
H3K36 dimethylation shapes the epigenetic interaction landscape by directing repressive chromatin modifications in embryonic stem cells
- Haifen Chen1,2,9,
- Bo Hu1,2,9,
- Cynthia Horth1,2,
- Eric Bareke1,2,
- Phillip Rosenbaum1,2,
- Sin Young Kwon3,4,
- Jacinthe Sirois3,4,
- Daniel N. Weinberg5,
- Faith M. Robison6,
- Benjamin A. Garcia6,
- Chao Lu7,8,
- William A. Pastor3,4 and
- Jacek Majewski1,2
- 1Department of Human Genetics, McGill University, Montreal, Quebec H3A 1B1, Canada;
- 2McGill University Genome Centre, Montreal, Quebec H3A 0G1, Canada;
- 3Department of Biochemistry, McGill University, Montreal, Quebec H3G 1Y6, Canada;
- 4The Rosalind & Morris Goodman Cancer Institute, McGill University, Montreal, Quebec H3A 1A3, Canada;
- 5Laboratory of Chromatin Biology and Epigenetics, The Rockefeller University, New York, New York 10065, USA;
- 6Department of Biochemistry and Molecular Biophysics, Washington University School of Medicine, St. Louis, Missouri 63110, USA;
- 7Department of Genetics and Development, Columbia University Irving Medical Center, New York, New York 10032, USA;
- 8Herbert Irving Comprehensive Cancer Center, Columbia University Irving Medical Center, New York, New York 10032, USA
-
↵9 These authors contributed equally to this work.
Abstract
Epigenetic modifications on the chromatin do not occur in isolation. Chromatin-associated proteins and their modification products form a highly interconnected network, and disturbing one component may rearrange the entire system. We see this increasingly clearly in epigenetically dysregulated cancers. It is important to understand the rules governing epigenetic interactions. Here, we use the mouse embryonic stem cell (mESC) model to describe in detail the relationships within the H3K27-H3K36-DNA methylation subnetwork. In particular, we focus on the major epigenetic reorganization caused by deletion of the histone 3 lysine 36 methyltransferase NSD1, which in mESCs deposits nearly all of the intergenic H3K36me2. Although disturbing the H3K27 and DNA methylation (DNAme) components also affects this network to a certain extent, the removal of H3K36me2 has the most drastic effect on the epigenetic landscape, resulting in full intergenic spread of H3K27me3 and a substantial decrease in DNAme. By profiling DNMT3A and CHH methylation (mCHH), we show that H3K36me2 loss upon Nsd1-KO leads to a massive redistribution of DNMT3A and mCHH away from intergenic regions and toward active gene bodies, suggesting that DNAme reduction is at least in part caused by redistribution of de novo methylation. Additionally, we show that pervasive acetylation of H3K27 is regulated by the interplay of H3K36 and H3K27 methylation. Our analysis highlights the importance of H3K36me2 as a major determinant of the developmental epigenome and provides a framework for further consolidating our knowledge of epigenetic networks.
Our genetic material, DNA, is wrapped around nucleosomes to form chromatin. Chromatin can be chemically modified to determine its functional properties. Such epigenetic modifications are deposited by enzymes in a finely controlled fashion. The individual components of a cell's epigenetic landscape are tightly interwoven (Strahl and Allis 2000; Janssen and Lorincz 2022). Steric changes introduced by modifications to some histone residues affect the probability of altering nearby or interacting residues. Chromatin modifying enzymes can be either recruited by or antagonized by specific epigenetic modifications. Some epigenetic “writers” such as de novo DNA methyltransferases have the ability to read the histone code. In turn, DNA methylation affects the binding affinity for many other chromatin writers (Soshnev et al. 2016). We are now aware that disturbing one component of the epigenetic puzzle has a domino effect on the entire interconnected system. Many well-founded individual pieces of evidence have been collected (Janssen and Lorincz 2022), but they are too often analyzed in isolation. There is a need for more systematic integrated analyses of those data sets to fully uncover the underlying relationships. Here, we borrow an approach from network analysis to isolate a small, highly interconnected epigenetic subnetwork and dissect it in detail.
The most fundamental epigenetic relationships exist at the level of an individual residue. A given lysine of a histone molecule can only be modified in one way at any given time: an H3K27 molecule can exist as H3K27, H3K27me1, H3K27me2, and H3K27me3, or H3K27ac: those states are mutually exclusive. The next level of complexity is exemplified by topological constraints introduced by nearby modification. It is well documented that the presence of higher-level methylation at lysine 36 (H3K36me2/3) hinders higher-level methylation at lysine 27 (particularly H3K27me3) (Schmitges et al. 2011; Yuan et al. 2011). Further relationships reflect the reader/writer properties of chromatin modifying enzymes and complexes. Polycomb repressive complex 2 (PRC2), the complex responsible for depositing H3K27 methylation, is capable of reading its own product (H3K27me2/3), which can further enhance its catalytic activity (Zhang et al. 2015; Holoch and Margueron 2017; Laugesen et al. 2019), while it is antagonized by the presence of H3K36 methylation on the neighboring lysine (Yuan et al. 2011; Finogenova et al. 2020). The enzymes involved in depositing H3K36me1/2 are also capable of “self-reading” (Sankaran et al. 2016). NSD1, the H3K36 methyltransferase that is central to this study, contains a PWWP domain that is capable of recognizing nucleosome modifications, however, its relationship to H3K27 methylation has not been elucidated.
Another level of complexity reflects the relationships between histone and DNA modifications. Several recent studies indicate that the PWWP domains of the de novo methyltransferases, DNMT3A and DNMT3B, are capable of recognizing H3K36 methylation and are preferentially recruited to regions marked by H3K36me2/3 (Baubec et al. 2015; Weinberg et al. 2019). There is also evidence that the maintenance methyltransferase, DNMT1, may be recruited to the chromatin by interaction with histone modifications (Rothbart et al. 2012; Nishiyama et al. 2020; Ren et al. 2020). The reverse mechanisms—how DNA methylation affects histone modifications—are not yet fully understood. There clearly exists an inverse relationship between H3K27me3 and DNA methylation. In undifferentiated cells, PRC2 nucleates at unmethylated CpG islands (Jermann et al. 2014; Holoch and Margueron 2017; Li et al. 2017). Full demethylation of the genome in embryonic stem cells is associated with increased H3K27me3 levels (Brinkman et al. 2012; Hagarman et al. 2013; Reddington et al. 2013; McLaughlin et al. 2019). However, in most cases the molecular basis of interactions between histone modifying proteins and DNA methylation has not been elucidated.
This work aims to consolidate the existing knowledge of the epigenetic interaction network and synthesize it in a unified framework to serve as a model for understanding broader sets of interactions. At the risk of being overly reductionist, we tackle only a subcomponent of the network, delimited by the space of modifications to H3K27, H3K36, and DNA methylation. There is solid evidence for strong interactions across this system (Li et al. 2021b), and we hope that, although we face only a small part of the overall challenge, this attempt will provide a basis for later exploring many additional connections. We focus on the central role of H3K36me2 in shaping the epigenetic landscape of embryonic stem cells (ESCs). In mouse ESCs (mESCs), H3K36me2 is highly abundant (Schwämmle et al. 2016), and the disruption of H3K36me2 deposition provides an exciting opportunity to observe its effect on related chromatin modifications. We use mESCs cultured in high methylation serum/LIF conditions as a model system (and unless otherwise stated, from here onward when we refer to mESCs, we mean cells cultured in serum/LIF conditions), because those cells are particularly amenable to epigenetic manipulation and a wealth of data is already publicly available and can be directly contrasted with additional data we generate here. Our broader aim is to establish a set of molecular mechanisms and rules governing those interactions to aid with understanding and predicting the behavior of this system, particularly as a result of perturbations occurring in disease and cancer.
Results
Distribution of H3K36, H3K27, and DNA methylation in mouse ESCs
We illustrate (Fig. 1A; Supplemental Fig. S1A,B) the normal genomic patterns of chromatin modifications in wild-type (WT) mESCs grown in conventional serum + LIF conditions. Methylation at the H3K36 residue, particularly H3K36me2/3, is typically associated with active euchromatic regions of the genome. The patterns of H3K36me3 in mammalian cells are consistent across all cell types (Roadmap Epigenomics Consortium et al. 2015), including mESCs. This modification is highly enriched within actively transcribed gene bodies, with preference for 3′ and exon-rich regions (McDaniel and Strahl 2017). The lower H3K36me2 mark has a broader genomic distribution, and in mESCs is present in nearly all the genome, although it is markedly higher in intergenic regions than expressed gene bodies, where it is upgraded to H3K36me3 (Weinberg et al. 2019). The genomic distribution of H3K36me1 has historically not been profiled partly owing to lack of reliable antibody, and because the functional significance of this modification is not clear, we also refrain from discussing it here. Methylation at the K27 position, particularly the higher states H3K27me3/2, is generally associated with transcriptionally inactive regions and facultative heterochromatin. In mESCs, H3K27me3 is focused mostly within peaks or narrow domains centered on unmethylated CGIs that serve as nucleation sites for the PRC2 complex (Fig. 1A,E). PRC2 spreads H3K27me3 outward from its nucleation sites (Højfeldt et al. 2018; Oksuz et al. 2018), but in mESCs the levels of H3K27me3 remain relatively low throughout most of the genome (Schwämmle et al. 2016), particularly in regions marked with H3K36me2 and H3K36me3. H3K27me2, in contrast, is able to spread away from the nucleation sites and form large domains that coincide with the distribution of H3K36me2 (Streubel et al. 2018), but it appears to be excluded from regions occupied by H3K36me3 (Supplemental Fig. S1A, active genes). Last, H3K27me1 has a broad genomic distribution, but it tends to be depleted from regions occupied by H3K27me2/3 and enhanced within active gene bodies where it coexists with H3K36me3 (Ferrari et al. 2014; Lavarone et al. 2019).
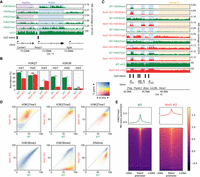
Distributions of H3K36me2/3, H3K27me1/2/3, and DNAme in mESCs and their reorganization in Nsd1-KO. (A) Genome browser representation of H3K36me2/3, H3K27me1/2/3, and CpG methylation (mCG) in wild-type mESCs, illustrating differences in epigenetic modifications around active versus inactive CpG islands (CGIs) and genes. The shaded areas indicate an inactive gene (purple) and an active gene (blue). The promoter of the silent Camkk1 gene on the left is an example of an unmethylated CGI serving as a PRC2 nucleation site resulting in an H3K27me3 peak. (B) Mass spectrometry (MS) analysis of H3K27 and H3K36 methylation levels (two replicates for each mark) in WT and Nsd1-KO, showing the gain of H3K27me3 and the loss of H3K36me2 in Nsd1-KO. Statistical significance levels are indicated based on t-test P-values: (****) [0, 0.0001); (***) [0.0001, 0.001); (**) [0.001, 0.01); (*) [0.01, 0.05); (ns) [0.05, 1]. (C) Genome browser representation of H3K36me2/3, H3K27me1/2/3, and CpG methylation (mCG) in WT (green) versus Nsd1-KO (red). Tracks are normalized using MS ratios. The shaded areas indicate active genes (blue) and an intergenic region (yellow). Genes transcribed at different levels are shown as examples, along with PRC2 nucleation sites (H3K27me3 peaks). The WT genomic distribution of H3K36me2 is broad, and H3K27me3 is much more localized. Nsd1-KO results in a drastic loss of H3K36me2 outside of transcribed genes, accompanied by nearly uniform spread of H3K27me3 into the regions vacated by H3K36me2. (D) Genome-wide correlation density plots of 10-kb binned ChIP-seq signal, MS normalized, shaded to indicate genic (blue) and intergenic (yellow) regions. In Nsd1-KO, the most drastic changes occur in intergenic regions and affect H3K36me2, H3K27me3, DNAme, and to a lesser degree H3K27me1. (E) Signal intensity plots (normalized by input and MS ratios) centered on transcription start sites (TSS) of inactive genes (n = 12,759) showing spreading of H3K27me3 in Nsd1-KO, indicated by the lower peak height and the higher enrichment in the surrounding regions in Nsd1-KO compared to WT. Replicate plots and additional synthesis of the data are in Supplemental Figure S1. Data used in this figure are generated in this study (Replicate 1).
The DNA methylome of mESCs is fairly uniform, with high (nearly 80%) DNAme levels throughout the genome, particularly in regions enriched for H3K36me2/3 (Fig. 1A), which is in line with previous studies demonstrating recruitment of de novo DNA methyltransferases DNMT3A/B to H3K36me2/3 marks (Baubec et al. 2015; Weinberg et al. 2019). DNAme is excluded from the majority of CGIs, reflecting the pluripotent state of mESCs and the limited extent of stable silencing of regulatory regions.
Nsd1-KO fully depletes intergenic H3K36me2 and allows nearly uniform H3K27me3 spread
At least five histone methyltransferases—SETD2, NSD1, NSD2, NSD3, and ASH1L—have been implicated in depositing H3K36me1/2, whereas only SETD2 has been convincingly shown to deposit H3K36me3. The activity and relative importance of each of those enzymes appears to be cell type–specific (Bennett et al. 2017; Farhangdoost et al. 2021; Li et al. 2021a). In mESCs, it has been shown that depletion of the NSD1 histone methyltransferase by siRNA reduces the levels of H3K36me2 and alters the patterns of lysine 27 methylation (Streubel et al. 2018). Here, we used CRISPR-Cas9 genome editing to create a full KO of Nsd1. We did not observe any increase in the numbers of dead cells or propensity to differentiate. However, we noted a slightly reduced proliferation in the Nsd1-KO mESCs. To study the effects of the deletion of this important epigenetic modifier on the epigenetic interaction network, we first performed tandem mass spectrometry analysis (Fig. 1B; Supplemental Tables S1, S2). We find that overall H3K36me2 levels are greatly reduced by a factor of nearly 5 (30% in WT compared with 6.3% in Nsd1-KO). Visual examination of coverage tracks indicates that intergenic domains of H3K36me2 are reduced to negligible levels (Fig. 1C), whereas genic H3K36me2 remains mostly unaffected, likely owing to the transcription-coupled activity of SETD2. This observation is corroborated by genome-wide analysis showing that regions with the greatest decrease are predominantly intergenic (Fig. 1D; Supplemental Fig. S1A,B). This result allows us to investigate the effect of depletion of H3K36me2 in a system where this mark normally occupies the majority of intergenic space.
Previous studies noted distinct patterns of H3K27me1/2/3 deposition in mESCs following PRC2 inhibitor treatment (Højfeldt et al. 2018). However, the mechanisms constraining H3K27me3 to nucleation sites of PRC2 and preventing it from exhibiting the broader distribution patterns of H3K27me1/2 have remained unclear. To definitively test whether and to what extent the antagonism by H3K36me2 may constrain H3K27me3 distribution, we examined the H3K27 methylation patterns in the absence of NSD1 and intergenic H3K36me2. Following the total removal of H3K36me2 from the intergenic space, H3K27me3 is able to spread nearly uniformly and form broad domains (Fig. 1C,E) and its levels increase by nearly 3.5-fold (Fig. 1B; Supplemental Tables S1, S2). As a result, those broad H3K27me3 domains become depleted of H3K27me1 as the lower mark is promoted to the highest methylation state (reduction of 1.5-fold), whereas H3K27me2 remains relatively unaffected (global decrease from 37% to 35%).
Loss of H3K36me2 leads to a decrease in DNAme by retargeting DNMT3A and redistributing de novo DNA methylation
We previously showed (Weinberg et al. 2019) the recruitment of DNMT3A by H3K36me2 and a reduction of intergenic DNAme subsequent to Nsd1-KO. Here, we further analyze the WGBS data to specifically investigate de novo DNA methylation and confirm that this decrease is indeed a direct result of change in DNMT3A activity. Although methylation levels of the CG dinucleotide reflect both de novo and maintenance activities, the CHH triplet cannot be symmetrically methylated and is thus not recognized by the maintenance DNMT1 methyltransferase. Although the levels of CHH methylation in mESCs are much lower than those of CpG methylation, they are measurable and are a direct metric of de novo methyltransferase activity.
In line with our previous observations, in Nsd1-KO cells the loss of intergenic H3K36me2 results in a massive redistribution of DNMT3A away from intergenic regions, and toward actively transcribed gene bodies (Fig. 2A,B), which are the only regions that retain H3K36me2/3. Profiling the levels of CHH methylation clearly shows that de novo DNA methylation is accordingly reduced in intergenic regions and enhanced in active gene bodies (Fig. 2A,B). This suggests that the reduction of intergenic CpG methylation is at least partly caused by a decrease in de novo methylation.
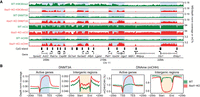
Nsd1-KO reduces DNAme (mCG) and redistributes DNMT3A and de novo DNAme (mCHH). (A) Genome browser representation of H3K36me2, DNMT3A, mCG, and mCHH in WT (green) versus Nsd1-KO (red), showing redistribution of H3K36me2 to genes, followed by DNMT3A and de novo DNAme. The shaded areas indicate H3K36me2-enriched regions in Nsd1-KO. (B) Aggregate signals of DNMT3A and mCHH around active genes (n = 15,850) and intergenic regions (n = 11,629), showing the enrichment (Nsd1-KO compared to WT) in active genes and depletion in intergenic regions. Data used in this figure are generated in this study.
DNA methylation has a limited effect on overall H3K27 methylation
Given the drastic effect of H3K36me2 reduction on H3K27me3 and DNAme, we turn our attention to the inverse relationships. There is considerable evidence in literature that DNAme inhibits the deposition of H3K27me3 (Jermann et al. 2014; Holoch and Margueron 2017). In some model systems, particularly in mESCs, this has been taken as indication that DNAme is the main obstacle keeping H3K27me3 from spreading into broad genomic domains (Brinkman et al. 2012; Hagarman et al. 2013; Reddington et al. 2013). Here, we take the opportunity to point out some finer intricacies behind this relationship. To directly compare the inhibitory effects of DNA methylation and H3K36me2, we generated two independent ChIP-seq replicates of H3K27me3 in Dnmt-TKO and Nsd1-KO mESCs. Those replicates were matched within two experimental batches to ensure compatibility and minimize effects of background noise and ChIP efficiency. First, we show (Fig. 3A) that the global changes in H3K27me levels resulting from loss of DNAme are considerably smaller than those resulting from depletion of H3K36me2. Most notably, H3K27me3 increases by 1.7-fold (compared with 3.5-fold increase in Nsd1-KO). Examination of ChIP-seq coverage tracks indicates that the changes of H3K27me1/2/3 distribution in Dnmt-TKO are much less pronounced than those in Nsd1-KO (Fig. 3B,C). Focusing on H3K27me3 (Fig. 3D–F), we find that the increase in H3K27me3 levels and the spread in H3K27me3 distribution is certainly detectable but relatively minor in Dnmt-TKO mESCs (which are devoid of any DNAme) compared to the Nsd1-KO (which shows a drastic loss of intergenic H3K36me2 along with a relatively limited reduction in intergenic DNAme). Thus, globally, the antagonistic effect of H3K36me2 appears to be a much stronger obstacle to H3K27me3 spread than DNAme. It may be expected that the largest effects of DNAme should occur in CpG-rich regions, particularly at CpG islands, which are also known to serve as nucleation sites for PRC2 (Jermann et al. 2014). Thus, although there is some limited incursion of H3K27me3 into regions that lose DNAme (Fig. 3B,D), much of the increase in H3K27me3 is manifested not as a “full spread” as in Nsd1-KO but is manifested by the appearance of new “peaks” within CpG-rich regions (Fig. 4A,B). To further illustrate this situation at CGIs, we show that in some cases in the TKO we observe the appearance of new nucleation sites (Fig. 4A,C left), whereas in many cases we observe a reduction of H3K27me3 at canonical PRC2 targets (Fig. 4C, right), as observed previously (Reddington et al. 2013; Dunican et al. 2020). This decrease is not general, because the majority of strong PRC2 targets remain occupied, including Hox gene clusters. However, the levels of H3K27me3, particularly at weaker nucleation sites, decrease (Fig. 4C, right). In view of new H3K27me3 peaks appearing at previously methylated CG-rich regions, it is likely that this decrease may be caused by redistribution of PRC2 away from its intended targets and into newly created ones owing to the evacuated DNAme (Reddington et al. 2013). A similar observation was made in 2i medium, where DNA is hypomethylated compared to serum culture (Supplemental Fig. S3C).
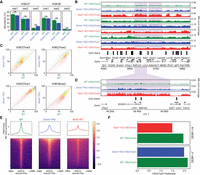
Depletion of DNAme has a limited effect on H3K27 methylation. (A) Mass spectrometry analysis of H3K27 and H3K36 methylation levels (two replicates for each mark) in WT and Dnmt-TKO, showing limited changes in Dnmt-TKO, except for slight H3K27me3 gain and H3K36me2 loss. P-values are indicated in the same way as in Figure 1B. (B) Genome browser representation of H3K27me1/2/3 in WT (green) versus Dnmt-TKO (blue) versus Nsd1-KO (red). Examples of regions with H3K27me3 spreading (in Nsd1-KO) are shaded. Note the much stronger effect of Nsd1-KO compared to Dnmt-TKO. (C) Genome-wide correlation density plots of 10-kb binned ChIP-seq signal, MS normalized, shaded to indicate genic (blue) and intergenic (yellow) regions, illustrating the global effect of DNA demethylation on H3K27me1/2/3 and H3K36me2. Aggregate plots of H3K27me across the genome centered on active genes are provided in Supplemental Figure S2A. (D) Highlighting the comparison of H3K27me3 in WT, Dnmt-TKO, and Nsd1-KO: although there is nearly uniform spread in Nsd1-KO, H3K27me3 increase in TKO is limited and more localized. The shaded area indicates an H3K27me3-spreading region in Nsd1-KO. (E) Signal intensity plot of H3K27me3 (normalized by input and MS ratios) centered on CGIs overlapping SUZ12 peaks (n = 4543), showing the localized spread in TKO and the nearly uniform spread in Nsd1-KO, indicated by the higher enrichment in the surrounding regions in KOs compared to WTs. The spread and increase of H3K27me3 upon Dnmt-TKO and Nsd1-KO are especially in inactive genes (i.e., outside of active genes), consistent with the observation in Supplemental Figures S1A and S2A (see the “H3K27me3” panel). Replicate plots are shown in Supplemental Figure S2B. (F) H3K27me3 “Peakiness score,” representing the average ChIP signal in the top 1% of 1-kb bins, confirming the spreading (lower Peakiness score) of H3K27me3 in Nsd1-KO, and the limited spread in Dnmt-TKO. Plots based on replicates are provided in Supplemental Figure S2C. Data used in this figure are generated in this study.
The DNAme-dependent H3K27me3 changes are more CpG-focused than those dependent on H3K36me2. (A) H3K27me3 in WT versus Dnmt-TKO versus Nsd1-KO, showing (in Dnmt-TKO) new peaks around CpG-rich regions and CGIs that were highly methylated and had no PRC2 binding in the WT (shaded areas). (B) Comparison of CpG density in regions gain/loss H3K27me3 in Nsd1-KO and Dnmt-TKO, indicating the H3K27me3 gain in TKO occurs in regions with elevated CpG density. P-values are based on t-test. A similar plot using replicates is provided in Supplemental Figure S3A. (C) Signal intensity plot of H3K27me3 (normalized by input and MS ratios) centered on highly methylated CGIs (n = 516) and unmethylated CGIs (n = 7400) in WT, respectively. In Dnmt-TKO H3K27me3 is gained within CGIs that were highly methylated in WT, but lost from its normal PRC2 nucleation sites, which had low DNAme in WT. Such changes do not occur in Nsd1-KO. Replicate plots are provided in Supplemental Figure S3B, and additional analysis in Supplemental Figure S3C,D. Data used in this figure are generated in this study, except EZH2 ChIP-seq data are from Kundu et al. (2017).
Modest effects of DNAme and H3K27me on H3K36me
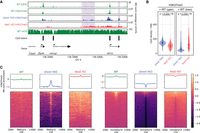
We next investigated the reciprocal relationship of DNAme and H3K27me on H3K36me2. In the Dnmt-TKO line where DNAme is completely depleted, we find that the levels of H3K36me2 show a moderate 1.4-fold decrease relative to the wild type (Fig. 3A). Genome-wide analysis of ChIP-seq signal (Figs. 3C, 5C) indicates that the decrease occurs uniformly across the genome. To study the effect of H3K27me, we used the Ezh2-KO mESC, where MS analysis (Fig. 5A) shows a total loss of H3K27me3, a sixfold reduction of H3K27me2 and no change in H3K27me1, as has been shown previously (Lavarone et al. 2019), whereas H3K36me2 levels show no change. Analysis of ChIP-seq signal also shows no notable change in the genome-wide distribution of H3K36me2 (Fig. 5B,C).
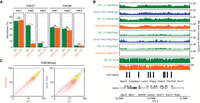
Effect of H3K27me2/3 loss on H3K36me2 is negligible. (A) Mass spectrometry analysis of H3K27 and H3K36 methylation levels (two replicates for each mark) in WT and Ezh2-KO, showing that deletion of Ezh2 results in total loss H3K27me3 and fivefold reduction in H3K27me2, but no significant reduction in H3K36me. P-values are indicated in the same way as in Figure 1B. (B) A representative 1-Mb region illustrating high-similarity epigenomic profiles of H3K36me2 in the Ezh2-KO and Dnmt-TKO. We also show that global DNA methylation is not visibly affected by the absence of H3K27me (Eed-KO, bottom). DNAme in Dnmt-TKO is completely depleted (data not shown). (C) Genome-wide correlation plots (10 kb) bins showing lack of change in global H3K36me2 distribution in Ezh2-KO and a small uniform decrease in Dnmt-TKO. All data in this figure are from this study, with the exception of DNAme in Eed-KO (Li et al. 2018).
Previous studies have shown that loss of H3K27me has very limited effect on DNAme and have described these effects particularly with respect to DNA methylation valleys (DMVs) that surround developmentally important genes (Li et al. 2018). We do not explore those trends further other than illustrating the general similarity of DNAme in WT and Eed-KO cells in Figure 5B. Overall, although removal of H3K36me2 results in massive spread and increase in H3K27me3 levels, the converse relationship is not true: in mESCs cultured in the serum/LIF conditions, the removal of H3K27me2/3 does not lead to a further spread of H3K36m2 or large-scale changes in DNAme.
The interplay of H3K36 and H3K27 methylation controls pervasive chromatin H3K27 acetylation
Several studies suggested a strong negative correlation, and perhaps a causative relationship, between lysine methylation and acetylation (Kuo and Allis 1998; Tie et al. 2009; Ferrari et al. 2014; Krug et al. 2019; Lavarone et al. 2019). Not only is the presence of these modifications on the same lysine residue chemically impossible, but there may also be an inhibitory effect of lysine methylation on histone acetyltransferase activity (Pasini et al. 2010). Recent work in mESCs described the effect of PRC2 mutations on H3K27ac levels and suggested that variation in H3K27ac is largely governed by changes in H3K27me2 (Ferrari et al. 2014; Lavarone et al. 2019). Here, we supplement those results and contrast the effect on H3K27ac of removal of PRC2 activity and removal of NSD1 activity (release of constraint on PRC2). We show (Fig. 6) that removal of H3K36me2 and spreading of H3K27me3 have the opposite effect on H3K27ac compared with removal of H3K27me (Eed-KO): there is a clear decrease in background acetylation levels in Nsd1-KO (Fig. 6A). This decrease is most pronounced in intergenic regions where H3K27me3 replaces H3K27me2 and H3K36me2 (Fig. 6B). Overall, the H3K27ac distribution becomes more restricted in Nsd1-KO and less localized in Eed-KO (Fig. 6C). Similar changes of H3K27ac as in Eed-KO were also observed in Ezh2-KO in 2i medium (Lavarone et al. 2019), where the further increase of H3K27ac in Ezh1/2-DKO suggests that the lowest methylation mark, H3K27me1, may also impede the deposition of H3K27ac (Supplemental Fig. S4A–D). Finally, we analyzed our data in view of recent findings that Nsd1-KO may lead to hyperacetylation of enhancers (Fang et al. 2021), but we found no evidence of this phenomenon. On the contrary, we observed positive association of H3K36me2 and H3K27ac levels at enhancers (Supplemental Fig. S4E), which is more in line with previous evidence implicating H3K36me2 in maintaining H3K27ac and enhancer activity (Lhoumaud et al. 2019; Farhangdoost et al. 2021; Rajagopalan et al. 2021).
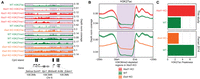
H3K27 methylation controls pervasive H3K27ac. (A) Comparison of H3K27 acetylation in Nsd1-KO versus WT and Eed-KO versus WT, showing the decrease in pervasive acetylation in Nsd1-KO and increase in Eed-KO. The shaded areas indicate H3K36me2-loss (or H3K27me3-spreading) regions in Nsd1-KO. The tracks are only normalized by read depth, not by any quantitative ratios, because no spike in or MS public data are available, and the distributions only reflect changes in levels between relevant regions. (B) Aggregate H3K27ac signal (read-depth normalized, not quantitatively) around H3K36me2-loss regions (n = 4361) in Nsd1-KO, showing the relative reduction in H3K27ac in Nsd1-KO and increase in Eed-KO, in those H3K36me2-loss regions compared to surrounding regions. (C) Peakiness scores of H3K27ac, showing the reduction of background H3K27ac (i.e., more restricted distribution or higher Peakiness score) in Nsd1-KO, and gain of background signal (less localized or lower Peakiness score) in Eed-KO. Comparison of H3K27ac in Ezh2-KO and WT (mESC in 2i medium) is shown in Supplemental Figure S4A–C. The ChIP-seq of H3K27ac and H3K27me in Nsd1-KO and WT are generated in this study, but those in Eed-KO and WT are from Ferrari et al. (2014).
Discussion
Our analysis highlights the central role of NSD1 and the chromatin modification H3K36me2 in shaping the mESC epigenome. We use the drastic rearrangement of the epigenome that follows the deletion of Nsd1 to provide the context for illustrating the general nature of epigenetic interactions. H3K36me2 is one of the most abundant histone modifications in mESCs, occupying at least 60% of intergenic regions where most subsequent changes occur. In terms of bulk genome-wide effects, the overall abundance of a mark is one of the most important parameters that influences downstream outcomes.
H3K36me2/3 are known to interfere with the activity of PRC2 (Alabert et al. 2020; Finogenova et al. 2020). Thus, removing intergenic H3K36me2 provides a much more favorable substrate for PRC2. The regions previously co-occupied by H3K36me2/27me2 are now easily converted to H3K27me3, resulting in its nearly fourfold increase. This happens at the expense of H3K27me1, which is upgraded to H3K27me3. The genic profiles of H3K36me and H3K27me modifications show relatively minor changes (Fig. 1D); however, as a result of its depletion in intergenic regions, genic enrichment of K27me1 is now more pronounced (Fig. 1C; Supplemental Fig. S1A).
Depleting H3K36me2 also removes a targeting mechanism for de novo methyltransferases (Shirane et al. 2020), particularly DNMT3A (Weinberg et al. 2019). We show that in the absence of NSD1, DNMT3A is now redistributed to genic regions which retain SETD2-deposited H3K36me2/3 (Fig. 2). As a result, de novo DNAme, represented by mCHH, increases in transcribed gene bodies, and decreases in intergenic regions. This retargeting of de novo methylation explains the substantial reduction of intergenic DNAme.
We also find that lack of NSD1 results in decrease of H3K27ac in regions that are now depleted of H3K36me2. This appears to mostly take the form of reduction of “pervasive” background acetylation that is loosely scattered throughout large domains. Based on earlier studies in mESCs (Ferrari et al. 2014; Lavarone et al. 2019), this is most likely a “secondary” effect of the spread of H3K27me3 into regions that are vacated by H3K36me2 and that no longer provide substrates for histone acetyltransferases. To what extent different levels of H3K27me prevent deposition of H3K27ac remains to be determined, but a reanalysis of Ezh2 versus Ezh1/2 mutant data in 2i medium (Lavarone et al. 2019) suggests that even H3K27me1 may be refractive to deposition of H3K27ac (Supplemental Fig. S4). It is also unclear whether the presence of H3K36me may have some primary effect on H3K27ac, but to date no plausible molecular mechanisms have been proposed.
Overall, our analysis of Nsd1 mutants and comparison with publicly available data shows that H3K36me2 plays a major role in shaping the mESC epigenome (schematic model and molecular dynamics in Fig. 7A,B). There is compelling evidence for molecular mechanisms governing this role, and the levels of H3K36me2 in those cells are normally very high. Our data suggest that DNA methylation plays a limited role in determining H3K27 and H3K36 states, and we argue that this is consistent with relatively low CpG density throughout the mouse genome (approximately 1 CpG per nucleosome) and very low levels (<2%) of non-CpG methylation. These relatively infrequent modifications may not have major effects on the genome-wide recruitment of chromatin modifying enzymes, and it may be expected that the effect of changes in DNA methylation should be greatest at CGIs and CpG-rich regions. We find this to be true in our analysis. The effect of H3K27 methylation on H3K36me and DNAme is also relatively limited. This is most likely the result of low levels and focused distribution of the most influential state, H3K27me3, in serum + LIF cultured mESCs.
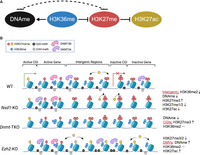
The H3K27, H3K36, and DNAme interaction network. (A) The principal interaction network in mESCs, with the central role of H3K36me2. The dashed line represents the weaker influence of the DNAme-H3K27me axis; however, the relative strengths of those interactions may differ across cell types. (B) Molecular cartoon illustrating the state of this three-component system across five distinct genomic compartments and showing how disturbing each of the components affects the system.
Recent literature indicates that in systems where H3K27me3 is more prevalent, changes in this modification have more pronounced effects. mESCs cultured in the 2i medium, thought to represent an earlier stage of pluripotency, are normally characterized by greatly reduced DNAme, reduced H3K36me2 and a much broader distribution of H3K27me3 (Oksuz et al. 2018; McLaughlin et al. 2019; van Mierlo et al. 2019; Alabert et al. 2020). It appears that in 2i medium, the absence of EED and H3K27me leads to considerable increase in intergenic H3K36me2 and DNAme. This has been interpreted as evidence of bidirectional antagonism between H3K36me and H3K27me (Alabert et al. 2020). However, no compelling molecular mechanisms of how methylation at lysine 27 influences modifications of lysine 36 have been proposed, and further studies are necessary.
Thus, within this relatively simple subsystem, it appears that depending on conditions, either K36 or K27 methylation can act as the main determinants of the global epigenetic profile. In the vast intergenic regions, DNA methylation is likely too sparse—even in very highly methylated mESCs—to influence chromatin-associated proteins. DNAme does, on the other hand, determine subtler features, such as nucleation of PRC2 sites at CGIs. Although globally less influential for bulk epigenome characteristic, such localized effects are likely to have considerable functional outcomes. The influence of H3K27ac appears somewhat akin to DNAme: this mark is globally too infrequent (present on <1% of nucleosomes) to have considerable direct bulk effects. However, at enhancers and promoters the high occupancy of H3K27ac may counteract deposition of H3K27me and DNAme (Tie et al. 2009).
We can speculate on the nature of the relative context-dependent importance of H3K36 and H3K27 methylation in shaping the epigenome. The most likely determinants are the relative strength and timing of activities of the respective writer enzymes/complexes: PRC2 versus H3K36 methyltransferases. In most cell types that have been studied, those two forces appear to counterbalance each other, subdividing the genome into clear domains of H3K27me3 and H3K36me2/3, where H3K36me2 often colocalizes with H3K27me2 (Streubel et al. 2018). However, we have come across at least two systems in which H3K27me3 and H3K36me2 appear to coexist in the same genomic locations: the head and neck cancer cell line FaDu (Farhangdoost et al. 2021) and prenatal prospermatogonia (Shirane et al. 2020), suggesting that in some cases the two forces may be evenly matched.
What are the next steps toward understanding the epigenetic network? Some ambitious steps have been taken toward computational modeling of the system (Chory et al. 2019; Alabert et al. 2020; Harutyunyan et al. 2020; Sandholtz et al. 2020; Lövkvist and Howard 2021). The conceptual frameworks already exist and can be further refined. However, before the models become useful for providing further insights into the behavior of the system, much more work is necessary to understand the underlying interactions. Even in a three-component system, distinguishing the relative importance of direct versus secondary indirect effects is not trivial. Adding more components will further complicate the picture. H3K4me3 may confound the relationship between DNAme and H3K27me3, because those two marks co-occur at bivalent promoters and H3K4me3 inhibits DNMT3A/B via their ADD domains (Noh et al. 2015). PRC1 and PRC2 show a strong cross talk through their H2AK119ub and H3K27me3 products, and H2AK119ub has recently been suggested to recruit DNMT3A through its UDR domain (Weinberg et al. 2021). H3K9me is known to influence DNAme through its interaction with UHFR and DNMT1 (Rothbart et al. 2012). This is not an exhaustive list of interactions that may indirectly affect such subnetworks. However, recent advances in tools such as degradable and inducible systems have already produced unprecedented insights into relative importance and timing of events (Højfeldt et al. 2018; Oksuz et al. 2018; Dobrinić et al. 2021). Along with combinatorial gene manipulations, fine-grained time series experiments performed in the most appropriate cellular contexts relevant to specific epigenetic components will be necessary to further understand the extended epigenetic interaction network.
Technical standardization is another key aspect necessary to ensure robustness of future epigenetic studies. Here, in addition to the in-house data of two replicates, we also incorporated public data sets to complement our findings. Because those public data came from different laboratories, we acknowledge that the variability in the experimental conditions and methods may affect some of the conclusions. Specifically, we must keep in mind that raw ChIP-seq signal is not quantitative and does not allow comparison of absolute modification levels across conditions. In our samples, we used exogenous Drosophila (Orlando et al. 2014) and synthetic nucleosome (EpiCypher) spike-ins (Supplemental Table S3). However, because of discrepancies (space limitations prevent us from discussing them here, but some of the problems are outlined in Dickson et al. 2020), we eventually opted for mass spectrometry normalization. In our experience, we found this method to perform most reliably, but more research on optimizing quantitative normalization is necessary, particularly to distinguish small changes in the levels of epigenetic modifications.
In this work, we focused on a small subset of the epigenetic interaction network. Understanding such relationships is important to realize the extent to which a single change may affect the entire interconnected system. It is also essential to understand the downstream functional consequences of epigenome dysregulation, in particular, its effect on transcription, gene expression levels, disease, and cancer. We have refrained from including functional outcomes in this study but, in closing, it is worth pointing out that transcription is not only a consequence but also a major force shaping the epigenome (Holoch et al. 2021), and in future studies it would benefit from consideration as a part of the interacting system rather than an outcome.
Methods
Culture of mouse ES cells
A1 mouse ES lines (background C57BL/6 × 129S4/SvJae F1), including Ezh2–/– and Nsd1–/– cells, were obtained from David Allis's laboratory (Weinberg et al. 2019). J1 control and Dnmt1/3a/3b triple knockout (TKO) lines were obtained from Dr. Amander Clark's laboratory, UCLA. The Dnmt1/3a/3b TKO line was originally generated by Dr. Masaki Okano, Riken (Tsumura et al. 2006). For comparison between Nsd1–/– and TKO, the cell lines were cultured in two replicates and maintained under standard serum/LIF conditions, with slight modifications detailed in Supplemental Methods.
Histone acid extraction, histone derivatization, and analysis of post-translational modifications by nano-LC–MS
We followed standard protocols established in the Garcia laboratory as previously described (Farhangdoost et al. 2021) and detailed in Supplemental Methods. Raw files were analyzed using EpiProfile 2.0 (Yuan et al. 2018).
ChIP-sequencing and analysis
We followed standard protocols for chromatin immunoprecipitation, similar to those previously used in Weinberg et al. (2021). Slightly different protocols were used in the two replicates of Nsd1-KO and TKO samples, and those differences are detailed in Supplemental Methods. Libraries were generated using KAPA Hyper Prep Kit according to manufacturer's instructions. ChIP libraries were sequenced using Illumina NovaSeq 6000 at 50-bp or 100-bp single-end reads. Reads were aligned to a combined reference of mm10 and dm6 using BWA (Li and Durbin 2009) version 0.7.17 with default parameters. All unmapped and multimapped reads were discarded by filtering out alignments with MAPQ less than 3 using SAMtools (Danecek et al. 2021) version 1.12. BEDTools (Quinlan and Hall 2010) version 2.22.1 was used to count reads in different sized bins (1 kb, 10 kb, and 100 kb) using “bedtools multicov –q 3 –bams $CHIP.bam –bed $REGION.”
Normalized ChIP-seq signals were obtained by dividing the total alignments (in millions) and additionally multiplying by MS abundance values when stated (Farhangdoost et al. 2021).
Coverage tracks (bamCoverage -b $BAM -o $OUTPUT.bigWig -of bigwig ‐‐normalizeUsing CPM ‐‐binSize 100 ‐‐blackListFileName $BL_bed) and aggregate matrices (computeMatrix scale-regions or reference-point ‐‐missingDataAsZero –skipZeros; “‐‐binSize 1000” for intergenic regions, and “‐‐binSize 500” for others) were generated using deepTools v3.1.0 (Ramírez et al. 2016). In particular, the ENCODE blacklist (Amemiya et al. 2019) was used.
The genome-wide correlation density plots were generated to capture the distribution of 10-kb genomic windows within a 2D space defined by MS-normalized ChIP-seq signals (in the range 0%–100%) per window for a specific modification across a pair of conditions. Data points with similar x and y values were grouped together using R (R Core Team 2021) library hexbin, producing a 50 × 50 grid subdividing the plot area. By assigning +1 to windows overlapping gene annotations, −1 to those that do not, and 0 to ambiguous cases, an average “genicity” value is computed for each hexbin. Additionally, the transparency of each hexbin is varied based on the number of constituent data points to convey density.
“Peakiness” scores were taken as the average read-depth normalized coverage of the top 1% most covered 1-kb windows across the genome, excluding those overlapping blacklisted regions.
Differential H3K27me3 regions for evaluation of CpG density were taken from the 100th and 1st percentile when ranking 10-kb bins based on H3K27me3 differences (Nsd1-KO − WT, TKO − WT).
H3K36me2 domains were called using the same approach as described in our previous paper (Weinberg et al. 2019), with those lost in Nsd1-KO generated by subtracting domains called in Nsd1-KO from those identified in WT, excluding intervals smaller than 50 kb in the final output.
RNA-seq and analysis
Total RNA was extracted from cell pellets (∼1 million cells) using miRNeasy Mini Kit (QIAGEN) with DNase digest using RNase-free DNase Set (QIAGEN) according to instructions from the manufacturer. Library preparation was performed with ribosomal RNA (rRNA) depletion according to instructions from the manufacturer (Epicentre) to achieve greater coverage of mRNA and other long noncoding transcripts. Paired-end sequencing (100 bp) was performed on the Illumina HiSeq 4000 platform.
Raw reads were aligned to mm10 genome build using STAR version 2.5.3a (Dobin et al. 2012). After alignment, we used featureCounts program (version 1.5.3) to count the reads for each gene from the GTF annotation
file (using the GENCODE version from UCSC) and expression levels were calculated as follows:
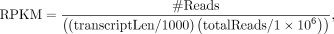 where #Reads is the read counts of the gene, transcriptLen is the transcript length, and totalReads is the total read counts.
We categorized all the genes into four groups based on their expression levels (RPKM), which were calculated by taking average
of replicates. The lowest group includes only genes with zero reads. The remaining three groups were divided with equal numbers
of genes. Here, the “Active genes” are referred to as the combination of the first two groups of genes with the higher expression
levels, and “Inactive genes” are the remaining two groups of genes with low expression. When comparing parental and Nsd1-KO, we took the intersection of genes in these two conditions.
where #Reads is the read counts of the gene, transcriptLen is the transcript length, and totalReads is the total read counts.
We categorized all the genes into four groups based on their expression levels (RPKM), which were calculated by taking average
of replicates. The lowest group includes only genes with zero reads. The remaining three groups were divided with equal numbers
of genes. Here, the “Active genes” are referred to as the combination of the first two groups of genes with the higher expression
levels, and “Inactive genes” are the remaining two groups of genes with low expression. When comparing parental and Nsd1-KO, we took the intersection of genes in these two conditions.
Whole-genome bisulfite sequencing and analysis
Whole-genome sequencing libraries were generated from 1000 ng of genomic DNA spiked with 0.1% (w/w) unmethylated λ DNA (Roche Diagnostics) and fragmented to 300- to 400-bp peak sizes using the Covaris focused ultrasonicator E210. Fragment size was controlled on a Bioanalyzer High Sensitivity DNA Chip (Agilent) and NxSeq AmpFREE Low DNA Library Kit (Lucigen) were applied. End repair of the generated dsDNA with 3′ or 5′ overhangs, adenylation of 3′ ends, adaptor ligation, and clean-up steps were performed per Lucigen's recommendations. The cleaned-up ligation product was then analyzed on a Bioanalyzer High Sensitivity DNA Chip (Agilent). Samples were then bisulfite converted using the EZ-DNA Methylation Gold Kit (Zymo Research) according to the manufacturer's protocol. DNA was amplified by nine cycles of PCR using the Kapa HiFi Uracil + Kit (Roche) DNA polymerase (KAPA Biosystems) according to the manufacturer's protocol. The amplified libraries were purified using Ampure XP Beads (Beckman Coulter), validated on Bioanalyzer High Sensitivity DNA Chips, and quantified by PicoGreen. Sequencing of the WGBS libraries was performed on the Illumina HiSeq X system using 150-bp paired-end sequencing.
Raw reads were submitted to Bismark (version 0.22.3) (Krueger and Andrews 2011) for mapping and methylation calling against mouse genome version mm10/GRCm38, discarding duplicate reads. Briefly, the methylation level at a CpG (similarly for CHH) site was the number of reads with that site methylated divided by the total number of reads covering the site. To ensure comparability of region DNA methylation levels across all samples, CpGs that overlapped with SNPs from dbSNPs or were located within the ENCODE blacklisted regions, termed Duke excluded regions (DER), were excluded and only CpGs covered by ≥5× in all samples were retained for the computation of DNA methylation levels.
Published data sets
H3K36me2/3 and DNMT3A in parental mESC and Nsd1-KO were published previously (Weinberg et al. 2019) under the NCBI Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/) accession number GSE118785. The ChIP-seq of H3K27me3 in mESC Dnmt-TKO and WT for Supplemental Figure S2C were from Brinkman et al. (2012) under GEO accession number GSE28254 and Hagarman et al. (2013), where data were obtained directly from the investigators. The ChIP-seq of EZH2 in mESC WT (Fig. 4A) was from Kundu et al. (2017) under GEO accession number GSE89949. The ChIP-seq of H3K27me3 (Marks et al. 2012) and WGBS data (Habibi et al. 2013) in mESC serum and 2i medium (Supplemental Fig. S3C) were downloaded from GEO (accession numbers: GSE23943, GSE41923). The WGBS data of mESC Eed-KO (Li et al. 2018) used in Figure 5B were obtained from GEO with accession number GSE102753. The ChIP-seq of H3K27ac and H3K27me in mESC Eed-KO and WT (Ferrari et al. 2014) used in Figure 6 were downloaded from GEO with accession numbers GSE39496 and GSE51006. The ChIP-seq of H3K27ac and H3K27me in mESC Ezh2-KO and WT (Lavarone et al. 2019) used in Supplemental Figure S4 were downloaded from GEO with accession number GSE116603.
Data access
The WGBS, ChIP-seq, and RNA-seq data generated in this study have been submitted to the NCBI Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/) under accession number GSE186506. Custom scripts used to generate all results in this study are available on GitHub (https://github.com/bhu/mesc_epigenome) and as Supplemental Code.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
J.M. is supported by funding from the United States National Institutes of Health (NIH) grant P01-CA196539; C.L. is supported by funding from NIH grant R35GM138181 and the Pew-Stewart Scholars for Cancer Research Award; W.A.P. is supported by funding from the Natural Sciences and Engineering Research Council of Canada (NSERC) grant RGPIN-2018-04856; B.A.G. is supported by NIH grants CA196539 and AI118891, and the St. Jude Children's Hospital Chromatin Consortium; B.H. is supported by the Canadian Institutes of Health Research (CIHR) Banting and Best Graduate Scholarship; and D.N.W. is supported by NIH grants T32GM007739 and F30CA224971.
Author contributions: J.M. conceived the project and wrote the manuscript. H.C. and B.H. performed the data analysis, interpreted and presented the results, and assisted with writing the manuscript. E.B. helped analyze the data and prepare the manuscript. C.H. oversaw laboratory experiments and helped prepare the manuscript. P.R. participated in data analysis. S.Y.K., J.S., and D.N.W. performed laboratory experiments. F.M.R. and B.A.G. performed and supervised mass spectrometry analysis. C.L., W.A.P., and J.M. supervised the work and helped with critical interpretation and organization of the results.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.276383.121.
-
Freely available online through the Genome Research Open Access option.
- Received November 17, 2021.
- Accepted March 30, 2022.
This article, published in Genome Research, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








