Süel et al. 2003; Russ et al. 2005; Socolich et al. 2005). The percentage of these key conserved residues is much smaller compared with the 20%–25% twilight zone boundary determined empirically using sequence alignment algorithms. AlphaFold2 somehow “learns” how to spot the key residues involved in 3D-fold through machine learning of hundreds of thousands of experimental structures. Protein structures make the level of conservation between these key residues more “visible” to topological-based structural alignment algorithms, whereas sequence-based algorithms are unable to pinpoint them for a possible weighted alignment.
Quantitatively measuring the sequence-structure spaces of the twilight zone has been a fundamental question in the field of evolutionary biology. In this study, we made the first attempt to determine the size of the twilight zone. Structural alignment detects considerably more homologous proteins than sequence alignment-based methods do (Esser et al. 2004). When we compared the H. sapiens proteome with the E. coli and M. jannaschii proteomes, we found that ∼16% and 7.5% of the protein-coding genes from the human genome were involved in the twilight zone above a structural similarity of 0.5, respectively. (Note that a gene could be involved in both the twilight zone and the midnight zone depending on the protein to which it is being compared.) What is interesting is that the number of detectable structural homologs in the midnight zone is even greater than the sum of those in the safe zone and the twilight zone. Thus, for most of the structural homologs detected, the divergent evolutionary relatedness cannot be convincingly inferred solely based on sequence information. The sequences of many evolutionarily related proteins may have diverged beyond the detection limit of sequence alignment, but they still remain within the detectable range by structural alignment, although convergent evolution could be an alternative explanation for some of them. It is worth noting that the scatter plot depicting the distribution of sequence identity and structural similarities above a sequence identity of 0.1 forms a reversed J-shaped curve for all three combinations of proteome comparisons (Fig. 3D–F). The sequence identity of about 0.1 appears to be a cutoff, under which structural similarity can be hardly greater than 0.6 (Fig. 3D–F). We speculated that this number could indicate the average percentage of the key residues involved in protein folding for each domain. Therefore, if the sequence identity is below this percentage number, the structural similarity has to be compromised in some regions.
An implication from the gene enrichment analysis based on structural alignment is that human proteins related to the central dogma are more likely to resemble M. jannaschii homologs than E. coli homologs, whereas those related to energy supply are more likely to resemble E. coli homologs than M. jannaschii homologs (Figs. 5, 6B, 7B–G). These results generally agree with a previous study that suggests eukaryotic genes related to genetic information processing originated from archaea and genes involved in metabolism have a bacterial origin (Brueckner and Martin 2020). The unique contribution of our analysis lies in our ability to illustrate the varying degrees of conservation for each pathway and identify which pathway stands out as the most conserved among all 186 KEGG pathways. Apparently, genetic information processing and metabolism are broad concepts, and different pathways or modules may have varying levels of structural conservation. In our analysis, glycolysis and gluconeogenesis are the most conserved pathways, and RNA transcription appears to be the least conserved by comparing genes from H. sapiens with those from five prokaryotic species (Fig. 7B–G). It is worth mentioning that the level of structural conservation for genes relating to fatty acid metabolism between H. sapiens and the two newly discovered archaeal species from the Lokiarchaeota phylum is much greater than that for the archaeal species from the Euryarchaeonta phylum. The findings suggest that the origin of the eukaryotic genes involved in lipid metabolism may not be as clear-cut as previously thought.
Another interesting finding is the identification of two ancestral GPCR-like signaling proteins in E. coli: dgcT and cdgI. It has been established that the expression of dgcT or cdgI repressed the swimming behavior of E. coli (Song et al. 2008; Sanchez-Torres et al. 2011). Both dgcT and cdgI contain a 7TM domain and a dinucleotide cyclase domain for synthesis or degradation of secondary messenger c-di-GMP. Because both proteins regulate swimming motility, it is conceivable that they are responsible for sensing extracellular stimuli and initiating intracellular signals through the secondary messenger. This is exactly what human GPCR proteins do, and the only difference is that the signaling cascade in E. coli lacks an intermediate G-protein. Incidentally, earlier research has shown that although GPCR and G-proteins are present in almost all metazoan species, some unicellular eukaryotes contain GPCR proteins but lack G proteins, suggesting the existence of an alternative link between GPCR and the signaling transduction pathway that does not rely on G-proteins in some microorganisms (de Mendoza et al. 2014). It should be noted that, in human cells, GPCR proteins normally signal downstream events through another secondary messenger, cAMP, a cyclic mononucleotide (Hilger et al. 2018). Nevertheless, the most similar human homolog of the dinucleotide cyclase domain from the two proteins is actually a mononucleotide cyclase protein, suggesting a structural similarity between the mononucleotide and dinucleotide cyclase domains. It is possible that during the evolution of the GPCR signaling pathway in eukaryotes, the cyclase domain somehow evolved from the dinucleotide cyclase to a mononucleotide cyclase. Alternatively, the dinucleotide cyclase domain in the GPCR-like protein of contemporary E. coli may have originated from a mononucleotide cyclase domain from an ancestral bacteria species. In any case, the identification of human GPCR homologs dgcT and cdgI provides a prototype for the GPCR-like signaling pathway in bacteria. Here, the structural comparison provides an important clue suggesting that the link between GPCR and cyclic nucleotide secondary messenger could be established without the involvement of G-proteins.
In addition to what has been discussed above, we have set up an online web server for the identification of structural homologs between species using the computed data presented here. Researchers may sometimes want to seek structural homologs for their protein of interest in another species. We hope that this online resource can be beneficial for the research community.
It is worth pointing out that convergent evolution may play a role when two proteins show structural similarity despite poor sequence identity or similarity. Protein structural information alone is insufficient to determine whether a small difference in protein structure is caused by convergent evolution. In addition, horizontal gene transfer may also give rise to high structural similarity between distantly related species, thereby causing potential issues in interpreting the phylogenetic relationship. Gene set–based analysis should partially mitigate the ambiguity caused by convergent evolution or horizontal gene transfer, because it is unlikely that convergent evolution would produce a set of proteins with the precise structural similarities required for the whole pathway to function. Additionally, considering the trend of multiple genes in a gene set as a whole can weaken the effect of horizontal gene transfer of one or two genes. Nevertheless, combining sequence information with structural information and comparing this information in multiple related species should be the ultimate solution for discounting the impact of convergent evolution or horizontal gene transfer.
In this study, we conducted comparisons between structural similarities and sequence similarities at the proteomic level. We defined individual domains based on protein structures and performed global sequence alignments for the entire sequences of each domain. Although proteomic-scale structural similarity searches have proven valuable in the detection of distantly related homologs, it is imperative to underscore several well-established local alignment tools. These tools use iterative model-based search methods and show high sensitivity in identifying distantly related homologs. Notable examples of such methods include HMMER (Eddy 2011) and PSI-BLAST (Altschul et al. 1997). Unlike traditional pairwise similarity search methods, these tools harness the wealth of information present in protein sequence databases to establish patterns or motifs within local peptide sequences, which are subsequently used to detect more homologous proteins. Essentially, these methods use intermediary sequences to infer similarities between genes that are distantly related. Although PSI-BLAST and HMMER are undoubtedly valuable and sensitive tools for homolog detection, they may occasionally yield false-positive hits due to the contamination of unrelated sequences in the position-specific scoring matrix (in the case of PSI-BLAST) or hidden Markov model (in the case of HMMER). This contamination can lead to deviations in the search results from the original homolog family, a phenomenon known as “homologous overextension” (Gonzalez and Pearson 2010). On the other hand, direct structural comparison does not suffer from contamination in motif establishment and serves as the gold standard for validating sequence alignment results.
To sum up, we used a combination of experimental structures and in silico modeled structures by AlphaFold2 to evaluate the evolutionary relationships of proteins between distantly related species. We characterized the twilight zone and revealed differential enrichment patterns of the structurally conserved proteins between H. sapiens and M. jannaschii or E. coli. One limitation of this study is that our evaluation of protein structures was restricted to a small number of representative species. Consequently, the protein structures we analyzed in this work do not sufficiently represent the diversity of protein structures present within each domain of life. These results only suggest the similarities in pathways between these particular species and cannot be readily extended to draw conclusions at the domain levels. An analysis featuring a larger number of species is necessary to draw more reliable conclusions at the domain level, as we cannot exclude the possibility of other bacterial or archaeal species bearing greater structural similarities than the ones investigated here for any given pathway. With the rapid accumulation of experimental structures in the database and improvement in computer modeling, constructing species phylogeny using protein structures from multiple species may become feasible in the near future, and phylogenetic trees based on structural similarities rather than sequence similarities ought to provide us with some more helpful information about the evolutionary history of life.
Methods
Selection and preprocessing of protein structures
Experimental protein structures were obtained from the PDB, and computer-modeled structures were from the AlphaFold2 database.
For the selection of experimental structures, we followed the criteria below: (1) The structure must be from either of the six species: H. sapiens, E. coli, M. jannaschii, Lokiarchaeum sp. GC14_75, C. Prometheoarchaeum syntrophicum, or C. vibrioides. (2) The protein for the structure should not be a chimeric protein or artificially designed construct. (3) The number of amino acids in a single chain should be more than 50 because structures smaller than this threshold often contain only one alpha helix or beta sheet. The topologies are too simple for making a meaningful structural comparison or identification of homologs. (4) The refinement resolution of the structure should be less than or equal to 2.8 Å. (5) The structure should contain only one protein entity because we intend to evaluate the protein fold rather than its conformation supported by a protein complex. (6) The structure should not contain any unnatural amino acids. Before structural analysis, water and ligands were removed for all the experimental structures, and a single chain was extracted from the protein structures.
The in silico modeled structures were obtained from the AlphaFold2 database. It is worth mentioning that the AlphaFold2-predicted structures contain intrinsically disordered regions. Unfolded interdomain linkers are also visible in the structures modeled by AlphaFold2. These regions were presented as sequences with low confidence scores. The Leiden algorithm (Traag et al. 2019), a graph-based community detection algorithm, was used to truncate the structures of multidomain proteins into single domains. Again, domains smaller than 50 aa were abandoned because the topologies of these structures were too simple. To map the distribution of structural similarity and estimate the size of the twilight zone, we excluded individual protein domains that contained fewer than three alpha helix and beta strands, such as the leucine zipper domain. This was necessary because the topological features of these domains were too simplistic, resulting in frequent alignment and an overestimation of the number of structural homologs. We determined the number of helix and beta strands in protein structures using the STRIDE software (Frishman and Argos 1995).
For a schematic representation of the workflow and methods used in this study, see Supplemental Figure S1.
Structural comparisons
We performed pairwise structural comparisons with TM-align, an algorithm for sequence-independent structural comparison (Zhang and Skolnick 2005). TM-align uses a heuristic dynamic programming alignment procedure for structural alignment. A TM-score was used for the evaluation of the similarities between structures. The score ranges between zero and one. Unlike RMSD, the score magnitude of the TM-score is independent of the size of the structure. A value of one indicates a perfect match, whereas a value of zero indicates an absolute no match. Those with a similarity score value >0.5 are generally considered to have the same protein fold, whereas two randomly related structures should have an average similarity score of around 0.17 (Zhang and Skolnick 2005; Xu and Zhang 2010). Note that the TM-score can be normalized to either of the two structures to be compared. We picked the smaller alignment score for all the structural comparisons.
The structural conservation score for each human protein was determined based on the TM-score between the protein of interest and its most similar homolog in the investigated species (E. coli or M. jannaschii). For a single-domain protein, the conservation score equals the TM-score. For multidomain proteins, the conservation score is the weighted average of the best TM-scores for each domain. The weights were assigned based on the number of amino acids in each domain.
Sequence comparisons
Pairwise amino acid sequence comparison of experimental structures from stratified sampling was performed with EMBOSS stretcher (Fig. 2C,D). Pairwise sequence comparison of AlphaFold2 modeled structures for characterization of twilight zones was performed with EMBOSS needle (Figs. 3, 4). EMBOSS stretcher implements the Myer–Miller algorithm (Myers and Miller 1988), and EMBOSS needle implements the Needleman–Wunsch algorithm (Needleman and Wunsch 1970). The former requires more time, whereas the latter needs more computer memory. The software suite EMBOSS version 6.6 was used to determine the sequence similarity or identity. The EMBOSS suite includes both the stretcher and needle programs. The EBLOSUM62 scoring matrix was used for the comparison of the sequences.
To perform sequence alignment using EMBOSS stretcher, we extracted the amino acid sequences from the PDB files of the protein structures to be compared. To perform sequence alignment using EMBOSS needle, we determined the sequence range for each domain using truncated AlphaFold2 structures with reference to the predicted alignment error matrix. Both programs were run with the default parameters for gap open penalty and gap extend penalty.
Gene set enrichment analysis and gene overrepresentation analysis
Gene set enrichment analysis was performed with GSEA 4.3.2 (Mootha et al. 2003; Subramanian et al. 2005). The number of permutations was set to 20,000. Built-in gene sets derived from the KEGG pathway database (Kanehisa and Goto 2000) and GO Cellular Component ontology (Ashburner et al. 2000; Carbon et al. 2009; The Gene Ontology Consortium 2020) were used for the enrichment analysis. Gene sets with fewer than 15 genes were excluded from the analysis. No upper limit was set to the size of the gene sets.
Gene overrepresentation analysis was performed with WebGestalt (Zhang et al. 2005; Wang et al. 2013, 2017; Liao et al. 2019). The built-in gene sets, KEGG pathways, were used for the analysis. The protein-coding genome was used as the reference genome.
Data access
Calculated structural conservation scores for each human protein-coding gene are available in Supplemental Table S4. Structural homologs for individual proteins can be queried at the Structural Homolog Finder database (http://micc.tmu.edu.cn/shf). The entire data set can be downloaded at the website. The structural homolog data used in this work can also be accessed in the Supplemental Material.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
This work was supported by the National Natural Science Foundation of China 32070711 (Y.F.) and the Tianjin Applied Basic Research Diversified Investment Foundation 21JCYBJC01490 (S.Z.). The computational requirements for this work were supported in part by the Tianjin Medical University High Performance Computing (HPC) Center's resources and personnel.
Author contributions: S.Z. and Y.F. conceived the project and implemented the data analysis. T.Z. and Y.F. generated the figures. Y.F. wrote the manuscript with editorial contributions from S.Z. and T.Z.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.277771.123.
- Received February 3, 2023.
- Accepted October 16, 2023.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see https://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








