
Proactive functional classification of all possible missense single-nucleotide variants in KCNQ4
- Honglan Zheng1,2,3,5,
- Xinhao Yan1,2,3,5,
- Guanluan Li1,2,3,
- Hengwei Lin1,2,3,
- Siqi Deng3,
- Wenhui Zhuang1,2,3,
- Fuqiang Yao1,2,
- Yu Lu4,
- Xin Xia1,2,3,
- Huijun Yuan4,
- Li Jin1 and
- Zhiqiang Yan1,2,3
- 1Human Phenome Institute, School of Life Sciences, Fudan University, Shanghai 200438, China;
- 2State Key Laboratory of Medical Neurobiology and MOE Frontiers Center for Brain Science, Institutes of Brain Science, Fudan University, Shanghai 200438, China;
- 3Institute of Molecular Physiology, Shenzhen Bay Laboratory, Shenzhen 518132, China;
- 4Institute of Rare Diseases, West China Hospital of Sichuan University, Chengdu 610041, China
-
↵5 These authors contributed equally to this work.
Abstract
Clinical exome sequencing has yielded extensive disease-related missense single-nucleotide variants (SNVs) of uncertain significance, leading to diagnostic uncertainty. KCNQ4 is one of the most commonly responsible genes for autosomal dominant nonsyndromic hearing loss. According to the gnomAD cohort, approximately one in 100 people harbors missense variants in KCNQ4 (missense variants with minor allele frequency > 0.1% were excluded), but most are of unknown consequence. To prospectively characterize the function of all 4085 possible missense SNVs of human KCNQ4, we recorded the whole-cell currents using the patch-clamp technique and categorized 1068 missense SNVs as loss of function, as well as 728 loss-of-function SNVs located in the transmembrane domains. Further, to mimic the heterozygous condition in Deafness nonsyndromic autosomal dominant 2 (DFNA2) patients caused by KCNQ4 variants, we coexpressed loss-of-function variants with wild-type KCNQ4 and found 516 variants showed impaired or only partially rescued heterogeneous channel function. Overall, our functional classification is highly concordant with the auditory phenotypes in Kcnq4 mutant mice and the assessments of pathogenicity in clinical variant interpretations. Taken together, our results provide strong functional evidence to support the pathogenicity classification of newly discovered KCNQ4 missense variants in clinical genetic testing.
Whole-exome sequencing is rapidly developing and expanding into the clinic, identifying many disease-related variants, most of which are missense single-nucleotide variants (SNVs) of uncertain significance (VUSs) (Collins 1999; Tennessen et al. 2012; Flannick et al. 2013; Lee et al. 2014; McLaughlin et al. 2014). However, because of the complexities and technical constraints of functional verification, the function of most missense variants is not defined. Functional uncertainty will ultimately lead to obstruction of diagnosis and treatment (Flannick et al. 2013; Manrai et al. 2016). Therefore, it is urgently necessary to study the function of missense variants in disease-related genes (Cutting 2014).
Mutations in a variety of ion channel genes can cause channelopathies, such as Brugada syndrome (BrS), long QT syndrome (LQTS), and epilepsy (Kass 2005; Ptáček 2015; Skinner et al. 2019). Because of the difficulty of recording ion channel activity, there is no large-scale functional classification of missense variants of any ion channel genes yet, and only dozens of missense variants in ion channel genes have been evaluated by automated high-throughput patch-clamp devices, such as KCNB1, KCNH2, KCNQ1, and SCN5A (Vanoye et al. 2018; Kang et al. 2019; Glazer et al. 2020; Ng et al. 2020).
Here, we studied human potassium voltage-gated channel subfamily Q member 4 (KCNQ4), which is crucial to hearing and can cause inherited deafness when mutations occur (Kubisch et al. 1999). KCNQ4 variants are linked to an autosomal dominant form of nonsyndromic progressive hearing impairment: DFNA2 in humans, a late-onset disease, with high-frequency hearing loss (Coucke et al. 1994; Van Camp et al. 1997; Kubisch et al. 1999). The KCNQ4 gene is one of the most commonly responsible genes for autosomal dominant nonsyndromic hearing loss (Hilgert et al. 2009; Oza et al. 2018). In addition, KCNQ4 variants are also associated with age-related hearing loss and noise-induced hearing loss (Van Eyken et al. 2006; Van Laer et al. 2006; Huang and Tang 2010; Peixoto Pinheiro et al. 2021). According to the Genome Aggregation Database (gnomAD) cohort, approximately one in 100 people harbors missense variants in KCNQ4 (only variants with minor allele frequency < 0.1% were included) (Lek et al. 2016). In the cochlea, KCNQ4 is mainly expressed in the basal membrane of sensory outer hair cells (OHCs), and Kcnq4-knockout mice show the degeneration of OHCs and exhibit progressive deafness (Kubisch et al. 1999; Jentsch 2000; Kharkovets et al. 2000, 2006; Beisel et al. 2005).
KCNQ4 variants identified in DFNA2 patients show loss of function when expressed alone and exert impaired heterogeneous channel function when coexpressed with wild type (WT) in the heterologous expression systems (Kubisch et al. 1999; Mencía et al. 2008; Kim et al. 2011; Gao et al. 2013; Jung et al. 2018). In this study, we set out to perform a functional classification of all possible 4085 missense SNVs of KCNQ4 to support the pathogenicity interpretation of KCNQ4 variants and assist the clinical diagnosis of hearing loss.
Results
Functional classification of 4085 KCNQ4 missense SNVs
Variants at multiple sites on the human KCNQ4 have been identified to be associated with DFNA2. Heterologous expression systems, such as Xenopus oocytes, HEK293T, and CHO-K1 cells, have been well established as reliable and convenient methods for determining the cellular and functional mechanisms of KCNQ4 variants in DFNA2, and the results generated in the heterologous expression systems are highly consistent with clinical pathogenicity interpretation (Kubisch et al. 1999; Mencía et al. 2008; Kim et al. 2011; Gao et al. 2013; Jung et al. 2018). Considering the convenience of the cell culture and minimal background currents after P/N leak subtraction, we used CHO-K1 cells to perform functional classification of variants in KCNQ4 on a large scale (Supplemental Fig. S1A).
To examine the effects of KCNQ4 variants on channel function, we constructed all possible 4085 KCNQ4 missense SNV plasmids, which is the sum of about four to seven substitutions for each of the 695 amino acids (Supplemental Table S1), and then recorded the whole-cell currents in CHO-K1 cells transfected with WT KCNQ4 or KCNQ4 variants (for a description of the overall scheme, see Supplemental Fig. S2). In contrast to untransfected CHO-K1 cells, cells transfected with KCNQ4 (vector: pIRES2-EGFP; nonfusion EGFP as a transfection marker) yielded outward currents traces under the voltage steps ranging from −100 mV to +40 mV, with a holding voltage of −80 mV (Supplemental Fig. S1A). Additionally, cells transfected with KCNQ4-EGFP (fusion EGFP at the C terminus), KCNQ4-Myc (with Myc tag in the extracellular domain), and KCNQ4-Myc-EGFP (with Myc tag in the extracellular domain and fusion EGFP at the C terminus) produced channel properties comparable to that of KCNQ4 (Supplemental Fig. S1). For the convenience of a potential follow-up study of the subcellular localization of WT KCNQ4 and variants, all the variant plasmids used in our experiments were inserted with a Myc tag in the extracellular domain as described in a previous study (Kim et al. 2011).
Then we performed electrophysiological recordings of all 4085 missense SNVs at different voltages when expressed alone (in the homozygous state) using patch-clamp recording (Supplemental Fig. S3). We calculated the two most important parameters for KCNQ4 channels: peak currents and half-maximal activation voltage (V1/2). To directly compare the channel properties of the WT and variants, we quantified differences in peak currents recorded at +40 mV and V1/2 relative to WT channels. The peak currents normalized to the WT of each variant (Imut/Iwt) were presented according to amino acid sequence and indicated with different colors (Fig. 1, left; for data set, see Supplemental Data Set A). Most of the variants located at the N terminus and C terminus produced currents similar to WT, whereas most of the variants located around the pore region showed reduced currents or no current, consistent with the pore region being the most critical part of channel function. In contrast, among the 3319 variants with an available V1/2, the V1/2 values of most variants were similar to that of the WT, and only a few variants showed large shifts in V1/2 in either the depolarizing or hyperpolarizing direction (Fig. 1, right; for data set, see Supplemental Data Set B). Variants with significant shifts in V1/2 were mainly located in the voltage-sensing region (S1–S4 transmembrane domain), which plays a key role in the regulation of channel gating.
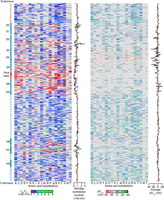
Sequence–currents and sequence–V1/2 maps for KCNQ4 missense SNVs. (Left) The average peak current values (normalized to WT) recorded at +40 mV of the 4085 missense SNVs when transfected alone were plotted in a heat map based on amino acid position along the KCNQ4 protein sequence (averaged from n ≥ 3 cells). The bar at the bottom shows the different current values with specific colors. Red and green indicate that an amino acid change results in low or high currents, whereas blue indicates currents similar to or close to the WT. The gray background indicates no data. Average normalized currents recorded at +40 mV summed by amino acid position are plotted to the right, showing the level of tolerance for any amino acid substitution. (Right) The shifts of the voltage-dependence of activation: V1/2 of the 3319 variants with currents enough to determine the V1/2. For 766 variants with severely reduced currents (<0.25 of the WT value in peak currents), V1/2 cannot be determined. ΔV1/2 indicates the change of V1/2 after mutation. ΔV1/2 = V1/2 in variants − V1/2 in WT. The V1/2 of WT is −17.10 ± 1.15 mV. Red indicates that sites show large shifts in activation V1/2 in the depolarizing direction, and green indicates that sites show large shifts in the hyperpolarizing direction.
The normalized currents recorded at +40 mV of all 4085 variants presented bimodal distribution, with one peak around one (WT) and the other close to zero (null variant) (Fig. 2A). We classified all variants based on the normalized peak currents measured at +40 mV using k-means clustering (Supplemental Methods). All 4085 missense SNVs were divided into three groups and named “reduced current,” “normal current,” and “enhanced current” based on the value of each cluster center (Fig. 2B). Each group contains 915, 3127, and 43 variants with the cluster centers of 0.09, 1.00, and 3.16, respectively (Fig. 2B; Supplemental Table S2). Two cutoffs were generated among these three groups: One cutoff for dividing “reduced current” and “normal current” was 0.54 of the WT value, and the other cutoff for dividing “normal current” and “enhanced current” was 2.08 of the WT value. The current distribution of 3127 variants classified as “normal current” by k-means clustering showed a Gaussian-like distribution with one peak near one, which is similar to the current distribution of WT KCNQ4 (Supplemental Fig. S4).
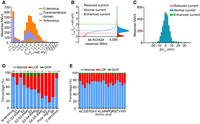
Functional classification of 4085 KCNQ4 missense SNVs when expressed alone. (A) The distribution of the normalized currents measured at +40 mV of 4085 variants in the homozygous state. (B) Raw normalized currents ranked for all 4085 KCNQ4 missense SNVs. The red dashed line indicates 0.54 of the WT value; gray indicates the WT value; and green indicates 2.08 of the WT value. Functional classification was based on k-means clustering. The curve of kernel density estimation is shown on the right. (C) The distribution of ΔV1/2 generated from 3319 variants with available V1/2. (D) The percentage of each functional phenotype in each domain. The number above each column represents the number of missense SNVs contained in the corresponding domain. (LOF) Loss of function, (GOF) gain of function. (E) The percentage of each functional phenotype of each type of amino acid after mutation.
V1/2 is an important parameter to reflect the gating properties of voltage-gated ion channels. Next, we examined V1/2 of all possible KCNQ4 missense SNVs when transfected alone, which might also cause KCNQ4 channel loss of function or gain of function. Overall, the V1/2 values of all variants showed a unimodal distribution, with the peak around the WT (Fig. 2C). The variants with >10-mV shifts in V1/2 relative to the WT channel were considered to have a strong effect on the channel gating properties, >10-mV rightward shifts indicating decreased channel function and >10-mV leftward shifts indicating increased channel function. Representative whole-cell current traces and functional properties from each type of variant are presented in Supplemental Figure S5. For 766 of 915 current-reduced variants, we did not calculate V1/2 because of severely reduced currents (<0.25 of the WT value in peak currents). Of the remaining 149 current-reduced variants (peak currents between 0.25 and 0.54 of the WT), 112 variants showed <10-mV shifts in V1/2, 27 showed >10-mV rightward shifts, and 10 showed >10-mV leftward shifts (Fig. 2C; Supplemental Table S3). A total of 2898/3127 variants with normal currents (peak currents between 0.54 and 2.08 of the WT) showed <10-mV shifts in V1/2, whereas 153/3127 had >10-mV rightward shifts and 76/3127 had >10-mV leftward shifts (Fig. 2C; Supplemental Table S3). Thirty of 43 current-enhanced variants (peak currents > 2.08 of the WT) had <10-mV shifts in V1/2, five of 43 variants had >10-mV rightward shifts, and eight of 43 had >10-mV leftward shifts (Fig. 2C; Supplemental Table S3).
For combined normalized peak currents with activation V1/2, all 4085 variants were classified as “loss of function,” “normal function,” and “gain of function” (Supplemental Table S3; Supplemental Methods). Together, we categorized 70.9% (2898/4085) of missense SNVs as normal function, 26.1% (1068/4085) as loss of function, and 2.9% (119/4085) as gain of function.
The functional properties of all missense SNVs were also presented and analyzed based on the amino acid position (Supplemental Fig. S6). At the N terminus (covering residues M1-G96, containing 564 missense SNVs), 88.5% (499/564) of missense SNVs had normal function, 9.6% (54/564) had loss of function, and only 2.0% (11/564) had gain of function (Fig. 2D; Supplemental Fig. S6C). There was an enrichment of current-enhanced variants at the end of the N terminus (Supplemental Figs. S3, S6A). In the transmembrane domain (covering residues W97-F335, containing 1402 missense SNVs), 43.7% (612/1402) of missense SNVs had normal function, 51.9% (728/1402) had loss of function, and 4.4% (62/1402) had gain of function (Fig. 2D; Supplemental Fig. S6C). In the S2–S3 linker, nine variants with significantly increased currents were classified as gain-of-function variants, which may regulate channel conformation (Supplemental Fig. S6A). In the S4 to S6 transmembrane domain, the number of loss-of-function variants had significantly increased, which may be due to the important roles of these domains in channel voltage sensing and pore forming (Fig. 2D; Supplemental Fig. S6). In particular, 83.4% (151/181) of missense variants in the pore region had loss of function, which was consistent with the important role of the pore region in ion channels (Fig. 2D; Supplemental Fig. S6); 70.6% (108/153) of variants with normal currents but >10-mV rightward shifts in V1/2 clustered in the transmembrane domain S1 to S4–S5 linker, which may participate in sensing voltage (Supplemental Fig. S6B). At the C terminus (covering residues E336-D695, containing 2119 missense SNVs), 84.3% (1787/2119) of variants showed normal function, 13.5% (286/2119) had loss of function, and only 2.2% (46/2119) had gain of function (Fig. 2D; Supplemental Fig. S6C). Loss-of-function and gain-of-function variants observed at the C terminus were mainly clustered in these key domains, such as helix A, B, C, and D, which are involved in channel gating, assembly, or trafficking (Fig. 1; Schwake et al. 2006; Howard et al. 2007; Haitin and Attali 2008; Chang et al. 2018).
Furthermore, we also classified the function of different amino acids at each site and found that mutating into different amino acids had varying effects on channel function. For instance, a change to aspartic acid or proline was most likely to reduce function, whereas a change to methionine or threonine was more likely tolerated (Fig. 2E; Supplemental Fig. S7); 96.3% (26/27) of missense SNVs mutating to basic amino acids with a positive charge (arginine, lysine, and histidine) in the pore region were loss of function (Supplemental Fig. S7A).
Structural basis of KCNQ4 missense SNVs
To display the current and V1/2 change trend and bias of KCNQ4 variants with its protein structure, the average normalized currents and the shifts in V1/2 indicated with different colors for all missense variants at each amino acid were overlaid on the known cryo-EM structure of human KCNQ4 (PDB: 7BYL) (Li et al. 2021). Seventy-one of 89 amino acids in the transmembrane domain with average normalized currents <0.5 of the WT value were located in the S5 domain, pore region, and S6 domain, regions that are the most important for KCNQ4 channel, showing that amino acid substitutions in these domains are more likely to be damaging (Fig. 3A,B; Supplemental Fig. S8A). Thirty-one amino acids with >10-mV rightward shifts in average V1/2 were located in the S1–S6 transmembrane domains, of which 16 amino acids were located in the S4 transmembrane domain and S4–S5 linker, which are responsible for channel voltage sensing and gating regulation (Fig. 3C,D; Supplemental Fig. S8B).
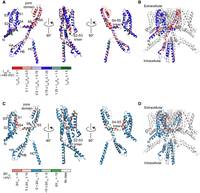
Structural basis of KCNQ4 missense SNVs. (A) Cartoon diagrams showing three different views of a KCNQ4 monomer denoting specific domains. Currents recorded at +40 mV were mapped onto the monomer structure (covering residues S74-Y367 and D524-G588). Variants were colored based on currents normalized to WT. The mean current of each amino acid is calculated by averaging the current values of all four to seven missense SNVs at this site. N-terminal front (M1-S73), S3–S4 linker (T193-A199), helix A–helix B linker (Y368-V523), and C-terminal end (R589-D695) residues are not available in the cryo-EM structure. Colors indicating the currents were drawn using PyMOL. (B) Normalized currents were mapped onto one subunit of the tetramer structure. (C) KCNQ4 protein monomer residues were colored by the shifts in activation V1/2. White indicates that the currents of all four to seven missense SNVs at this site are small, and the V1/2 cannot be determined. (D) Activation V1/2 was mapped onto one subunit of the tetramer structure.
Functional properties of the heterogeneous KCNQ4 channels
Most DFNA2-associated KCNQ4 missense variants are present in a heterozygous state and show decreased heterogeneous channel function when coexpressed with WT (Kubisch et al. 1999; Kim et al. 2011; Jung et al. 2018). We further performed research to mimic the effect of KCNQ4 variants in heterozygous DFNA2 patients and only studied the variants in the heterozygous state if they showed significant functional differences from WT in the homozygous state, including 1068 loss-of-function variants and 119 gain-of-function variants. Of these variants, 229 variants with normal peak currents but >10-mV shifts in V1/2 in the homozygous state were also tested for the functional properties in the heterozygous state. Although it is unclear whether a large shift in V1/2 can cause DFNA2, it is also worthy to explore. For example, it has been found that variants that promote positive or negative effects on channel gating in KCNQ2 can cause human epilepsy (Soldovieri et al. 2007; Miceli et al. 2013, 2015; Millichap et al. 2017). It is possible that KCNQ4 variants could produce DFNA2 by affecting channel gating. We assessed the functional properties of heterogeneous channels by cotransfecting these variants with WT (KCNQ4-Myc-mCherry) at a 1:1 ratio in CHO-K1 cells. Cells transfected with KCNQ4-Myc-mCherry, cotransfected KCNQ4-Myc-EGFP with vector (WT + vector 1:1), and cotransfected KCNQ4-Myc-mCherry with KCNQ4-Myc-EGFP (WT + WT 1:1) generated current magnitudes and V1/2 similar to WT (Supplemental Fig. S1).
First, we cotransfected 1068 loss-of-function variants with WT channels at a 1:1 ratio, including 915 variants with reduced currents and 153 variants with normal currents but large rightward shifts in V1/2 when expressed alone. The heterozygous channel currents obtained from coexpressing 915 current-reduced variants with WT were generally lower than WT currents, showing a unimodal distribution (Fig. 4A; Supplemental Fig. S9; for data set, see Supplemental Data Set C). Then we defined the functional classification of heterogeneous channels based on the heterozygous current distribution of 25 known pathogenic/likely pathogenic and 28 known benign/likely benign variants. The curation guidelines for variants as pathogenic/likely pathogenic and benign/likely benign without our functional data will be introduced in detail in the next section. We used the normalized current of 0.77 as the classification threshold, the midpoint between mean currents of known pathogenic/likely pathogenic (mean = 0.50) and benign/likely benign variants (mean = 1.04) in the heterozygous state (Fig. 4B). A total of 516/915 variants in the heterozygous state showed reduced or only partially rescued currents (Imut + wt/Iwt + wt ≤ 0.77) relative to WT, suggesting that these variants inhibited the function of heterogeneous channels to varying degrees and may be potentially pathogenic variants, which could be caused by a variety of factors, such as dominant-negative effects and haploinsufficiency (Fig. 4C). A total of 399/915 variants in the heterozygous state showed currents greater than the threshold (Imut + wt/Iwt + wt > 0.77), indicating that the heterogeneous channel function can be fully rescued by a functional allele of WT KCNQ4 and these variants may not cause hearing loss (Fig. 4C); 16.7% (9/54) variants at the N terminus and 35.2% (93/264) at the C terminus showed impaired or only partially rescued heterozygous channel function, whereas 69.3% (414/597) in the transmembrane domain showed impaired or only partially rescued heterozygous channel function (Fig. 4D). Especially, in the pore region, a variant hotspot in KCNQ4 associated with DFNA2 (Van Hauwe et al. 2000), 82.1% (119/145) of current-reduced variants showed impaired heterozygous channel function. In contrast, the vast majority of V1/2 generated from heterogeneous channels were similar to the WT (Fig. 4E; for data set, see Supplemental Data Set C). A total of 858/915 variants had <10-mV shifts in V1/2 in the heterozygous state, 46 had >10-mV rightward shifts, and only 11 had >10-mV leftward shifts. Likewise, known pathogenic/likely pathogenic and benign/likely benign variants in the heterozygous state almost show similar V1/2 to that of WT, indicating that large shifts in V1/2 may not be a common cause of DFNA2 (Fig. 4F). The results of electrophysiological recording in vitro alone may not suffice to down-classify variants with large shifts in V1/2 in the heterozygous state as a benign or pathogenic category, and more phenotypic and human genetics studies are needed for further pathogenicity classification.
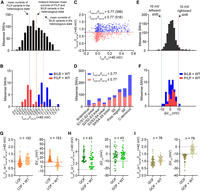
Functional properties of loss-of-function and gain-of-function variants in the heterozygous state. (A) The distribution of the normalized peak currents in the heterozygous state of 915 current-reduced variants. The black dashed line indicates the mean currents of known pathogenic/likely pathogenic variants in the heterozygous state, and gray indicates the mean currents of known benign/likely benign variants in the heterozygous state. The red solid line indicates the midpoint between the mean currents of known pathogenic/likely pathogenic and known benign/likely benign variants in the heterozygous state. (B) The distribution of the normalized peak currents in the heterozygous state of 25 known pathogenic/likely pathogenic and 28 known benign/likely benign variants. (P) Pathogenic, (LP) likely pathogenic, (B) benign, and (LB) likely benign. (C) Plots for comparing normalized currents of expressing 915 current-reduced variants alone and coexpressing with WT. The red dashed line indicates 0.77 of the WT value. There are 516/915 variants with normalized peak currents ≤ 0.77 and 399/915 variants with normalized peak currents > 0.77 in the heterozygous state. (D) The number of variants with normal or impaired currents in the heterozygous state in each domain. (E) The distribution of V1/2 obtained from 915 current-reduced variants in the heterozygous state. (F) The distribution of V1/2 obtained from 25 known pathogenic/likely pathogenic and 28 known benign/likely benign variants in the heterozygous state. (G) Normalized peak currents (left) or V1/2 activation (right) for expressing 153 loss-of-function variants (normal currents and large rightward shifts in V1/2 when expressed alone) versus coexpressing with WT. (H) Normalized peak currents or V1/2 activation for expressing 43 current-enhanced variants versus coexpressing with WT. (I) Normalized peak currents or V1/2 activation for expressing 76 gain-of-function variants (normal currents and large leftward shifts in V1/2 when expressed alone) versus coexpressing with WT.
In addition, the peak currents of coexpressing the remaining 153 loss-of-function variants with WT were still similar to that of the WT, and the V1/2 of 119 variants can be rescued by coexpressing with WT channels (Fig. 4G; for data set, see Supplemental Data Set D). So far, the pathogenicity for variants with activation curves shifting toward more positive potential in the heterozygous state is unclear, and no carrier has been reported, which requires further investigation.
Further, we measured the heterogeneous channel function composed of 119 gain-of-function variants and WT, including 43 variants with enhanced currents and 76 variants with normal currents but large leftward shifts in V1/2 when expressed alone. The peak currents of coexpressing 43 current-enhanced variants with WT were still clustered at more than two of WT + WT (Fig. 4H, left; for data set, see Supplemental Data Set E). A total of 4/43 variants had a > 10 mV leftward shift when cotransfected with WT channels, indicating enhanced voltage sensitivity of the channel (Fig. 4H, right). In contrast, the peak currents and V1/2 of coexpressing 76 current-normal variants measuring large leftward shifts in V1/2 with WT were almost normal (Fig. 4I; for data set, see Supplemental Data Set F). There is no evidence to show the phenotypes of humans carrying KCNQ4 variants with significantly increased currents or large leftward shifts in V1/2, thus more genetic or experimental evidence and mouse model verification are necessary.
Hearing evaluation of Kcnq4 point mutant mice
To verify the functional classification of heterogeneous channels and investigate the correlation between the functional properties of heterogeneous channels in vitro and hearing phenotype in vivo, we chose five variants (p.G287R, p.L249R, p.D272Y, p.S201C, and p.A154T) with no channel function when expressed alone and with varying degrees of reduced currents when coexpressed with WT to construct the knock-in (KI) point mutant mouse strains (Fig. 5A; Supplemental Fig. S10). These five variants produced successively increasing currents at +40 mV when coexpressed with WT, which were 0.47, 0.61, 0.75, 0.92, and 1.04 of the WT, respectively. Variants p.G287R, p.L249R, and p.D272Y showed impaired heterozygous channel function based on our functional classification in vitro, whereas variants p.S201C and p.A154T produced heterozygous channel function similar to WT. Then we introduced these variants (equivalent to mouse p.G288R, p.L250R, p.D273Y, p.S202C, and p.A155T) into the mouse Kcnq4 gene, using CRISPR-Cas9 genome editing technology (Supplemental Fig. S11; Supplemental Table S4).
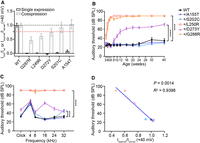
Correlation between in vitro functional data and auditory phenotypes in Kcnq4 mutant mice. (A) Comparison of the normalized currents of expressing WT, p.G287R, p.L249R, p.D272Y, p.S201C, p.A154T alone, and coexpressing with WT (n ≥ 3, mean ± SEM). The black dotted line indicates the WT value, and the red indicates 0.77 of the WT value. (B) ABR measurement of WT and five heterozygous mutant mouse strains at 16 kHz at different ages. At least three mice were measured in each strain and each age. Data were analyzed by two-way ANOVA with a Dunnett correction. (****) P < 0.0001, compared with WT. (C) ABR measurement of WT and five heterozygous mutant mouse strains at the age of 16 wk at different frequencies. (D) Correlation between in vitro functional data and hearing phenotypes in WT and five Kcnq4 mutant heterozygous mouse strains result in R2 = 0.9398 and P = 0.0014 by Pearson's correlation.
To evaluate the hearing ability of the Kcnq4 mutant KI mice, we performed auditory brainstem response (ABR) analysis in all five mutant mouse strains at different ages. As expected, for these five variants with nearly no current in the homozygous state, homozygous Kcnq4G288R, Kcnq4L250R, Kcnq4D273Y, and Kcnq4S202C KI mice developed severe deafness from the age of 3 wk, and homozygous Kcnq4A155T mice showed severe deafness with an ABR threshold reaching ∼80 dB SPL from 16th wk compared with WT mice (Supplemental Fig. S12A).
We further tested whether the auditory phenotypes of heterozygous mutant mice were correlated with the varied functional properties of heterogeneous channels in vitro. Two heterozygous KI mouse strains carrying the equivalents (p.G288R and p.L250R) of the human p.G287R and p.L249R variants that exerted high degrees of inhibiting effects on the function of the heterogeneous channels in vitro, based on our patch-clamp data, were nearly completely deaf at an early age of 6 wk across all frequencies (Fig. 5B; Supplemental Fig. S12). A heterozygous KI mouse strain carrying the equivalent (p.D273Y) of the human p.D272Y variant that exerted mildly impaired heterozygous channel function, showed progressively declined hearing ability from the age of 8–10 wk and displayed a significant threshold shift of ∼36 dB SPL at 16 wk at 16 kHz compared with WT mice of the same age (Fig. 5B,C; Supplemental Fig. S12). No hearing impairment was observed in the other two heterozygous KI mouse strains carrying the equivalents (p.S202C and p.A155T) of the human p.S201C and p.A154T variants, which produced currents comparable to those of WT in the heterozygous state (Fig. 5B,C; Supplemental Fig. S12). Taken together, the results of ABR measurement in vivo were consistent with the results of currents obtained from coexpressing variants with WT in vitro.
In summary, we observed a strong correlation between the peak currents measured from coexpressing these five variants with the WT channel at +40 mV and the ABR threshold of the heterozygous mice (Fig. 5D; Supplemental Fig. S13). Indeed, the hearing phenotypes of these mutant mice are consistent with the in vitro functional classification, which provide evidence that variants with impaired heterozygous channel function may cause hearing loss in vivo.
Pathogenicity interpretation of KCNQ4 missense variants observed in the clinic
Next, we tested whether our recording results are consistent with the clinical pathogenicity classification of KCNQ4 missense variants and assessed the impact of our patch-clamp data on variant pathogenicity interpretation. A total of 509 KCNQ4 missense variants were collected from multiple data sources, such as the Deafness Variation Database (DVD) (Azaiez et al. 2018), ClinVar (Landrum et al. 2016), the 1000 Genomes Project (The 1000 Genomes Project Consortium 2012), and gnomAD (Lek et al. 2016). Based on the specified American College of Medical Genetics and Genomics/Association for Molecular Pathology (ACMG/AMP) variant interpretation criteria for KCNQ4 missense variants, these 509 variants were classified into five categories: pathogenic, likely pathogenic, VUSs, likely benign, and benign (Supplemental Fig. S14; Supplemental Methods). Without our patch-clamp data, 22 variants were curated as benign, six were likely benign, 12 were likely pathogenic, 13 were pathogenic, and 456 were VUSs (Supplemental Table S5).
The functional properties of variants curated as benign/likely benign were functionally normal or near-normal (Fig. 6A–D; Supplemental Fig. S15; Supplemental Table S6). A total of 24/25 variants curated as pathogenic/likely pathogenic had no current in the homozygous state and were functionally impaired in the heterozygous state (Figs. 4A,B, 6A,C,D; Supplemental Fig. S15; Supplemental Table S7). These results were highly concordant with the clinical pathogenicity interpretation of ACMG/AMP rules. Meanwhile, we also verified that most synonymous variants showed normal function and that null mutants showed loss of function and impaired heterozygous channel function (Supplemental Fig. S16; data set presented in Supplemental Data Sets G,H). In addition, most variants curated as benign showed allele frequency exceeding the maximum cutoff of 0.1% in at least one population, and our functional data confirmed that variants with higher allele frequencies in the general population are more likely to function normally (Fig. 6E). Furthermore, to validate the evidence strength of our functional data, we calculated the odds of pathogenicity (OddsPath) presented by the Clinical Genome Resource (ClinGen) Sequence Variant Interpretation (SVI) Working Group using 25 pathogenic/likely pathogenic and 28 benign/likely benign variant controls (Brnich et al. 2020). The result showed that our patch-clamp assay for the KCNQ4 functional testing met BS3 and PS3 criteria (Supplemental Table S8).
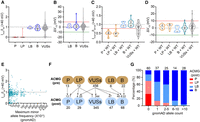
Pathogenicity interpretation of KCNQ4 clinical missense variants with functional data. (A) Violin plots comparing normalized peak currents at +40 mV when expressed alone for KCNQ4 missense variants observed in the clinic, colored by curated classification based on ACMG criteria without our functional data (13 pathogenic, 12 likely pathogenic, six likely benign, 22 benign, and 456 VUSs). (B) Distribution of the shifts in V1/2 (ΔV1/2) for six likely benign, 22 benign, and 394 VUSs with measurable V1/2. (C,D) Violin plots comparing normalized peak currents (C) and ΔV1/2 (D) for coexpressing variants interpreted as pathogenic, likely pathogenic, likely benign, benign, and VUSs with WT. The functional properties of VUSs in the heterozygous state only contained 75 variants showing reduced-current variants in the homozygous state. The red dashed line in C indicates 0.77 of the WT value. (E) Relationship between maximum minor allele frequency in any population and normalized peak currents recorded at +40 mV for 335 variants found in gnomAD database (p.H455Q with high allele frequency was not included). (F) Variants were classified based on ACMG/AMP criteria and reclassification with our functional data as BS3 and PS3 evidence. (Top) Variants were classified without our functional data. (Bottom) Variants were reclassified with our functional data. (G) Pathogenicity interpretation varied with the different gnomAD allele counts.
Further, we incorporated patch-clamp data as BS3 and PS3 evidence to reclassify clinical missense variants. Combined with functional evidence in this study, 28 variants that were initially classified as benign or likely benign were all reclassified as benign, and 13 variants that were initially classified as pathogenic were still pathogenic, whereas of 12 variants that were initially classified as likely pathogenic, seven were reclassified as pathogenic, four were still likely pathogenic, and one was VUS with normal function (Fig. 6F; Supplemental Table S5). Collectively, these results showed that our patch-clamp data serve as strong functional evidence and exert important implications for the reclassification of likely benign and likely pathogenic variants.
Then we analyzed the potential application of our functional evidence to reclassify all the discovered KCNQ4 missense variants with unknown significance; 82.9% (378/456) of initially classified VUSs had normal currents, 16.4% (75/456) had reduced currents, and 0.7% (3/456) had enhanced currents when expressed alone (Fig. 6A). Also, 93.7% (369/394) of variants curated as VUSs with detectable V1/2 showed <10-mV shifts (Fig. 6B). A total of 35/75 current-reduced variants produced impaired heterozygous channel function (Fig. 6C,D). After incorporating our patch-clamp data, 40/456 VUSs were reclassified as benign, 47 were likely benign, 25 were likely pathogenic, and 344 were still VUSs (Fig. 6F; Supplemental Table S5). Overall, the pathogenicity of 112/456 VUSs (24.6%) were reclassified, and the remaining would be interpreted when more sequencing data are available and more DFNA2 pedigrees are collected. Also, we found the pathogenicity interpretation results varied according to the frequency in the population, suggesting that variants with higher allele counts in gnomAD were more likely to be classified as benign variants (Fig. 6G). Together, our functional data will be immediately useful to provide strong functional evidence to support the interpretation of newly discovered KCNQ4 missense SNVs found in the clinic or the general population.
Conventional computational predictions of variant deleteriousness, such as CADD (Kircher et al. 2014), REVEL (Ioannidis et al. 2016), and EVE (Frazer et al. 2021), are frequently used in interpreting clinical variants. Pathogenic or likely pathogenic variants showed relatively high predictive scores, whereas benign or likely benign variants showed a wide distribution of scores (Supplemental Fig. S17). Many variants with currents similar to WT had high predictive scores and were predicted to be deleterious. These results indicate that computational predictions may not be highly specific and misclassify many normal function variants as loss of function. Further, these computational methods cannot predict the heterozygous channel function of KCNQ4 variants and cannot mimic DFNA2 patients’ genotypes.
Discussion
The ACMG/AMP guidelines describe a standard process based on a series of weighted criteria that can be scored to assess the possibility of pathogenicity for classifying variants into five categories: pathogenic, likely pathogenic, uncertain significance, likely benign, and benign (Richards et al. 2015). Functional data (PS3: well-established functional studies supportive of a damaging effect; BS3: well-established functional studies show no damaging effect) are strong evidence to classify variants. The OddsPath for our patch-clamp assay in KCNQ4 variants shows that our functional data are enough to serve as PS3 and BS3 criteria (Supplemental Table S8). The pathogenicity interpretation of newly discovered KCNQ4 missense SNVs would be deciphered with a series of criteria proposed by ACMG/AMP, with our functional data as strong evidence PS3 and BS3. We performed functional classification mainly based on peak currents, followed by V1/2. In addition to these two parameters, functional testing of ion channels also needs to consider multiple other parameters, such as reversal potential, activation kinetics, and deactivation kinetics. However, pathogenic and benign variants could be nearly perfectly separated based on the peak currents, and the hearing phenotypes of Kcnq4 point mutant mice also support functional classification in vitro, suggesting that our functional classification mainly based on peak currents is practicable.
Previous studies have shown that decreased channel currents in heterologous expression systems are one of the strongest predictors of KCNQ4 variants associated with DFNA2 (Kubisch et al. 1999; Mencía et al. 2008; Kim et al. 2011; Gao et al. 2013; Jung et al. 2018). Despite the many advantages of functional testing in a heterologous expression model, there are some limitations. It is possible that some variants show different properties in cultured cells compared to hair cells. First, at the mRNA level, such as the splicing effects, overexpressing cDNA in CHO-K1 cells failed to measure the effects of variants on splicing. Second, at the protein level, for example, cultured cells may lack protein–protein interactions or post-translational modification in hair cells.
ACMG/AMP guidelines for genetic hearing loss state that missense variants in the pore-forming region (amino acids Y270-R297) of KCNQ4 are eligible for moderate evidence (PM1) (Oza et al. 2018). Of 165 variants located in residues Y270-R297, 138 variants showed loss of function and 114 showed impaired heterozygous channel function (Supplemental Figs. S3, S9). A total of ∼30% of variants showed no effect on heterozygous channel function in this region, suggesting that it may not be directly considered that all variants located in the hotspot regions meet the PM1 criteria. Variants that do not meet PM1 criteria can be excluded based on our functional data.
Patch-clamp recording of large-scale functional testing of variants in ion channel genes is difficult and time-consuming. We are the first to analyze the function of thousands of missense variants in ion channel genes. Because of the difficulties and time-consuming nature of patch-clamp recording, the number of recorded cells for some variants is limited. There may be some limitations in the functional testing of some variants with mildly reduced or mildly increased currents, which may overlook some subtle changes in channel properties because of the small number of recorded cells.
Each KCNQ family member plays an important role in different tissues, and variants in KCNQ family genes can cause a variety of channelopathies (Jentsch 2000; Soldovieri et al. 2011). A large number of KCNQ1 and KCNQ2 variants have been observed in the LQTS and epilepsy patients, respectively. The protein sequences are highly conserved among KCNQ family channels. Our functional classification for all possible missense SNVs of KCNQ4 is informative to predict the functional changes of the corresponding missense SNVs in other KCNQ gene family members. The number of the same missense SNVs for each KCNQ family member at the corresponding position is 1933 for KCNQ1, 2044 for KCNQ2, 2064 for KCNQ3, and 2519 for KCNQ5. Collectively, the functional classification of 4085 missense SNVs in KCNQ4 provides a rich resource for analyzing the functions of KCNQ4 and KCNQ family ion channels.
Furthermore, many variants have been found in other voltage-gated ion channel genes, which have a high pathogenicity rate, such as SCN1A, SCN5A, KCNH2, and KCNB1 (Lai and Jan 2006; Kapplinger et al. 2009, 2010; Kang et al. 2019; Symonds and McTague 2020). Summarily, our work shows the possibility for the comprehensive functional classification of all potential missense SNVs in clinically disease-related voltage-gated ion channels.
Methods
Plasmid constructs and point mutations
Complementary DNAs of human KCNQ4 (NP_004691.2, isoform a) were purchased from GeneCopoeia (EX-U0210-M98-5). WT KCNQ4 and variants used in patch-clamp recordings were fused with EGFP at the C terminus as a transfection marker. For the potential possible study of the subcellular localization of WT and variants, a modified c-Myc tag was inserted in the extracellular domain of the KCNQ4 channel as described in the previous study (Kim et al. 2011). All KCNQ4 missense variant clones used in the electrophysiological recording were generated from the WT KCNQ4 CDS with a modified c-Myc tag in the plasmids EX-U0210-M98-5. For the cotransfection experiment, EGFP was substituted with mCherry by a homologous recombination method and verified by Sanger sequencing. Additionally, full-length cDNAs encoding WT KCNQ4 were also engineered in the vector pIRES2-EGFP as a control.
All possible KCNQ4 4085 missense SNVs were calculated through MATLAB based on the codon sequence of KCNQ4 (NM_004700.4). Point-mutated plasmids were constructed based on PCR and homologous recombination and were Sanger-sequenced to confirm that the mutation site was included and no other mutations were introduced.
Detailed descriptions of the procedures are provided in the Supplemental Methods (plasmid construct and point mutations).
CHO-K1 cell culture and transfection
Chinese hamster ovary (CHO) cells were cultured in DMEM/F12 medium (Gibco) and supplemented with 10% fetal bovine serum (Gibco) and penicillin (50 IU/mL)/streptomycin (50 μg/mL; Gibco) at 37°C with 5% CO2. Cells were transfected with WT or KCNQ4 variant plasmids using Lipofectamine 3000 reagent (Thermo Fisher Scientific) according to the manufacturer's instructions. Electrophysiological experiments were performed 24 h after transfection.
Detailed descriptions of the procedures are provided in the Supplemental Methods (CHO-K1 cell culture and transfection).
Electrophysiological recordings
KCNQ4 currents were measured in CHO-K1 cells, using the whole-cell patch-clamp technique. Currents were recorded using Axopatch 200B and MultiClamp 700B amplifier (Axon Instruments) and Digidata 1550B (Axon Instruments). Currents were sampled at 10 kHz. All data were low pass filtered at 1 kHz. Before acquiring currents, the cell membrane capacitance (Cm) and series resistance (Rs) were compensated using a circuit of the patch-clamp amplifier. Series resistances were compensated 70%–90% and no less than 15 MΩ. All current trace data were analyzed using Clampfit 10.6 (Molecular Devices). For whole-cell recording, borosilicate glass pipettes (BF150-86-10, Sutter Instrument) were pulled using a Flaming/Brown micropipette puller (P-97, Sutter Instrument). Patch electrodes with 4- to 7-MΩ resistance were used. The bath solution for the whole-cell patch-clamp contained (in mM): 145 NaCl, 4 KCl, 1.8 CaCl2, 0.5 MgCl2, 10 HEPES, 5 D-glucose (pH 7.4), with NaOH. The pipette solution contained (in mM): 140 KCl, 1 MgCl2, 10 HEPES, 10 EGTA, 1 CaCl2, 4 K2ATP (pH 7.2), with KOH.
The step pulse protocol used for KCNQ4 channel recordings started from a holding potential of −80 mV, depolarized for 1 sec from −100 mV to +40 mV with 20-mV increments, and followed by a tail pulse at −50 mV for 0.5 sec. P/N leak subtraction was performed. The current–voltage relationship was generated by the average current of the last 25 msec of the activation process and fitted according to the step potential. Continuous curves were generated by plotting the normalized tail current against membrane potentials using the Boltzmann function I/Imax = 1/{1 + [exp(V1/2–Vm)k]}, where I is the magnitude of the tail current, Imax is the maximum current magnitude, V1/2 is the half-activation voltage, Vm is the membrane potential, and k = RT/zF is the slope factor.
Detailed descriptions of the procedures are provided in the Supplemental Methods (electrophysiological recordings).
Data exclusion and repeat criteria for electrophysiological recordings
Current amplitudes were analyzed from recording cells that satisfied the following quality control criteria: seal resistance ≥ 500 MΩ, series resistance ≤ 15 MΩ, and cell capacitance between 5 and 25 pF. The capacitances of the cells we chose to record were mostly ∼8–15 pF. Data were also removed from the analysis if the following problems existed: (1) electrically unstable cells (baselines were unstable); (2) cells with high leak currents resulted from the loss of seal; (3) cells with reduced currents resulted from poor cell state (WT as the control); (4) cells with obvious current run-down.
Detailed descriptions of the procedures are provided in the Supplemental Methods (data exclusion and repeat criteria for electrophysiological recordings).
Variant functional classification
k-Means clustering was used to divide all 4085 missense SNVs into three groups based on the magnitude of the normalized peak current values (Imut/Iwt) recorded at +40 mV of each variant. Two thresholds (0.54 and 2.08) were obtained to distinguish each type of variant. Variants with >10-mV shifts in V1/2 relative to the WT channel were considered to have a strong effect on the channel gating properties. Combining the normalized peak currents and half-activation voltage properties, we classified all 4085 variants into three categories: loss of function, normal function, and gain of function.
Detailed descriptions of the procedures are provided in the Supplemental Methods (variant functional classification).
Generation of Kcnq4 KI point mutant mice and genotyping
All point mutant KI mice (p.G288R, p.L250R, p.D273Y, p.S202C, and p.A155T) in a C57BL/6J background were obtained by CRISPR-Cas9-mediated genome engineering (Cyagen Biosciences). Mice were genotyped by PCR using tail genomic DNA. All primers for genotyping are described in Supplemental Table S4.
Detailed descriptions of the procedures are provided in the Supplemental Methods (generation of Kcnq4 knock-in point mutant mice and genotyping).
ABR measurement
For each genotype group, three to five mice (C57BL/6J) of either sex were tested by ABR measurement from 3 to 40 wk of age. After anesthetization with xylazine (i.p., 10 mg/kg) and ketamine (i.p., 100 mg/kg), mice were placed in a soundproof chamber on a heating pad (Homeothermic Monitoring System, Harvard Apparatus) to maintain their body temperature. Subdermal needle electrodes were placed at the vertex (active), ipsilateral ear at the same side as stimulus delivery (reference), and base of the tail (ground). Stimuli were produced by SigGenRZ software, and responses were recorded by BioSigRZ (Tucker-Davis Technologies). Scalp potential was band-pass filtered over a 20-msec epoch between 0.03 and 3 kHz. At each level, 512 responses were averaged. ABRs were elicited by pure tone pips (5-msec duration, 21 times per second) and click via a speaker (TDT) placed 5 cm away from the vertex. ABR wave II was used to determine the threshold from 90 to 0 dB SPL at frequencies of 4 kHz, 8 kHz, 16 kHz, 24 kHz, and 32 kHz. Thresholds (dB) were determined by reducing the sound intensity in 5-dB steps until no detectable response.
Classification criteria for variant pathogenicity interpretation
A total of 509 KCNQ4 missense variants were collected from multiple data sources including the DVD (Azaiez et al. 2018), ClinVar (Landrum et al. 2016), the 1000 Genomes Project (The 1000 Genomes Project Consortium 2012), and gnomAD (Lek et al. 2016). Following the universal ACMG/AMP variant interpretation guidelines and expert specification for genetic hearing loss (Richards et al. 2015; Oza et al. 2018), these 509 variants were manually classified into five categories: pathogenic (13), likely pathogenic (12), uncertain significance (456), likely benign (six), and benign (22). ACMG/AMP criteria used for KCNQ4 variant pathogenicity classification are shown in Supplemental Figure S14. Variant pathogenicity classification pre- and postfunctional data are presented in Supplemental Table S5.
Detailed descriptions of the procedures are provided in the Supplemental Methods (classification criteria for variant pathogenicity interpretation).
Statistical analyses
The obtained values were expressed as means ± SEM. One-way ANOVAs and two-way ANOVAs were used to assess statistical significance. The data were plotted with GraphPad Prism 8.0.2 and MATLAB. The peak current value and half-activation voltage of WT and variants were obtained by customized MATLAB codes (Supplemental Codes). The k-means clustering was analyzed by the Statistical Program for Social Science (SPSS).
Data access
All raw data generated in this study have been submitted to MAVEdb (https://www.mavedb.org) under accession number urn:mavedb:00000094-a, with data displaying after processing is complete, and submitted as a Supplemental Data Set. MATLAB scripts used in this study are available as Supplemental Codes and provided at GitHub (https://github.com/Yan-lab-KCNQ4/KCNQ4).
Competing interest statement
The authors declare no competing interests.
Acknowledgments
We thank Fangyi Chen (Southern University of Science and Technology) and Yuchen Song for helping write the code to calculate the current value. We also thank Daniel L. Minor Jr., Jing Cheng, Xiaomei Luo, Binxin Xu, and Wenyong Fan for comments. This work was supported by funds from the China Brain Project (2021ZD0203304), Shenzhen Science and Technology Program (RCJC20210609104631084), National Key R&D Program of China Project (2021YFA1101302, 2017YFA0103900, 2016YFA0502800), National Natural Science Foundation of China (31571083, 31970931), Program for Professor of Special Appointment (Eastern Scholar of Shanghai, TP2014008), Shanghai Municipal Science and Technology Major Project (No. 2018SHZDZX01), ZJLab and Shanghai Center for Brain Science and Brain-Inspired Technology, and the Shanghai Rising-Star Program (14QA1400800).
Author contributions: Z.Y. conceived the project and designed the experiments. H.Z., X.Y., G.L., W.Z., and F.Y. generated variant plasmids. H.Z., X.Y., G.L., W.Z., F.Y., and H.L. performed the patch-clamp recordings. H.L. and S.D. performed the auditory brainstem response measurement. H.Z., W.Z., and Z.Y. designed the experiments. H.Z., X.Y., and Z.Y. analyzed the data with the assistance of G.L., H.L., S.D., and W.Z. Y.L. and H.Y. helped analyze the interpretation of variants in the clinical databases. L.J. provided guidance on the project. H.Z., X.Y., S.D., H.L., and Z.Y. wrote the manuscript. All authors discussed the results and commented on the manuscript.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.276562.122.
- Received January 8, 2022.
- Accepted June 21, 2022.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see https://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








