
Genetic, epigenetic, and environmental mechanisms govern allele-specific gene expression
Abstract
Allele-specific expression (ASE) is a phenomenon in which one allele is preferentially expressed over the other. Genetic and epigenetic factors cause ASE by altering the final composition of a gene's product, leading to expression imbalances that can have functional consequences on phenotypes. Environmental signals also impact allele-specific expression, but how they contribute to this cross talk remains understudied. Here, we explored how genotype, parent-of-origin, tissue, sex, and dietary fat simultaneously influence ASE biases. Male and female mice from a F1 reciprocal cross of the LG/J and SM/J strains were fed a high or low fat diet. We harnessed strain-specific variants to distinguish between two ASE classes: parent-of-origin-dependent (unequal expression based on parental origin) and sequence-dependent (unequal expression based on nucleotide identity). We present a comprehensive map of ASE patterns in 2853 genes across three tissues and nine environmental contexts. We found that both ASE classes are highly dependent on tissue and environmental context. They vary across metabolically relevant tissues, between males and females, and in response to dietary fat. We also found 45 genes with inconsistent ASE biases that switched direction across tissues and/or environments. Finally, we integrated ASE and QTL data from published intercrosses of the LG/J and SM/J strains. Our ASE genes are often enriched in QTLs for metabolic and musculoskeletal traits, highlighting how this orthogonal approach can prioritize candidate genes. Together, our results provide novel insights into how genetic, epigenetic, and environmental mechanisms govern allele-specific expression, which is an essential step toward deciphering the genotype-to-phenotype map.
Deciphering the genotype-to-phenotype map remains a fundamental quest in biology. Gene expression is a promising focal point, because it is an intermediate step between DNA sequence and gross phenotype. Gene expression itself is a complex trait regulated by genetic, epigenetic, and environmental factors (Pastinen 2010). Recent efforts to characterize gene expression have often only interrogated changes in total expression levels and assumed that genes are biallelically expressed (i.e., both alleles are equally expressed), which may mask underlying regulatory mechanisms. Epigenetic changes and gene-by-environment interactions can also alter gene expression patterns in a dynamic and tissue-specific manner without modifying the underlying nucleotide sequence; these effects are missed in a typical sequence-based method. Thus, an allele-specific approach is required to directly measure how cis-regulatory variation impacts gene expression and to tease it apart from trans-acting factors that affect both chromosomes (Bonasio et al. 2010). Allele-specific expression (ASE) is a phenomenon in which a gene's expression diverges from biallelic. In diploid organisms, one allele is preferentially expressed over the other. Previous findings estimate that 30%–56% of genes show evidence of allelic imbalance, indicating allele-specific effects have widespread impacts on gene regulation (Ge et al. 2009; Keane et al. 2011; Castel et al. 2020). Depending on a gene's function, these expression imbalances can lead to phenotypic variation with functional consequences.
ASE can be divided into two classes: sequence or parent-of-origin (Fig. 1). Sequence-dependent ASE occurs when the alleles are differentially expressed based on their haplotype or nucleotide identity. These patterns are thought to be largely driven by cis-acting genetic variants in coding and noncoding regions, such as a premature stop codon that truncates one allele's transcript or a variant in a promoter region that prevents transcription factors from binding (Keane et al. 2011; Rivas et al. 2015). They can also occur further away from the gene yet still impact its expression, such as motif variations for long-range enhancers or in the sequence context of DNA methyltransferase substrates (Wienholz et al. 2010; Cavalli et al. 2016). Stochastic transcriptional bursts can also cause transient fluctuations in which allele is expressed, resulting in random monoallelic expression that is inconsistent across cells or organisms (Deng et al. 2014; Reinius and Sandberg 2015).

Evaluating ASE across tissues and environmental contexts. We partitioned ASE into its parent-of-origin and sequence effects, then compared ASE patterns across metabolic tissues, in response to dietary fat and between sexes. An example of parent-of-origin-dependent ASE is when the maternal allele (red) is preferentially expressed over the paternal allele (blue), regardless of which haplotype contributed it. An example of sequence-dependent ASE is when the LG/J allele is preferentially expressed over the SM/J allele, regardless of which parent contributed it.
In contrast, parent-of-origin-dependent ASE occurs when the alleles are differentially expressed based on which parent contributed it, regardless of the underlying sequence. These patterns are examples of parent-of-origin effects (POEs), a broader class of epigenetic phenomena that manifest as phenotypic differences according to maternal or paternal inheritance (Lawson et al. 2013). The best characterized POE mechanism is genomic imprinting, an extreme case of parent-of-origin-dependent ASE in which one parent's allele is silenced via selective epigenetic marks, such as DNA methylation and histone modifications (Umlauf et al. 2004; Barlow and Bartolomei 2014; Inoue et al. 2017). Known imprinted genes compose ∼1% of the human and mouse genomes, yet play important roles in development, metabolism, and cognition (Reik and Walter 2001). Partial imprinting is another, more subtle, form of parent-of-origin-dependent ASE that has been documented but is less understood (Wolf et al. 2008; Morcos et al. 2011). Random monoallelic expression can also produce transient and nonheritable parental biases in individual cells through different mechanisms than imprinting (Gimelbrant et al. 2007; Morcos et al. 2011). Other sources of POEs include maternal effects and gene-specific trinucleotide expansions, but these do not involve ASE (Hager et al. 2008).
Comprehensive atlases of how ASE patterns vary between tissues and developmental stages have been generated in human and mouse models (Babak et al. 2015; Leung et al. 2015; Andergassen et al. 2017; Castel et al. 2020). These studies reveal that both parental and sequence biases are not always consistent between tissues, indicating that tissue-specific genetic and epigenetic features can mediate allelic imbalances. Additionally, trans-acting environmental factors have been shown to interact with cis-regulatory variants to modulate the magnitude of ASE effects in human and rice models (Buil et al. 2015; Moyerbrailean et al. 2016; Knowles et al. 2017; Shao et al. 2019). Imprinted genes can also be responsive to environmental exposures during fetal development (Kappil et al. 2015). Together, these findings suggest that a complicated cross talk between genetic variants, epigenetic changes, and environmental signals underlies allele-specific gene regulation, but this model needs to be further investigated.
Here, we explored how allelic genotype, parent-of-origin, tissue type, sex, and dietary fat simultaneously work together to influence ASE patterns in a F1 reciprocal cross of the LG/J and SM/J inbred mouse strains. These strains are uniquely suited for gene-by-environment studies owing to their divergent genetic backgrounds and variable responses to dietary nutrition (Ehrich et al. 2003; Nikolskiy et al. 2015; Miranda et al. 2019; Carson and Lawson 2020). They are also the progenitors of the longest-running advanced intercross line (AIL). An AIL is a multigenerational population produced by intercrossing two inbred strains beyond the F2 generation while minimizing relatedness between mates (Darvasi and Soller 1995). Since its inception, the LG/J x SM/J AIL has been extensively used to map quantitative trait loci (QTL) related to metabolic, musculoskeletal, behavioral, and physiological traits (Ehrich et al. 2005; Lawson et al. 2010, 2011a,b; Lionikas et al. 2010; Cheverud et al. 2011; Parker et al. 2014; Gonzales et al. 2018; Cordero et al. 2019). To probe whether ASE imbalances contribute to complex traits, we integrated our ASE results with these AIL mapping studies and highlighted how this orthogonal approach can prioritize candidate genes. Untangling the genetic, epigenetic, and environmental mechanisms that govern allele-specific gene regulation is crucial to improving our ability to predict phenotypes from genotypes.
Results
Partitioning allele-specific expression into parent-of-origin and sequence effects
We measured ASE in a F1 reciprocal cross of the LG/J and SM/J inbred strains. Briefly, LG/J mothers were mated with SM/J fathers and vice versa, resulting in F1 offspring who are genetically equivalent but differ in the allelic direction of inheritance. Male and female F1 mice were fed either a high or low fat diet. We obtained RNA-seq data from three metabolically relevant tissues: hypothalamus (HYP), white adipose (WAT), and liver (LIV). We analyzed nine environmental contexts per tissue: high fat (H), low fat (L), females (F), males (M), high fat females (HF), high fat males (HM), low fat females (LF), low fat males (LM), and all contexts collapsed (All). Together, these 27 “tissue-by-context” analyses (three tissues × nine contexts) allowed us to explore how tissue type and environmental signals influence ASE (Fig. 1).
The LG/J and SM/J genomes differ by >5 million single-nucleotide polymorphisms (SNPs) and indels (Nikolskiy et al. 2015). To be informative for ASE, a strain-specific variant must be located within a gene's exome. In this F1 model, informative variants are located in 55% of expressed genes in our three tissues and 47% of all genes in the mm10 reference genome (Supplemental Fig. S1). We harnessed those strain-specific variants to map sequencing reads to their chromosome of origin. Overall, 9016 genes had detectable ASE in at least one tissue-by-context analysis (∼37% of all expressed genes) (Supplemental Fig. S2). Next, we identified 2853 genes with significant ASE biases (∼6%) and classified them into two patterns: parent-of-origin-dependent (unequal expression based on parental origin) and sequence-dependent (unequal expression based on nucleotide identity) (Fig. 1; Supplemental Table S1).
Both classes of ASE patterns are prevalent and distinct
Across our 27 tissue-by-context analyses, we identified 271 genes with significant parent-of-origin-dependent ASE (Supplemental Tables S2–S4). HYP had the greatest number of genes (229), followed by WAT (35), then LIV (27). Fourteen genes were expressed in multiple tissues, but the majority were tissue-specific (Fig. 2A). In HYP, the most genes were detected in the “All” context. However, in WAT and LIV, more genes were only detected in a specific diet, sex, and/or diet-by-sex context; those expression biases were missed when contexts were collapsed (Fig. 2B). Two hundred fourteen genes (79%) were paternally biased, 55 genes (20%) were maternally biased, and two genes (1%) switched their ASE bias direction across the cohorts (Fig. 2C). This heavy paternal skew is driven by a 672-kb cluster of 171 small nucleolar RNAs (snoRNAs) in the Prader-Willi/Angelman syndrome (PWS/AS) domain on Chromosome 7 that are only expressed in HYP (Supplemental Fig. S3). Parentally biased ASE genes also included protein-coding genes, microRNAs, long noncoding RNAs, long interspersed noncoding RNAs, and pseudogenes (Fig. 2D).
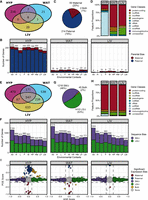
Both classes of ASE patterns are prevalent and distinct. (A) Venn diagram of all parent-of-origin-dependent ASE genes across tissues. (B) Number of significant parentally biased genes in each tissue-by-context analysis (maternal, red; paternal, blue): (All) all contexts; (H) high fat; (L) low fat; (F) females; (M) males; (HF) high fat females; (HM) high fat males; (LF) low fat females; and (LM) low fat males. (C) Summary of ASE biases across all analyses. (D) Proportions of gene classes in each tissue. (E) Venn diagram of all sequence-dependent ASE genes across tissues. (F) Number of significant sequence-biased genes in each tissue-by-context analysis (SM/J, purple; LG/J, green). (G) Summary of ASE biases across all analyses. (H) Proportions of gene classes in each tissue. (I) Parent-of-origin effect (POE) versus allelic genotype effect (AGE) scores in the “All” context of each tissue. Dots represent individual genes and are color coded by their ASE bias direction: (red) maternal; (blue) paternal; (purple) SM/J; (green) LG/J; and (yellow) both ASE classes. Most genes have no bias (gray). Dashed lines indicate significant score thresholds.
We identified 2673 genes with significant sequence-dependent ASE across our 27 tissue-by-context analyses (Supplemental Tables S2–S4). WAT had the greatest number of genes (1495), followed by LIV (1486), then HYP (1264). Although some genes’ biases were tissue-specific, 1657 genes (62%) were biased in multiple tissues (Fig. 2E). In all tissues, the most genes were detected in the “All” context, then the diet or sex contexts, and finally the diet-by-sex contexts, likely reflecting the sample sizes and power available in each cohort (Fig. 2F). As shown in Figure 2G, 1218 genes (46%) were SM/J biased, 1410 genes (53%) were LG/J biased, and 45 genes (2%) switched their expression bias direction across the cohorts. Sequence-biased ASE genes were predominantly protein-coding genes (∼80%) but also included pseudogenes, immunoglobulins, and various noncoding RNAs (long noncoding, long interspersed noncoding, micro, ribosomal, small interfering, and small nucleolar) (Fig. 2H).
Parent-of-origin and sequence-dependent ASE patterns were often mutually exclusive. In each tissue, genes with extreme parental biases typically had weak sequence biases and vice versa (Fig. 2I; Supplemental Fig. S4). However, 61 genes in HYP and three genes in WAT had both parental and sequence biases, likely because of epigenetic regulatory mechanisms affected by allelic variation. Both ASE patterns occurred genome wide (Supplemental Fig. S5). Parentally biased genes tended to cluster in known imprinted regions, like the Ube3a/Snrpn, Meg3, Peg3/Usp29, and H13/Mcts2 domains (Supplemental Fig. S6). Sequence-biased genes were spread diffusely across the genome, but occasionally clustered in discrete regions potentially controlled by the same regulatory element (Supplemental Fig. S7).
Parent-of-origin-dependent ASE recapitulates canonical imprinting patterns
To evaluate whether parent-of-origin-dependent ASE is influenced by tissue or environmental context, we characterized the expression profiles of those 271 genes across our 27 tissue-by-context analyses. For each analysis, we quantified the direction and magnitude of each gene's parental bias by calculating a POE score from the mean allelic bias of each F1 reciprocal cross. POE scores ranged from −1 (completely maternally expressed) to +1 (completely paternally expressed); a score of 0 indicated biallelic expression (Methods). In each analysis, a gene could be expressed in three possible ways: significant parental bias, biallelic (expressed but with no allelic bias), or not expressed. We sorted the 271 parentally biased ASE genes into three expression profiles: tissue-independent, tissue-dependent, and context-dependent (Fig. 3A–D; Supplemental Fig. S3).
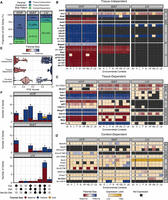
Parent-of-origin-dependent ASE patterns fall into three expression profiles. (A) Proportion of ASE genes per tissue with each parental bias profile. Heatmaps of ASE profiles across analyses: (B) tissue-independent, (C) tissue-dependent, and (D) context-dependent. A subset of the 271 genes is shown, including those validated with pyrosequencing. Genes are color coded by their expression pattern in each tissue-by-context analysis. Shades of red and blue indicate their degree of maternal or paternal bias, respectively (POE scores). If genes are not biased, shades of yellow indicate their biallelic expression levels (log-transformed total counts). Black indicates genes are not expressed. Bolded genes are canonically imprinted. The y-axis is grouped and sorted by chromosomal position. Supercolumns denote tissues: (HYP) hypothalamus; (WAT) white adipose; and (LIV) liver. Subcolumns denote environmental contexts: (All) all contexts; (H) high fat; (L) low fat; (F) females; (M) males; (HF) high fat females; (HM) high fat males; (LF) low fat females; and (LM) low fat males. (E) POE scores for each parental bias profile. Vertical lines indicate mean POE scores. Dots represent individual ASE genes. (F) UpSet plots of the significant sex, diet, and/or sex-by-diet effects of context-dependent genes in each tissue. Bar height and color indicate how many genes with each parental bias: (red) maternal; (blue) paternal; and (yellow) direction switching.
We identified 183 tissue-independent genes (67%), defined as a consistent parental bias in every tissue they were expressed. Within a given tissue, they were parentally biased in most environmental contexts (Fig. 3B). Next, we identified 15 genes (6%) as tissue-dependent. These genes showed a parental bias across most contexts in one or two tissues, but were biallelically expressed (no bias) across contexts in the other tissue(s) (Fig. 3C). Here, we distinguish between tissue-specific gene expression (not expressed in certain tissues) and tissue-dependent ASE (biased expression only in certain tissues).
All 198 tissue-independent/-dependent genes were related to genomic imprinting, the best characterized mechanism of parent-of-origin-dependent ASE. Twenty-nine genes were canonically imprinted (bolded in Fig. 3, defined in Methods), and the other 169 genes were various noncoding RNAs located within known imprinted domains. For example, 156 genes were in a cluster of 171 paternally biased snoRNAs in the PWS/AS domain on Chromosome 7 (Supplemental Fig. S3). As expected with imprinting, these genes were extremely biased toward one parent's allele; their mean POE scores were −0.82 and 0.71 for maternally and paternally biased genes, respectively (Fig. 3E). HYP had the highest proportion of these genes (85%) among our three adult tissues, even if the aforementioned HYP-specific snoRNA cluster is excluded (52%). WAT had the second highest proportion (49%), but few of these genes were expressed in LIV (26%) (Fig. 3A). These findings are consistent with the previously reported dichotomy of imprinting levels between neural and non-neural adult tissues (Babak et al. 2015) as well as imprinting's role in development and cognition (Barlow and Bartolomei 2014). Furthermore, our data replicated 25 of 96 genes previously found to be imprinted in these tissues (Babak et al. 2015), comprising nearly all bolded genes in Figure 3B–D. Five genes had sequence biases instead and 11 genes were biallelically expressed, potentially a result of methodology differences between the studies. We could not compare the imprinting status of the remaining 55 genes because 34 had no informative variants in our F1 model and 21 no longer appeared in the mm10 reference assembly.
We validated the expression profiles of two canonically imprinted genes (Peg3 and Grb10) by pyrosequencing (Supplemental Fig. S8). Paternally expressed 3 gene (Peg3) was paternally biased in all three tissues, regardless of context. Its locus had 60 strain-specific variants, but none were predicted as functional. Peg3 functions as a DNA-binding transcriptional repressor to control fetal growth rates, maternal caring behaviors, and tumor growth. It is only expressed from the paternal allele in most tissues, especially the placenta and brain (Thiaville et al. 2013; He and Kim 2014). Growth factor receptor bound protein 10 (Grb10) was paternally biased in HYP but maternally biased in WAT and LIV. Its locus had 154 strain-specific variants, but none were predicted as functional. Grb10 encodes an adapter protein that interacts with receptor tyrosine kinases to impact insulin signaling and growth hormone pathways (He et al. 1998). It has a documented pattern of maternal expression in most adult mouse tissues, but paternal expression in the brain (Plasschaert and Bartolomei 2015).
Dietary environment and sex influence parent-of-origin-dependent ASE in a partially imprinted manner
Finally, we classified 73 genes (27%) as context-dependent, meaning they had a parental bias only in certain environmental contexts within a tissue, but biallelic expression (no bias) in other contexts and/or tissues (Fig. 3D). Only three genes were canonically imprinted, and 18 genes were noncoding RNAs located in known imprinted domains (including 14 snoRNAs in the PWS/AS domain). The remaining 52 genes had no clear connection to genomic imprinting, yet they showed significant parent-of-origin-dependent ASE in certain context(s). These genes had more subtle allelic biases than the tissue-independent/-dependent genes; their mean POE scores were −0.39 and 0.39 for maternally and paternally biased genes, respectively (Fig. 3E). These patterns are consistent with partial imprinting, in which the two parental alleles are differentially expressed in a less extreme manner than the uniparental expression associated with genomic imprinting (Wolf et al. 2008; Morcos et al. 2011). LIV had the highest proportion of these genes among the three tissues (74%). WAT had the second highest proportion (51%), whereas HYP had the lowest proportion (15%) (Fig. 3A). LIV is also a tissue where more parentally biased genes were detected in the diet- and/or sex-specific contexts than the “All” context (Fig. 2B). LIV is responsive to environmental factors, especially diet, given its central roles in digestion and detoxification (Trefts et al. 2017). Taken together, these findings suggest a mechanism of parent-of-origin-dependent ASE outside of traditional imprinting that is sensitive to environmental perturbations.
To further explore how sex and/or dietary fat can alter parental biases, we calculated individualized POE scores for each context-dependent gene and modeled how they vary across diet, sex, and diet-by-sex contexts (Supplemental Tables S5, S6). These categories were not mutually exclusive; across all three tissues, most genes were significant for more than one effect. Nonetheless, when we intersected these significant gene lists, we found that each tissue showed a distinct pattern of context-dependent parental biases (Fig. 3F; Supplemental Fig. S9). For example, LIV had similar proportions of significant sex, diet, and diet-by-sex effects (each 75%–80%); 60% of its genes were significant for all three effects. Most of WAT's genes had a significant sex effect (83%), but diet or diet-by-sex effects were much less common (44% and 28%, respectively). Finally, 63% of HYP's genes had a significant sex effect, whereas 40%–45% had significant diet or diet-by-sex effects.
We validated the expression profiles of two context-dependent genes (Apob and Slc22a3) by pyrosequencing (Supplemental Fig. S10). Apolipoprotein B (Apob) had a significant diet effect in WAT, reflected in maternal biases in the HF and F contexts. Apob was biallelically expressed in the remaining WAT contexts and all contexts in LIV and HYP. Its locus had 126 variants, including seven nonsynonymous SNPs (six, LG/J genome). Apob produces the main component of lipoproteins, which transport lipids in the blood (Olofsson and Borèn 2005). Maternal-specific associations between APOB variants and adiposity traits have been found in humans (Hochner et al. 2015). Apob expression also differs between high and low fructose diets in mice livers (Sud et al. 2017). Solute carrier family 22 (organic cation transporter), member 3 (Slc22a3) had significant diet and diet-by-sex effects in LIV, reflected in maternal biases in the HF and HM contexts. Slc22a3 was biallelically expressed in the remaining LIV contexts and all contexts in WAT and HYP. Its locus had 285 strain-specific variants, but none were predicted as functional. Slc22a3 is a transporter that eliminates organic cations from cells (Kekuda et al. 1998). It has been reported as maternally expressed in liver and extraembryonic tissues, but biallelic elsewhere (Babak et al. 2015). Slc22a3 is also differentially expressed in kidneys between high fat and chow-fed mice (Gai et al. 2016).
Sequence-dependent ASE arises from haplotype-specific genetic variation
Next, we similarly characterized the expression profiles of the 2673 sequence-biased genes across our 27 tissue-by-context analyses to evaluate whether sequence-dependent ASE is also influenced by tissue or environmental context. For each analysis, we quantified the direction and magnitude of each gene's sequence bias by calculating an allelic genotype effect (AGE) score from the mean allelic bias of each F1 reciprocal cross. AGE scores ranged from −1 (completely SM/J expressed) to +1 (completely LG/J expressed); a score of 0 indicated biallelic expression (Methods). In each analysis, a gene could be expressed in three possible ways: significant sequence/allelic genotype bias, biallelic (expressed but with no bias), or not expressed. We sorted the 2673 sequence-biased ASE genes into three expression profiles: tissue-independent, tissue-dependent, and context-dependent (Fig. 4A–D).
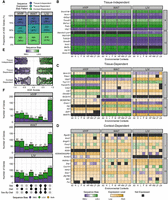
Sequence-dependent ASE patterns fall into three expression profiles. (A) Proportion of ASE genes per tissue with each sequence bias profile. Heatmaps of ASE profiles across analyses: (B) tissue-independent, (C) tissue-dependent, and (D) context-dependent. A subset of the 2673 genes are shown, including those validated with pyrosequencing. Genes are color coded by their expression pattern in each tissue-by-context analysis. Shades of purple and green indicate their degree of SM/J or LG/J bias, respectively (AGE scores). If genes are not biased, shades of yellow indicate their biallelic expression levels (log-transformed total counts). Black indicates genes are not expressed. The y-axis is grouped and sorted by chromosomal position. Supercolumns denote tissues: (HYP) hypothalamus; (WAT) white adipose; and (LIV) liver. Subcolumns denote environmental contexts: (All) all contexts; (H) high fat; (L) low fat; (F) females; (M) males; (HF) high fat females; (HM) high fat males; (LF) low fat females; and (LM) low fat males. (E) AGE scores for each sequence bias profile. Vertical lines indicate mean AGE scores. Dots represent individual ASE genes. (F) UpSet plots of the significant sex, diet, and/or sex-by-diet effects of context-dependent genes in each tissue. Bar height and color indicate how many genes with each sequence bias: (purple) SM/J; (green) LG/J; and (yellow) direction switching.
We identified 605 genes (23%) as tissue-independent, meaning they had a consistent sequence bias in every tissue they were expressed. Within a given tissue, they had a sequence bias in most environmental contexts (Fig. 4B). These genes were strongly biased toward one strain's allele; their mean AGE scores were −0.79 and 0.78 for SM/J and LG/J biased genes, respectively (Fig. 4E). All three tissues had a similar proportion of these genes (29%–35%) (Fig. 4A). These patterns likely reflect the vast genetic variation accumulated between the LG/J and SM/J backgrounds over the decades, whereby a cis-acting variant impacts one strain's allelic function wherever that gene is expressed, regardless of tissue type.
We validated the expression profiles of two tissue-independent genes (Eef1a1 and Tubb2a) by pyrosequencing (Supplemental Fig. S11). Eukaryotic translation had a LG/J bias in all three tissues, regardless of context. Its locus had 30 strain-specific variants, including two nonsynonymous SNPs in the SM/J genome. Eef1a1 delivers aminoacylated transfer RNAs to the elongating ribosome during protein synthesis and has crucial roles in protein degradation and other cellular processes. It is abundantly and ubiquitously expressed in most tissues (Mateyak and Kinzy 2010; Li et al. 2013). Tubulin, beta 2A class IIA (Tubb2a) had a SM/J bias in all three tissues, regardless of context. Mutations in this gene are rare and often nonviable; however, its locus had one SNP in the LG/J genome. Tubb2a is a tubulin isoform that binds GTP to create microtubules, which are critical for the cytoskeleton organization, intracellular trafficking, and mitotic cell division. It is also ubiquitously expressed in most tissues, especially the brain (Hammond et al. 2008; Rice et al. 2008).
Sequence-dependent ASE can be mediated by tissue-specific features
Next, we identified 684 genes (25%) as tissue-dependent. These genes showed a sequence bias across most contexts in one or two tissues, but were biallelically expressed (no bias) across contexts in the other tissue(s) (Fig. 4C). Here, we again distinguish between tissue-dependent ASE (biallelic in some tissues) and tissue-specific gene expression (not expressed in some tissues). These genes were moderately biased toward one strain's allele; their mean AGE scores were −0.57 and 0.55 for SM/J and LG/J biased genes, respectively (Fig. 4E). All three tissues also had a similar proportion of these genes (27%–32%) (Fig. 4A). These patterns show that sequence-dependent ASE is not solely a result of genetic variation; here, tissue-specific epigenetic factors likely interact with cis-acting variants to influence allelic frequency.
We validated the expression profiles of two tissue-dependent genes (Upp2 and Mettl7b) by pyrosequencing (Supplemental Fig. S12). Uridine phosphorylase 2 (Upp2) had a strong SM/J bias across contexts in HYP, a weaker SM/J bias in WAT, and biallelic expression in LIV. Its locus had 1043 strain-specific variants, including five nonsynonymous SNPs in the LG/J genome. Upp2 catalyzes the phosphorolysis of uridine into uracil and ribose-1-phosphate during nucleoside metabolism (Johansson 2003). In mice, it is predominantly expressed in liver and weakly expressed in brain (Roosild et al. 2011). Methyltransferase like 7b (Mettl7b) had a LG/J bias across contexts in WAT, biallelic expression in LIV, and no expression in HYP. Its locus had 77 strain-specific variants, including one nonsynonymous SNP in the SM/J genome. Mettl7b is an akyl thiol methyltransferase implicated in several cancers (Maldonato et al. 2021). It is highly expressed in lipid droplets, particularly liver and adipose tissue (Turró et al. 2006).
Sequence-dependent ASE is sensitive to sex and dietary environments
Finally, we classified 1384 genes (52%) as context-dependent. These genes had a sequence bias only in certain environmental contexts within a tissue, but biallelic expression (no bias) in other contexts and/or tissues (Fig. 4D). These genes had more subtle allelic biases than the other two profiles; their mean AGE scores were −0.32 and 0.34 for SM/J and LG/J biased genes, respectively (Fig. 4E). LIV had the most context-dependent genes (616) and HYP had the fewest (483), but overall all three tissues comprised a similar proportion of these genes (38%–41%) (Fig. 4A). These patterns suggest that environmental factors can interact with genetic variation to influence the final allelic composition of a gene's product.
To further explore how sex and/or dietary fat can alter sequence biases, we calculated individualized AGE scores for each context-dependent gene and modeled how they vary across diet, sex, and diet-by-sex contexts (Supplemental Tables S5, S6). When we intersected these gene lists, we found that each tissue showed a similar pattern of context-dependent sequence biases (Fig. 4F; Supplemental Fig. S13). Significant sex effects were the most prevalent in each tissue, ranging from 32% of genes in HYP, 43% in WAT, to 51% in LIV. Diet effects were slightly less common but still widespread: 30% of genes in HYP, 43% in WAT, and 48% in LIV. Finally, diet-by-sex effects were also pervasive in each tissue, comprising 29% of genes in HYP, 33% in WAT, and 41% in LIV. These categories were not mutually exclusive; most genes were significant for more than one effect across the three tissues.
We validated the expression profiles of two context-dependent genes (Ifi205 and Gas1) by pyrosequencing (Supplemental Fig. S14). Interferon activated gene 205 (Ifi205) had significant sex and diet-by-sex effects in WAT, reflected in strong LG/J biases in the three female-related (HF, LF, F) and “All” contexts. Ifi205 was biallelically expressed in the remaining WAT contexts and all LIV contexts. Its locus had 254 strain-specific variants, including 10 nonsynonymous SNPs in the SM/J genome. Ifi205 binds DNA in response to interferon signaling, which activates the immune system (Albrecht et al. 2005). Significant sex differences have been reported in mice, in which females have higher total Ifi205 expression levels than males (Cao et al. 2018). Growth arrest specific 1 (Gas1) had significant diet and sex effects in LIV, reflected in strong SM/J biases in the three low fat diet-related (LF, LM, L), female, and “All” contexts. Gas1 was biallelically expressed in the remaining high fat diet and male contexts in LIV and all contexts in HYP and WAT. Its locus had 57 strain-specific variants, although none were predicted as functional. Gas1 encodes a membrane glycoprotein that binds and regulates sonic hedgehog during development (Lee et al. 2001). Gas1 is differentially expressed between high and low selenium diets in mice ovaries, suggesting its expression may be sensitive to dietary environment (Qazi et al. 2021).
Sequence-dependent ASE genes can switch the direction of their allelic biases
Finally, we found 45 sequence-dependent ASE genes with inconsistent patterns of allelic biases. These genes showed significant ASE in opposite directions among the tissues and/or environmental contexts (Fig. 5). For 44 of the 45 genes, such direction switching occurred at the tissue level: for example, a gene may have a LG/J bias in one tissue, a SM/J bias in another tissue, and sometimes even no bias (biallelic) in the third tissue. Four genes had tissue-independent ASE, or a sequence bias across contexts in every expressed tissue (albeit in different directions). Nineteen genes had context-dependent ASE, or a sequence bias only in certain environmental contexts that switched direction across tissues. One gene (Cidec) had a context-dependent switch in ASE direction within the same tissue, discussed further below. The remaining 22 genes had a combination of context- and tissue-dependent ASE patterns: such genes had a sequence bias in one direction across contexts in one or two tissues, but a context-dependent sequence bias in the opposite direction in another tissue. Overall, these genes had moderate allelic biases; their mean AGE scores were −0.38 and 0.40 for SM/J and LG/J biased genes, respectively. These dynamic direction-switching patterns confirm that sequence-dependent ASE is not solely a result of genetic variation. Otherwise, the same variant causing ASE in one tissue would also be present in any other cell expressing that gene. These patterns hint at epigenetic regulatory elements interacting with genetic variation in a tissue-specific manner to influence the final allelic frequency.

Forty-five ASE genes switch their sequence bias direction across conditions. Heatmap of ASE profiles for the 45 genes with significant sequence biases in opposite directions, including those validated with pyrosequencing. Genes are color coded by their expression pattern in each tissue-by-context analysis. Shades of purple and green indicate their degree of SM/J or LG/J bias, respectively (AGE scores). If genes are not biased, shades of yellow indicate their biallelic expression levels (log-transformed total counts). Black indicates genes are not expressed. The y-axis is grouped and sorted by chromosomal position. Supercolumns denote tissues: (HYP) hypothalamus; (WAT) white adipose; and (LIV) liver. Subcolumns denote environmental contexts: (All) all contexts; (H) high fat; (L) low fat; (F) females; (M) males; (HF) high fat females; (HM) high fat males; (LF) low fat females; and (LM) low fat males.
We validated the expression profiles of two direction-switching genes (Stab2 and Ociad2) with pyrosequencing (Supplemental Fig. S15). Stabilin 2 (Stab2) had a LG/J bias in all contexts in LIV but a SM/J bias across most contexts in HYP and WAT. Its locus had 1019 strain-specific variants, including 14 nonsynonymous SNPs in the SM/J genome. Stab2 binds to hyaluronic acid and mediates its transportation inside the cell (Zhou et al. 2002). It is predominantly expressed in two isoforms in the liver and spleen, but weakly expressed in the brain and adipose tissue (Falkowski et al. 2003). OCIA domain containing 2 (Ociad2) had significant sex and sex-by-diet effects in WAT and LIV. These are reflected in LG/J biases in the L, F, and “All” contexts in WAT, but SM/J biases in the LF, L, F, and “All” contexts in LIV. Ociad2 was biallelically expressed in the remaining contexts of both tissues and in HYP. Its locus had 118 strain-specific variants, although none were predicted as functional. Ociad2 regulates cell migration and is moderately expressed in several tissues, including the brain and liver (Sinha et al. 2018). It is highly expressed in female ovarian tumors (Nagata et al. 2012), but otherwise sex or diet effects on Ociad2 expression have not been explored.
ASE genes are enriched in QTLs for metabolic and anatomical traits
Mapping studies in the LG/J x SM/J advanced intercross line have identified hundreds of QTLs associated with a constellation of complex traits. Such approaches are agnostic to tissue and often implicate regions with many genes, making it difficult to pinpoint which ones are phenotypically relevant. In contrast, ASE patterns are gene-specific and vary across tissues and environments. If there is a functional difference between alleles, then an expression imbalance in the right conditions can have phenotypic consequences and manifest as QTL signals. To probe this, we integrated ASE and QTL data from populations derived from the same founder strains at various degrees of intercrossing (Fig. 6A; Supplemental Table S7).
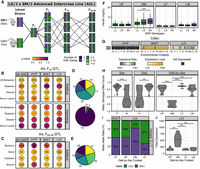
Integrating ASE and QTL data reveal Cidec as a candidate gene for insulin levels. (A) Breeding scheme for the LG/J x SM/J advanced intercross line. We calculated enrichment of tissue-specific ASE gene sets (x-axis) in trait-specific AIL QTL sets (y-axis) among sequence-dependent ASE genes in additive F16 QTL (top) and parent-of-origin-dependent ASE genes in imprinting F16 QTL (bottom) (B), and sequence-dependent ASE genes in additive F50–56 QTL (C). Circle color corresponds to the enrichment P-value. Numbers indicate the total overlapping ASE genes in each QTL set. (D,E) Tally of how many AIL QTLs contain each range of ASE genes: 0 (purple) to more than 10 (yellow). Pie charts match their adjacent ASE gene and QTL set intersections. (F) The AIL F16 QTL Ddiab6d showed context-dependent additive effects. Box plots of serum insulin levels across SNP rs6393943 genotypes in F16 mice (LL, LG/J homozygous; LS, heterozygous; SS, SM/J homozygous). The x-axis is grouped by diet-by-sex context: (HF) high fat females; (HM) high fat males; (LF) low fat females; and (LM) low fat males. Horizontal bars denote mean phenotypes. (***) P ≤ 0.001; assessed by Student's t-test. (G) Cidec had a context-dependent switch in sequence bias direction within LIV. ASE heatmap of Cidec across tissues-by-context analyses (for full description, see Fig. 4 or 5). (H) Cidec ASE biases had significant sex, diet, and diet-by-sex effects. Violin plots display individual AGE scores for Cidec (y-axis) across environmental contexts (x-axis). Horizontal bars denote mean AGE scores. Dots are color coded by their sequence bias. (**) P ≤ 0.01, (***) P ≤ 0.001; assessed by ANOVA (sex, diet) or Tukey's post hoc tests (diet-by-sex). (I) Cidec ASE profile in LIV and WAT was validated by pyrosequencing. Bar graphs denote mean allelic ratios (y-axis) in select cohorts (x-axis) and are color coded by allele (LG/J, green; SM/J, purple). (J) Cidec had significantly higher expression in HM. Violin plots of total expression levels in LIV (y-axis) for each diet-by-sex context (x-axis). (**) P ≤ 0.01; assessed by Student's t-test.
Previously, 127 QTLs influencing metabolic traits were mapped in the AIL F16 generation (Lawson et al. 2010, 2011b; Cheverud et al. 2011). These studies incorporated the same environmental contexts (sex and dietary fat) used here, traced the parental origin of each marker allele, and characterized the context-dependent additive and imprinting effects influencing metabolic traits. Overall, sequence-dependent ASE genes were significantly enriched in the 83 F16 QTLs with additive effects (P = 0.01) (Fig. 6B). We compared the QTL sets for each metabolic trait category with the full ASE gene sets for each tissue. QTLs for obesity traits were enriched for HYP, WAT, and LIV genes (P < 0.01). QTLs for diabetes traits were enriched for HYP genes (P = 0.01), but not WAT or LIV. QTLs for serum lipid traits were enriched for WAT genes (P = 0.02), but not HYP or LIV (Supplemental Fig. S16). These findings reflect the discrete roles of each tissue in metabolism physiology and illustrate that ASE patterns can inform which tissues are phenotypically relevant. Conversely, parent-of-origin-dependent ASE genes (excluding the 171 snoRNA cluster in the PWS/AS domain) were not enriched in the 76 F16 QTLs with imprinting effects (P = 0.47) (Fig. 6B). Tissue-specific ASE gene sets were also not enriched in the QTL sets for any metabolic trait category (P > 0.05) (Supplemental Fig. S16), supporting previous work showing parental biases are often not the direct source of imprinting QTL effects (Macias-Velasco et al. 2022).
Additionally, 149 QTLs influencing behavioral, musculoskeletal, and metabolic traits were mapped in later generations (F50–56) of the same AIL population (Gonzales et al. 2018; Cordero et al. 2019). These studies did not capture environmental contexts or parental origin, but still explored how additive effects impact complex traits in a context-independent manner. Overall, sequence-dependent ASE genes were significantly enriched in additive F50–56 QTLs for muscle weights (P = 0.03), bone sizes (P = 0.01), and diabetes-related traits (P = 0.01) (Fig. 6C). These results are consistent with the history of the LG/J and SM/J founder strains: they were independently selectively bred for large and small body sizes, so it is not surprising that strain-specific variation can induce ASE biases in their F1 cross that often occur in/near regions relevant for morphometric traits. Furthermore, sequence-dependent ASE genes were not enriched in additive F50–56 QTLs for behavioral traits related to locomotor activity and prepulse inhibition (P = 0.42) (Fig. 6C). Tissue-specific ASE gene sets were also not enriched in behavioral QTLs, even for the brain region HYP (P = 0.79) (Supplemental Fig. S17), suggesting the metabolic tissues profiled here are not relevant for these behavioral phenotypes.
Although sequence-biased genes were located in additive AIL QTL more often than expected, individual QTL typically contained few ASE genes (Supplemental Table S7). Additive F16 QTLs overlapped a mean of 75 genes each, yet 51% of those QTLs contained only one to five sequence-dependent ASE genes. Thirty-seven percent of QTLs contained more than six ASE genes and merely 12% had no ASE genes (Fig. 6D). Similarly, additive F50–56 QTLs overlapped a mean of 61 genes each, but 36% of those QTLs contained only one to five sequence-dependent ASE genes. Forty-five percent of QTLs contained more than six ASE genes and 19% had no ASE genes (Fig. 6E). In contrast, imprinting F16 QTLs overlapped a mean of 62 genes each, but 92% of them did not contain any parent-of-origin-dependent ASE genes (Fig. 6D). Only two imprinting F16 QTLs contained more than 10 ASE genes and they both overlapped the cluster of 171 snoRNAs on Chromosome 7.
Cidec has a context-dependent switch in ASE direction and is a candidate gene for insulin levels
Because ASE genes often comprise a subset of all genes within a QTL, we propose that incorporating ASE patterns can dissect QTL etiology and prioritize candidate genes in relevant tissues. Here, we present an example of the utility of this orthogonal approach. Ddiab6d was an AIL F16 QTL on Chromosome 6 associated with serum insulin, basal glucose, and glucose tolerance levels that showed additive effects in a context-dependent manner (Lawson et al. 2011b). In high fat males, AIL F16 mice homozygous for the SM/J allele had significantly higher insulin levels than the homozygous LG/J mice (P < 0.001). There were no significant differences in insulin levels between homozygote genotypes in the high fat females, low fat females, or low fat male cohorts (Fig. 6F). The Ddiab6d locus spanned 14.7 Mb and contained 100 genes, complicating efforts to pinpoint the causal mechanisms of this additive effect.
However, only four sequence-dependent ASE genes were located in the Ddiab6d locus: Brpf1, Cidec, Irak2, and Syn2. Cell death-inducing DFFA-like effector c (Cidec) had a diet- and sex-dependent switch in bias direction within the same tissue, which was a notable exception to the tissue-level ASE direction switches reported above. In LIV, Cidec had a strong SM/J bias in the HM context, yet a LG/J bias in the HF, LF, and F contexts (Fig. 6G). It was biallelically expressed in the remaining LIV contexts and across all WAT contexts, but not expressed in HYP. Cidec ASE biases also had significant sex, diet, and diet-by-sex effects in LIV (P < 0.01) (Fig. 6H). We validated the context-dependent switch in ASE bias direction with pyrosequencing (Fig. 6I). Cidec was also expressed in LIV at a significantly higher level in HM than the other diet-by-sex contexts (P < 0.01) (Fig. 6J).
Cidec promotes and regulates lipid storage in liver and adipose tissue (Xu et al. 2012). Cidec expression is sensitive to diet composition and sex hormones; for example, male mice had higher total hepatic expression than females when fed a Western diet (i.e., high cholesterol and saturated fats) (Herrera-Marcos et al. 2020). In another study, high fat-fed mice had increased Cidec expression in liver that was also associated with improved insulin sensitivity (Iv et al. 2015). The liver plays an important role in regulating homeostatic insulin levels and removing it from the bloodstream (Tokarz et al. 2018). Hepatic insulin clearance is an emerging component of type 2 diabetes and metabolic syndrome, as impaired hepatic insulin clearance is correlated with increased waist circumference, blood pressure, fasting glucose, triglycerides, and insulin secretion (Pivovarova et al. 2013; Najjar and Perdomo 2019). Together, these data implicate Cidec as a candidate gene for Ddiab6d whose allelic composition in the liver is sensitive to metabolically relevant environmental factors and can influence insulin levels.
Discussion
Allele-specific expression imbalances caused by genetic and epigenetic variation have widespread functional consequences on complex traits, but how environmental signals contribute to this cross talk remains understudied. Here, we explored how allelic genotype, parent-of-origin, tissue type, sex, and dietary fat simultaneously impact ASE biases in an adult mouse F1 reciprocal cross. We present a genome-wide map of parent-of-origin and sequence-dependent ASE patterns across three metabolically relevant tissues and nine environmental contexts. The granularity of our analyses revealed that both ASE classes are highly dependent on tissue and environmental context. We identified 2853 genes with significant parental and/or sequence biases and sorted them into three expression profiles: tissue-independent, tissue-dependent, and context-dependent. We also found 45 genes with inconsistent ASE biases that switched direction across tissues and/or contexts. Although the breadth of these patterns precluded a detailed discussion of each gene, we validated examples of each expression profile to show how these allelic imbalances could manifest and lead to potential functional consequences.
“Tissue-independent” genes are strongly biased wherever they are expressed. Most parent-of-origin-dependent ASE genes (67%) have this expression profile—all of which are related to genomic imprinting, a well-characterized epigenetic phenomenon that results in uniparental expression and is often conserved across tissues (Barlow and Bartolomei 2014). In contrast, only 23% of sequence-dependent ASE genes have this expression profile, likely owing to cis-acting genetic variants in coding regions that severely affect one allele's function whenever that gene is expressed. These patterns conform to the conventional ASE mechanism whereby the genetic and epigenetic processes that influence allelic composition will always do so wherever a gene is expressed, in a manner impervious to tissue type or environmental signals (Lo et al. 2003; Babak et al. 2015). However, we found this is not always the case.
“Tissue-dependent” genes are moderately biased in one direction in some tissues, but are biallelically expressed or biased in the opposite direction in other tissues. Tissue-specific gene expression (simply not expressed in certain tissues) is not considered here, because we are specifically interested in cases in which a gene has one allelic ratio in one tissue (e.g., 80% SM/J, 20% LG/J) and a different ratio in another tissue (e.g., 50% SM/J, 50% LG/J). Merely 6% of parent-of-origin-dependent ASE genes have this expression profile, and all are located in known imprinted domains. Twenty-five percent of sequence-dependent ASE genes have this profile, demonstrating that sequence biases are not solely attributable to genetic sources. Instead, both patterns may be explained by variation in tissue-specific regulatory features, such as enhancers or RNA-binding proteins. Such tissue-specific factors can then interact with cis-acting variants to produce a tissue-dependent allelic imbalance. This is likely the case for genes with inconsistent biases between tissues, for which the variation in one allele may be favorable in certain tissues’ regulatory landscape but the opposite allele is more favorable elsewhere (Leung et al. 2015; Andergassen et al. 2017).
Finally, “context-dependent” genes are subtly biased in one direction in certain environmental contexts, but are biallelically expressed or biased in the opposite direction in other contexts and tissues. We found this expression profile is quite prevalent in both classes of ASE. Twenty-seven percent of parent-of-origin-dependent ASE genes have diet- and/or sex-specific biases in a less extreme manner than the other profiles. Most genes have no clear connection to imprinting, suggesting another mechanism for parental ASE outside of traditional imprinting that is sensitive to environmental perturbations (Wolf et al. 2008; Morcos et al. 2011). More than half of sequence-dependent ASE genes (52%) have diet- and/or sex-specific biases, indicating these extrinsic and intrinsic factors interact with genetic variation to cause a sequence bias. Both patterns suggest a model in which environmental signals may interact more efficiently with one allele over the other, leading to shifting and inconsistent allelic proportions in response to environmental cues (Shao et al. 2019). Often, these genes are not significantly biased in our “all contexts” analysis, only in a specific environmental cohort. This highlights the necessity of studying gene-by-environment interactions, as such effects are obscured when multiple contexts are collapsed together.
We can only detect ASE in genes with strain-specific variants in transcribed regions, which is a fraction of all expressed genes. In particular, imprinted genes that are crucial for development may be under tight evolutionary control and not have variants; thus, we are unable to assess their ASE status. Although our exact findings are limited to this F1 mouse model, the broad patterns nevertheless show the complexity of ASE and its contribution to complex traits. Traditional mapping studies connect genotypes to phenotypes, but are agnostic to tissue and often do not consider environment. Expression QTL studies connect genotypes to total gene expression, but assume biallelic expression. Tissue- and context-dependent ASE of both classes can bridge these approaches and pinpoint which tissues and/or environments are relevant for a phenotype. A gene may be expressed at the same level in two cohorts, but its allelic composition could differ owing to ASE. If there is a functional difference between its alleles, then an expression imbalance in the right conditions could have phenotypic consequences. We integrated ASE and QTL data from populations derived from the same founder strains at various degrees of intercrossing. Tissue-specific ASE genes are enriched in QTLs for metabolic and musculoskeletal traits, yet consist of a small portion of all genes within the QTL. Incorporating these dynamic ASE patterns with orthogonal evidence will help us decipher the genotype-to-phenotype map.
Methods
F1 reciprocal cross mouse model and RNA sequencing
We obtained LG/J and SM/J founders from The Jackson Laboratory and generated F1 reciprocal crosses by mating LG/J mothers with SM/J fathers (LxS) and vice versa (SxL). F1 offspring were weaned into sex-specific cages at 3 wk and randomly placed on either a high fat diet (42% kcal from fat; Teklad TD88137) or an isocaloric low fat diet (15% kcal from fat; Research Diets D12284). They were fed ad libitum. The F1 crosses did not differ in weight gained in any diet-by-sex cohort (P > 0.05) (Supplemental Fig. S18). At 20 wk, F1 mice were euthanized with a sodium pentobarbital injection followed by cardiac perfusion with phosphate-buffered saline. We harvested hypothalamus (HYP), liver (LIV), and reproductive white adipose (WAT) tissue, which were flash frozen in liquid nitrogen and stored at −80°C until RNA extraction. All procedures were approved by the Institutional Animal Care and Use Committee at Washington University School of Medicine.
We sequenced 32 samples per tissue, representing four mice from each sex, diet, and F1 cross cohort. We extracted total RNA from WAT and HYP using the RNeasy Lipid Tissue Mini Kit (QIAGEN) and from LIV using a standard TRIzol-chloroform procedure. Samples were selected based on sufficient NanoDrop RNA concentrations (Thermo Fisher Scientific) and RNA integrity scores ≥8.0 (Agilent). We constructed RNA-seq libraries with the Ribo-Zero rRNA Removal Kit (Illumina), checked their quality with the Bioanalyzer DNA 1000 assay (Agilent), and sequenced them at 100-bp paired-end reads on an Illumina HiSeq 400. After sequencing, reads were demultiplexed and assigned to individual samples.
Allele-specific expression mapping
Mapping ASE in F1 heterozygotes is vulnerable to reference genome alignment bias (Degner et al. 2009; Wang and Clark 2014). Previously, LG/J and SM/J reference genomes were created by combining strain-specific SNPs and indels with the GRC38.72-mm10 reference template (Nikolskiy et al. 2015). Customized gene annotations were created by adjusting Ensembl definitions (GRCm38.72) for indexing differences owing to strain-specific indels. To mitigate this concern, we combined those LG/J and SM/J genomes into a custom pseudogenome and aligned RNA-seq reads to both strains simultaneously. We mapped reads uniquely using the two-pass mapping strategy in STAR v.2.7.2b (Dobin et al. 2013). Briefly, splice junctions are collected during a first round and used to inform a second round of mapping. By not allowing multimapping, we only retained reads that uniquely cover strain-specific variants so we could assign their allelic origin; reads covering identical regions between the parental strains were discarded. Alignment summaries are provided in Supplemental Table S8 and Supplemental Figure S19.
Next, we assigned each aligned read to a gene using BEDTools v.2.27.1 (Quinlan and Hall 2010) and our strain-specific Ensembl annotations. We assessed ASE at the gene level because not all exons within a given gene contained informative variants. Gene-level allele-specific counts were then upper quartile normalized (Supplemental Fig. S20) and filtered to remove lowly expressed genes (total normalized counts < 20). We retained a total of 9171 genes in HYP, 9745 genes in WAT, and 8026 genes in LIV.
Library complexity
Insufficient library complexity can hamper ASE detection (Wang and Clark 2014). To measure each library's complexity, we fit the distribution of LG/J allele expression biases (the proportion of total allele-specific read counts with the LG/J haplotype) to a beta-binomial distribution using the VGAM package (Yee 2010). We estimated the shape parameters (α, β) of the beta-binomial distribution and calculated the overdispersion parameter (ρ) as ρ = 1/(1 + α + β). Lower values of ρ (<0.075) indicate a library is sufficiently complex. One WAT and one LIV library had poor complexity and were removed from further analyses (Supplemental Fig. S21).
Determining biased allele-specific expression
For each “tissue-by-context” analysis, we required a gene to be expressed in ≥75% of biological replicates per F1 reciprocal cross. This conservative sample size threshold retained only genes expressed in most biological replicates in a given cohort, allowing us to confidently conclude their expression status. We intentionally removed sporadically expressed genes, because they may result in false or transient biases.
We adapted a published regression model (Takada et al. 2017) to jointly estimate parent-of-origin (PO) and allelic genotype (AG) effects on ASE. First, we assigned two binary variables
to each gene's allele-specific counts based on their allelic origin. For the PO term, maternal alleles received a 0 and paternal
alleles received a 1; for the AG term, LG/J alleles received a 0 and SM/J alleles received a 1 (Supplemental Fig. S22). Next, we added a term representing the RNA-seq library barcodes so that both allele-specific read counts (two separate
rows in our data set) for an individual mouse are treated as a pair. This paired-sample design helps distinguish between cases
in which a gene is not expressed in a particular sample (i.e., both alleles’ read counts are zero) and an extreme expression
bias (i.e., only one allele's counts are zero), which results in a lower biological coefficient of variation and lower overall
dispersion (Supplemental Fig. S22). Our final regression model equation was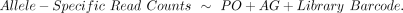 Finally, we fit a negative binomial generalized linear model (GLM) and conducted likelihood ratio tests in edgeR (Robinson et al. 2010; McCarthy et al. 2012) to estimate the POE and AGE on ASE.
Finally, we fit a negative binomial generalized linear model (GLM) and conducted likelihood ratio tests in edgeR (Robinson et al. 2010; McCarthy et al. 2012) to estimate the POE and AGE on ASE.
Next, we quantified the direction and magnitude of each gene's expression biases. For each sample, we calculated a gene's
allelic bias as the proportion of total counts with the LG/J haplotype (Lbias) or SM/J haplotype (Sbias). Using the mean allelic biases of each F1 cross, we constructed POE and AGE scores per gene as follows: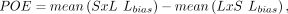
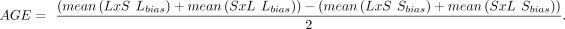 POE scores ranged from −1 (completely maternally expressed) to +1 (completely paternally expressed). Similarly, AGE scores
ranged from −1 (completely SM/J expressed) to +1 (completely LG/J expressed). Scores of 0 for both indicated biallelic expression.
Full summary statistics for each analysis are provided in Supplemental Tables S2–S4.
POE scores ranged from −1 (completely maternally expressed) to +1 (completely paternally expressed). Similarly, AGE scores
ranged from −1 (completely SM/J expressed) to +1 (completely LG/J expressed). Scores of 0 for both indicated biallelic expression.
Full summary statistics for each analysis are provided in Supplemental Tables S2–S4.
Estimating significance thresholds
Adjusting for multiple tests in ASE analyses is challenging. Allelic biases are often correlated for genes within and between regions, breaking any independence assumptions. We used a permutation approach to estimate our significance thresholds. For each tissue-by-context analysis, we generated a permuted null distribution of both AGE and POE terms. Over several iterations, we randomly shuffled the allele-specific read counts for all genes, reran our GLM analyses, and recalculated the POE/AGE scores. Instead of choosing an arbitrary number, we continued to add new iterations until the null model was “stable.” After every iteration, we added those permuted results to the overall null model and evaluated the difference in mean permuted likelihood ratios. We defined “stability” as when the difference in mean permuted likelihood ratios for both terms fluctuated by <|0.001| for 10 consecutive iterations (Supplemental Fig. S23). In other words, the null model was considered “stable” once incorporating another iteration did not shift the overall distribution of test statistics (mean iterations per analysis = 51). Summary statistic comparisons between the real and permuted data sets are provided in Supplemental Figures S24–S26.
Next, we built an empirical cumulative distribution function (ECDF) from each term's permuted P-values for each tissue-by-context analysis (Supplemental Fig. S27). We fit each term's raw P-values to its respective ECDF to compute the adjusted P-values, that is, the proportion of tests from the null model that are more extreme (smaller P-values) than a test from the real model. We also calculated the 5th and 95th quantiles of the permuted POE and AGE scores in each analysis; the more extreme value of each score became our critical thresholds. We deemed adjusted P-values ≤ 0.05 as statistically significant and real scores beyond their critical threshold as biologically significant. Parent-of-origin-dependent ASE genes had significant POE P-values and POE scores; sequence-dependent ASE genes had significant AGE P-values and AGE scores (Supplemental Fig. S28; Supplemental Table S1).
Total expression mapping
During allele-specific mapping, reads were not aligned if they covered homozygous regions between the parental strains. We saved those unmapped reads and aligned them separately to the LG/J and SM/J genomes using a standard multimapping approach in STAR v.2.7.2b. We assigned each aligned read to a gene using BEDTools v.2.27.1 and our strain-specific Ensembl annotations. Next, we averaged the read counts for both genomes; because those reads covered homozygous regions, they mapped to each genome similarly. We combined the averaged “multimapped” counts with the raw “allele-specific” counts from the previous mapping step, resulting in raw “total expression” counts. Finally, gene-level counts were upper quartile normalized and filtered to remove lowly expressed genes (total counts < 10). We retained a total of 25,926 genes in HYP, 26,450 genes in WAT, and 24,076 genes in LIV.
Characterizing ASE profiles: tissue-independent, tissue-dependent, and context-dependent
We sorted both classes of ASE genes into three expression profiles. Tissue-independent genes had a significant bias in every expressed tissue. Within a tissue, the gene must be biased in five or more of the nine environmental contexts (i.e., most, but not all, contexts). This flexibility allowed for genes that may have true ASE but were excluded because of our conservative sample size requirements; these genes could appear as biallelic because they still passed our other filtering thresholds. Tissue-dependent genes had a significant bias in some tissues and no bias in others. The gene must be biased in more than five of the nine contexts in one or two tissues, but biallelic in more than five of the nine contexts in the other tissue(s). Both profiles allowed for genes to not be expressed in some tissues, because we wanted to distinguish between tissue-specific gene expression and tissue-dependent ASE. Finally, context-dependent genes had a significant bias only in certain environmental contexts and no bias elsewhere. The gene must be biased in five or more of the nine contexts within a tissue, but biallelic in the other contexts and/or tissues. All significant ASE genes fit into one of these expression profiles; no genes were unclassified.
Evaluating environmental context dependency
For each context-dependent gene, we calculated each sample's allelic bias as the proportion of total read counts with the
LG/J haplotype (Lbias) or the SM/J haplotype (Sbias). We constructed individualized POE or AGE scores per sample per gene as follows: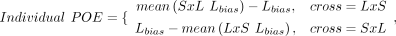
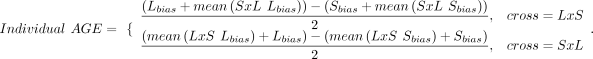 POE scores ranged from −1 (completely maternal) to +1 (completely paternal); AGE scores ranged from −1 (completely SM/J) to
+1 (completely LG/J). Scores of 0 indicated biallelic expression or not expressed.
POE scores ranged from −1 (completely maternal) to +1 (completely paternal); AGE scores ranged from −1 (completely SM/J) to
+1 (completely LG/J). Scores of 0 indicated biallelic expression or not expressed.
Next, we used ANOVA models to test whether a gene's allelic biases (individualized scores) were influenced by sex, diet, and/or their interaction. We considered FDR-corrected P-values ≤ 0.1 to be significant (Supplemental Table S5). For genes with significant diet-by-sex interactions, we conducted Tukey's post hoc tests to identify significant differences among diet-by-sex cohorts (adjusted P ≤ 0.05) (Supplemental Table S6).
Chromosomal location enrichment
We performed overrepresentation analysis for chromosomal locations with the WEB-based GEne SeT AnaLysis Toolkit v2019 (Liao et al. 2019). For each tissue-by-context analysis, we analyzed the list of genes with significant biases against the background of all genes with detectable ASE. A Benjamini–Hochberg FDR-corrected P-value ≤ 0.1 was considered significant.
Canonically imprinted genes
We defined genes as “canonically imprinted” if they appeared in the Geneimprint mouse database (https://www.geneimprint.com, as of May 2020) and/or a PubMed search with imprinting-related terms.
Pyrosequencing
We validated two to three examples of each expression profile (total = 13 genes). We sorted genes in each profile by total expression level. To minimize noise during pyrosequencing, we prioritized genes that were highly expressed and statistically significant (for context-dependent examples), but excluded those with high expression variance between biological replicates. For each gene, we identified the strain-specific SNPs within exons and designed primer sets to flank the variants using Geneious Prime 2020.0.4 (https://www.geneious.com) (Kearse et al. 2012). Wherever possible, target regions were 150- to 200-bp long and spanned an exon–exon junction to avoid genomic DNA contamination. We verified the specificity of each primer set in silico with Geneious and in vitro with PCR and Sanger sequencing. All primer sequences are provided in Supplemental Table S9.
We extracted total RNA from the HYP, WAT, and LIV of one mouse per F1 cross in each diet-by-sex cohort using the RNeasy Lipid Tissue Mini Kit (QIAGEN). The cDNA of each gene target was reverse transcribed and PCR amplified with the PyroMark OneStep RT-PCR Kit (QIAGEN) using one biotinylated (reverse) and one nonbiotinylated (forward) primer. The biotinylated single-stranded PCR products were purified with Streptavidin Sepharose High Performance beads (Cytiva) and hybridized to sequencing primers (same as forward) on the PyroMark Vacuum Prep Workstation (QIAGEN). Finally, we performed pyrosequencing with the allele quantification program on the PyroMark Q24 system (QIAGEN). The PyroMark Q24 software quantified the allelic ratio of the variable position(s) in each gene's assay. We calculated the mean allelic ratios of each variant in each tissue-by-context cohort.
Integrating ASE and QTL data
The genotypes, phenotypes, and QTL mapping methods for the LG/J x SM/J advanced intercross line are described elsewhere (Lawson et al. 2010, 2011b; Cheverud et al. 2011; Gonzales et al. 2018; Cordero et al. 2019). The F16 QTL were originally mapped to the NCBI37/mm9 reference genome, so we converted those intervals to their GRCm38/mm10 coordinates with the UCSC Genome Browser's liftOver tool (https://genome.ucsc.edu) (Lee et al. 2022). We also extracted the ASE gene positions from the GRCm38/mm10 assembly annotation.
We analyzed the two ASE classes separately: sequence-dependent ASE genes were intersected with F16 and F50–56 additive QTL, and parent-of-origin-dependent ASE genes were intersected with F16 imprinting QTL (Supplemental Table S7). We counted how many ASE genes overlapped with each QTL using BEDTools v.2.27.1. We sorted each AIL generation's QTL based on their associated trait category and compared those trait-specific QTL sets with the tissue-specific ASE gene sets. Next, we conducted enrichment analyses to determine whether ASE genes are overrepresented in AIL QTLs. We randomly shuffled the QTL windows around the genome and tallied how many ASE genes overlapped with regions of those lengths by chance. These permutations were repeated more than 10,000 iterations to establish a null model. We calculated a Z-score based on the null model and the real ASE/QTL overlap, then sampled a normal distribution to obtain a P-value. We considered P-values ≤ 0.05 to be significantly enriched.
Data access
All raw RNA sequencing data generated in this study have been submitted to the NCBI BioProject database (https://www.ncbi.nlm.nih.gov/bioproject/) under accession number PRJNA753198.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
This work was supported by the Washington University Department of Genetics, the Washington University Diabetes Research Center (C.F.S.: P30DK020579), the National Science Foundation Graduate Research Fellowship Program (C.L.S.: DGE1745038, DGE2139839), the National Institute of Diabetes and Digestive and Kidney Diseases (H.A.L.: K01DK095003), the National Institute of Environmental Health Sciences (H.A.L.: U24ES026699), and the National Human Genome Research Institute (J.F.M.-V.: T32GM007067).
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.276193.121.
-
Freely available online through the Genome Research Open Access option.
- Received September 10, 2021.
- Accepted April 14, 2022.
This article, published in Genome Research, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








