
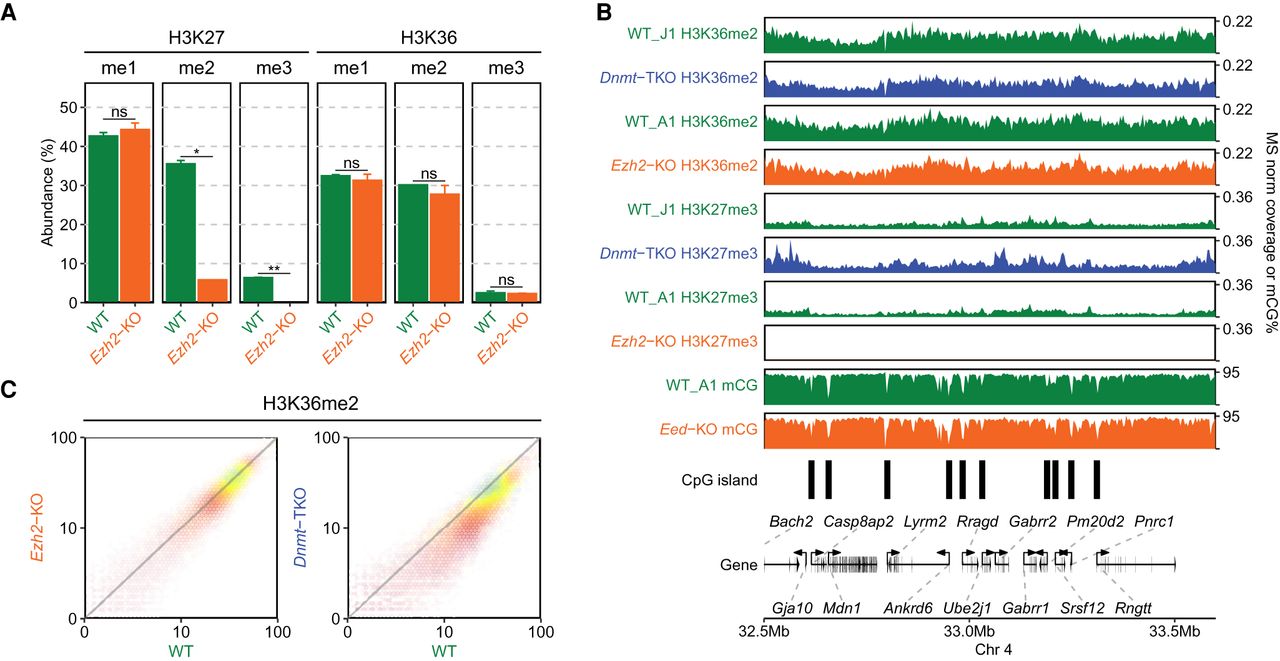
Effect of H3K27me2/3 loss on H3K36me2 is negligible. (A) Mass spectrometry analysis of H3K27 and H3K36 methylation levels (two replicates for each mark) in WT and Ezh2-KO, showing that deletion of Ezh2 results in total loss H3K27me3 and fivefold reduction in H3K27me2, but no significant reduction in H3K36me. P-values are indicated in the same way as in Figure 1B. (B) A representative 1-Mb region illustrating high-similarity epigenomic profiles of H3K36me2 in the Ezh2-KO and Dnmt-TKO. We also show that global DNA methylation is not visibly affected by the absence of H3K27me (Eed-KO, bottom). DNAme in Dnmt-TKO is completely depleted (data not shown). (C) Genome-wide correlation plots (10 kb) bins showing lack of change in global H3K36me2 distribution in Ezh2-KO and a small uniform decrease in Dnmt-TKO. All data in this figure are from this study, with the exception of DNAme in Eed-KO (Li et al. 2018).








