
Variations in antibody repertoires correlate with vaccine responses
- Yana Safonova1,2,3,
- Sung Bong Shin4,
- Luke Kramer5,
- James Reecy5,
- Corey T. Watson2,
- Timothy P.L. Smith4 and
- Pavel A. Pevzner1
- 1Computer Science and Engineering Department, University of California at San Diego, San Diego, California 92093, USA;
- 2Department of Biochemistry and Molecular Genetics, University of Louisville School of Medicine, Louisville, Kentucky 40202, USA;
- 3Department of Computer Science, Johns Hopkins University, Baltimore, Maryland 21218, USA;
- 4U.S. Meat Animal Research Center, USDA-ARS, Clay Center, Nebraska 68933, USA;
- 5Department of Animal Science, Iowa State University, Ames, Iowa 50011, USA
Abstract
An important challenge in vaccine development is to figure out why a vaccine succeeds in some individuals and fails in others. Although antibody repertoires hold the key to answering this question, there have been very few personalized immunogenomics studies so far aimed at revealing how variations in immunoglobulin genes affect a vaccine response. We conducted an immunosequencing study of 204 calves vaccinated against bovine respiratory disease (BRD) with the goal to reveal variations in immunoglobulin genes and somatic hypermutations that impact the efficacy of vaccine response. Our study represents the largest longitudinal personalized immunogenomics study reported to date across all species, including humans. To analyze the generated data set, we developed an algorithm for identifying variations of the immunoglobulin genes (as well as frequent somatic hypermutations) that affect various features of the antibody repertoire and titers of neutralizing antibodies. In contrast to relatively short human antibodies, cattle have a large fraction of ultralong antibodies that have opened new therapeutic opportunities. Our study reveals that ultralong antibodies are a key component of the immune response against the costliest disease of beef cattle in North America. The detected variants of the cattle immunoglobulin genes, which are implicated in the success/failure of the BRD vaccine, have the potential to direct the selection of individual cattle for ongoing breeding programs.
Introduction
The challenge of identifying variations in immunoglobulin genes that affect vaccine response
Although vaccination is a primary tool to control the spread of viral and bacterial diseases, the success of vaccines at the population level does not always translate to protection at the individual level. Figuring out why a vaccine fails in some individuals is important both during the vaccine development stage (to inform changes in the vaccination protocols) and the vaccine administration stage (to identify a subpopulation with a poor vaccine response). A promising approach to understanding why a vaccine succeeds in some individuals and fails in others is to analyze the germline variations in the immunoglobulin (IG) loci of individuals with successful/failing antibody responses to the vaccine.
Antibodies are not encoded in the germline genome but rather result from somatic genomic recombinations called VDJ recombinations (Tonegawa 1983). This process affects an IG locus containing the families of the variable (V), diversity (D), and joining (J) genes (referred to as IG genes) by selecting one V, one D gene, and one J gene, and concatenating them together to generate one of the antibody chains. Further diversity of antibodies is generated by the class-switch recombination and somatic hypermutations (SHMs) (Dudley et al. 2005). There are three types of IG loci in mammalian species (including cows): heavy chain, kappa light chain, or lambda light chain. In this work, we focus on the heavy chain (IGH) locus only. In addition, we follow IMGT nomenclature guidelines for IG gene symbols (https://www.imgt.org/IMGTScientificChart/Nomenclature/IMGTnomenclature.html).
The expression quantitative trait loci (eQTL) analysis links variation in gene expression to genotypes. Although eQTL analysis has greatly contributed to the dissection of the genetic basis of disease and vaccine response (Franco et al. 2013; Bhalala et al. 2018), the IG loci remain virtually untouched by eQTL studies (Watson et al. 2017). eQTL studies usually start from generating an n × m genotype matrix that contains information about each of m markers (e.g., SNPs) in each of n individuals and an n × k phenotype (expression) matrix that contains information about expression levels of each of k genes in each of n individuals. Generating analogs of the genotype and phenotype matrices in immunogenomics studies is a more complex task than in traditional eQTL studies.
First, whereas the set of genes in eQTL studies is fixed and shared by all individuals, the antibody repertoire is composed of a virtually unlimited set of proteins, and there are typically few antibodies shared between any two individuals. Thus, given an antibody repertoire represented as a repertoire sequencing (Rep-Seq) data set, it is not clear how to define the phenotype matrix. One possibility is to consider each germline gene in the IG locus (e.g., a V gene, a D gene, or a J gene) and to define the usage of this gene as the fraction of antibodies that originated from this gene among all antibodies in the Rep-Seq data set. Our goal is to identify usage QTLs (IgQTLs) that link variation in usage to a genotype.
Second, eQTL studies are usually based on RNA-seq and whole genome sequencing (WGS) data, whereas immunosequencing studies generate Rep-Seq data about antibodies. Thus, the genotype matrix in immunosequencing studies has to be inferred from Rep-Seq data alone because the WGS data is typically not available. Although inference of alleles of V, D, and J genes from Rep-Seq data is a well-studied problem (Gadala-Maria et al. 2015, 2019; Corcoran et al. 2016; Safonova and Pevzner 2019; Bhardwaj et al. 2020), the existing allele inference tools, primarily developed for naive repertoires, often fail in the case of more complex antigen-stimulated repertoires that represent the primary goal of IgQTL studies. Also, in contrast to allele inference tools that attempt to infer SNPs and ignore frequent SHMs, IgQTL studies should account for both SNPs and frequent SHMs because they may play equally important roles in vaccine responses.
Bovine respiratory disease
We conducted a personalized immunogenomics study of 204 calves to analyze the efficacy of the bovine respiratory disease (BRD) vaccine, the largest time-series immunosequencing data set generated so far across all species, including human. Because cattle production accounted for $67 billion in 2018 in the United States (Economic Research Service, U.S. Department of Agriculture 2021), maintaining cattle health is an important direction of agricultural studies. The BRD is the costliest disease of beef cattle in North America (Taylor et al. 2010). Although vaccination reduces the risk of BRD, losses from BRD remain substantial and individuals respond very differently to the BRD vaccine (Kramer et al. 2017). In order to understand links between variants in cattle IG genes and antibody responses to the vaccine, we generated four Rep-Seq data sets (taken before and after the BRD vaccination) for each of 204 calves.
Ultralong cattle antibodies
The evolution of the cattle IGH locus has resulted in a loss of many functional V genes, thus reducing the diversity of the cattle antibody repertoire (Haakenson et al. 2018). To compensate for the reduced VDJ recombination diversity, cattle have developed antibodies with ultralong CDR3s that have a unique mechanism of structural diversification (Dong et al. 2019). We refer to antibodies with CDR3s longer than 50 amino acids as ultralong antibodies (for comparison, the average length of human CDR3s is only 15 aa). Although nonconventional recombination processes (such as D-D fusions and V gene replacement) can generate ultralong human antibodies (including broadly neutralizing human antibodies against HIV-1 [Yu and Guan 2014]), they are rare in human antibody repertoires, typically accounting for <1% of all antibodies (Safonova and Pevzner 2019). In contrast, ultralong antibodies account for ∼10% of cattle antibodies (Wang et al. 2013).
The vast majority of ultralong cattle antibodies are generated by the VDJ recombination of the same V, D, and J genes: IGHV1-7, IGHD8-2, and IGHJ2-4 (Wang et al. 2013). An unusually long IGHD8-2 gene (148 nt) contributes to all ultralong CDR3s and enables their structural diversification. This gene encodes four cysteines that form disulfide bonds and turn the CDR3 loop into a complex protein structure called a knob (Wang et al. 2013). IGHD8-2 also contains many codons that differ from the cysteine codons by a single nucleotide. SHMs often result in new cysteines that form new disulfide bonds, thus extending the diversity of the knob structures.
Ultralong cattle antibodies open new therapeutic opportunities (Muyldermans and Smider 2016) and may even neutralize various strains of HIV (Sok et al. 2017). Human antibodies target the HIV envelope glycoprotein (Env) presented on the virus surface. However, highly mutated Env proteins are often covered by glycans, making them a hard-to-reach target for human antibodies. Ultralong cattle antibodies penetrate glycans and directly target the conservative sites that are unreachable for human antibodies because they are buried inside the Env protein. Although ultralong cattle antibodies have great pharmaceutical potential in terms of targeting unusual antigens (Burke et al. 2020), their role in the native immune response remains unclear.
The challenge of finding IgQTLs
Previous immunosequencing studies have succeeded in linking variants in the human IGH locus to disease and vaccination efficacy (Thomson et al. 2008; Lingwood et al. 2012; Avnir et al. 2016; Parks et al. 2017, Lee et al. 2021, Mikocziova et al. 2021) but have not yet resulted in a software tool addressing the above-mentioned complications in finding IgQTLs. We thus developed the IgQTL tool for detecting both variants of germline V genes and their frequent SHMs for downstream analysis of antibody repertoire-specific QTLs in the V genes.
Results
Rep-Seq data sets and antibody titers
We analyzed Rep-Seq data sets for 204 purebred American Angus calves vaccinated against BRD. Each animal was initially vaccinated at day 0 and then given a booster vaccination 3 wk later (Fig. 1A; see also “Sample preparation” in Methods). For each animal, four blood samples were collected: 3 wk prior to vaccination (referred to as “–3”), at the moment right after the first vaccination (referred to as “0”), 3 wk postvaccination (referred to as “+3”), and 6 wk postvaccination (referred to as “+6”). From each blood sample, IgG transcripts were extracted and sequenced (see “Repertoire sequencing” in Methods). Because the same primers were applied to all samples, we contend that the same primer selection bias exists across all samples in the study, and therefore comparisons across samples were valid as the source of bias remained constant. In the initial manuscript describing our bovine Rep-Seq efforts, replicate data were presented and indicated relatively minor variation between replicate Rep-Seq of the same RNA sample (Larsen and Smith 2012). Thus, we argue that the sample size is sufficient that biases introduced at this step would not substantively affect the conclusions of the study.
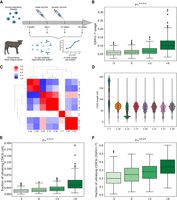
Overview of study design and characteristics of antibody repertoires. (A) An overview of the study design. Two hundred four calves were vaccinated against BRD, and their expressed antibody repertoires were sequenced at four time points pre- and postvaccination. Serum from each of the sequenced animals was also assayed for BRD-specific antibody titers at the same time points. (B) The distribution of IGHV1-7 usage at four time points. Here (and further), each box shows the quartiles of the distribution. The whiskers show the rest of the distribution, except for outliers found using a function of the interquartile range implemented by the Seaborn package in Python. P-values have the following notations: (ns) ≥ 0.05, (*) P < 0.05, (**) P < 0.01, (***) P < 0.001, (****) P < 0.0001. (C) The matrix shows the Pearson's correlations between gene usages computed across all high-usage V genes at time point “−3.” Correlation values vary from −1 (blue) to 1 (red). Statistically insignificant correlations (P ≥ 0.05) are shown as white cells. (D) The histogram of the distributions of the CDR3 lengths for eight highly used cattle V genes. The histogram is computed for individual 14,007. (E) The distribution of the fraction of ultralong CDR3s in all CDR3s at four time points. (F) The distribution of the fraction of ultralong CDR3s in CDR3s derived from IGHV1-7 at four time points.
Serum from each of the sequenced animals was also assayed for BRD-specific antibody titers at the same time points (Kramer et al. 2017) to quantify pre-existing immunity (e.g., resulting from maternal antibodies that are passed through milk from the dam to the calf) and as a measure of vaccine success (see “Antibody titers” in Methods). All calves were at the same farm and processed through the same facilities at the same time.
High-usage cattle V genes
Each Rep-Seq data set was aligned to 13 cattle V genes from the international ImMunoGeneTics information system (IMGT) (Lefranc et al. 2009; see “Preprocessing Rep-Seq data” in Methods). For each V gene, we computed its usage in an individual as the fraction of sequences with distinct CDR3s aligned to this V gene in the corresponding Rep-Seq data set. We did not take into account the number of reads contributing to each distinct CDR3 because these numbers can reflect PCR artifacts rather than the amount of initial mRNA molecules (Shlemov et al. 2017). We defined the average usage of a V gene as its average usage across all individuals and all time points. Only eight out of 13 V genes had an average usage exceeding 0.01: IGHV1-7, IGHV1-10, IGHV1-14, IGHV1-17, IGHV1-20, IGHV1-21, IGHV1-27, and IGHV1-30. We refer to them as high-usage V genes and limit further analysis to these genes only.
IGHV1-7 is the only V gene with a statistically significant increase in usage after vaccination
We first assessed differences in usage between pre- and postvaccination for all V genes. We found that only IGHV1-7 has significantly higher usage after the vaccination (Supplemental Table S1). Figure 1B shows that the usage of the IGHV1-7 gene is significantly increased at time points “+3” and “+6” (P-value = 8.17 × 10–115), suggesting that vaccination triggered the production of antibodies derived from IGHV1-7. Henceforth, we used the linear mixed effect model to estimate P-values (referred to simply as “P”) for repeated measures (representing four time points), unless a different method is specified.
Analysis of Rep-Seq data sets collected at the time point “–3” revealed that each V gene but IGHV1-7 has positive or negative correlation of usages with other IGHV genes. Figure 1C shows that V genes form three groups based on their usage, consisting of seven positively correlated genes: G1 = (IGHV1-10, IGHV1-14), G2 = (IGHV1-17, IGHV1-20, IGHV1-30), and G3 = (IGHV1-21, IGHV1-27). IGHV1-7 is the only gene with an independent usage profile. These correlations are consistent across time points “0,” “+3,” and “+6.” We conjecture that this is explained by an association of IGHV1-7 with ultralong CDR3s and their special role in the adaptive immune response. Figure 1C illustrates that the usages in groups G1, G2, and G3 anticorrelate.
BRD vaccination triggers the increased production of ultralong antibodies
Figure 1D shows that for all high-usage cattle V genes, except for IGHV1-7, the mean CDR3 length does not exceed 75 nt. It also illustrates that, unlike other V genes, IGHV1-7 has a bimodal distribution of CDR3 lengths, and the second mode is represented by ultralong CDR3s. On average, 21% of sequences derived from IGHV1-7 have ultralong CDR3s at time point “–3” and 99% of ultralong CDR3s across all individuals are derived from IGHV1-7. The latter observation agrees with findings reported by Walther et al. (2013), Wang et al. (2013), Deiss et al. (2019), and Dong et al. (2019).
Figure 1E illustrates that both initial and booster vaccinations significantly increase the fraction of ultralong CDR3s in all CDR3s (across all V genes) at time points “+3” and “+6” (P = 3.61 × 10–107). Figure 1F shows that the fraction of ultralong CDR3s in all CDR3s derived from IGHV1-7 also increases after vaccinations (P = 1.19 × 10–119). These observations suggest that the vaccination triggers the production of antibodies derived from IGHV1-7.
Antibody titers correlate with fractions of ultralong CDR3s
Some calves have pre-existing immunity because they either were previously exposed to the BRD-causing virus or have maternal antibodies specific to BRD. This pre-existing immunity (as well as cross-reactivity of antibodies) may affect titers at the initial time point “–3.” Downey et al. (2013) demonstrated that the decay rate of maternal antibodies is rather low and that there is a threshold effect: The calves do not respond to the vaccine if the level of maternal antibodies exceeds a threshold and only respond when this level drops. Also, the impact of calf ages on antibody titers to BRD was shown to be insignificant (Supplemental Fig. S1).
Figure 2A shows that, on average, the booster vaccination increased neutralizing antibody titers. Figure 2B shows the Pearson's correlation r between antibody titers at four time points and illustrates that they correlate at points “–3” and “0” (r = 0.78, P = 5.83 × 10–43), “–3” and “+3” (r = 0.43, P = 1.53 × 10–10), and “0” and “+3” (r = 0.43, P = 2.29 × 10–10). In contrast, antibody titers from time points “–3” and “0” anticorrelate with final titers at the time point “+6” (r = –0.42, P = 3.34 × 10–10 and r = –0.4, P = 1.78 × 10–9, respectively). This suggests that pre-existing immunity to BRD antigens may be suboptimal, preventing development of a successful immune response to the BRD vaccine. Impacts of suboptimal antibody responses caused by pre-existing immunity were reviewed by Zimmermann and Curtis (2019) (for various antigens) and Iwasaki and Yang (2020) (for SARS-CoV-2) and discussed in the Supplemental Method (“The Relations Between Pre- and Post-Vaccination Immunity to the BRD Vaccine in Calves”).
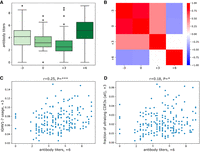
Antibody titer statistics. (A) The distribution of antibody titers at four time points. Titers at time point “−3” show the number of BRD-specific antibodies without any antigen stimulation. Titers at time point “0” represent immediate memory responses triggered by the vaccine. Titers at time points “+3” and “+6” reflect antibodies produced as a result of the vaccination. Details of the titer analysis are described in Kramer et al. (2017). (B) The matrix shows the Pearson's correlations between antibody titers at four time points. Correlation values vary from –1 (blue) to +1 (red). Statistically insignificant correlations (P ≥ 0.05) are shown as white cells. (C) Antibody titers at time point “+6” versus usages of IGHV1-7 at time point “+3.” (D) Antibody titers at time point “+6” versus fractions of ultralong CDR3s in all CDR3s at time point “+3.” The Pearson's correlations (r) and P-values (P) are shown at the top of panels C and D.
We have not found any statistically significant correlations between the titers and the usages of all high-usage V genes, except for IGHV1-7. Both the usage of IGHV1-7 (Fig. 2C) and the fraction of ultralong CDR3s (Fig. 2D) at the time point “+3” correlate (albeit weakly) with final titers at the time point “+6” (r = 0.25, P = 0.0004, and r = 0.18, P = 0.0125, respectively). These observations support our hypothesis that antibodies with ultralong CDR3s play an important role in recognizing the vaccine antigens.
The IgQTL pipeline
To reveal the associations between germline/somatic variants and features of cattle antibody repertoires (gene usages, antibody titers, and fractions of ultralong antibodies), we developed the IgQTL tool. IgQTL takes Rep-Seq reads and antibody titers (if available) as an input and consists of the following steps (Fig. 3A):
-
Generating the phenotype matrix containing information about gene usages, fractions of ultralong CDR3s, antibody titers, etc.
-
Finding germline and somatic variations (GSVs).
-
Generating and clustering a genotype matrix to reveal subjects with common genotypes.
-
Finding statistically significant genotype–phenotype associations.
-
Identifying the most consequential GSVs with respect to phenotypes.
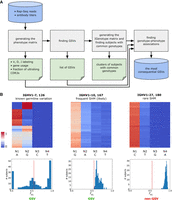
Overview of IgQTL method. (A) The IgQTL pipeline. Gray rectangles show various steps of the IgQTL pipeline. The input (Rep-Seq reads and antibody titers) and the final output (the most consequential GSVs) are shown in blue. Intermediate output is shown in green. (B) The procedure for finding GSVs (126, IGHV1-7, A/G) and (167, IGHV1-10, G/A), as well as non-GSVs (180, IGHV1-27, C/T). Heat maps in the upper row show the fractions of the nucleotides across all subjects varying from 0 (blue) to 1 (red). Columns are arranged according to the sum of fractions across all subjects. N1 and N2 correspond to the first and the second columns, respectively. Histograms in the lower row show distributions of fractions fN1 across 204 animals. The red vertical line in each histogram corresponds to freq = 0.55.
Below we applied IgQTL to reveal the genotype–phenotype associations in the cattle immunosequencing study.
Generating the phenotype matrix
The phenotype matrix is defined as a matrix with 204 rows, where each column represents either the usage of one of the V genes, or the fraction of ultralong antibodies, or an antibody titer. In the case of the cattle immunosequencing data set, IgQTL forms a 204 × 14 phenotype matrix that represents usages of all high-usage V genes (the first eight columns), the fraction of ultralong antibodies among all antibodies in the repertoire (ninth column), the fraction of ultralong antibodies derived from IGHV1-7 among all antibodies derived from IGHV1-7 (10th column), and the antibody titers at four time points (the last four columns).
Finding germline variations and frequent SHMs in the V genes
Previous studies have revealed associations between germline variations in V genes and variation in their usages and antibody titers (Thomson et al. 2008; Lingwood et al. 2012; Avnir et al. 2016; Parks et al. 2017, Lee et al. 2021, Mikocziova et al. 2021). However, these studies did not consider the impact of frequent SHMs that, similarly to germline variations, may be associated with variation in gene usage. Because the Rep-Seq data, obtained from IgG antibodies, represent mature antibody responses, below we analyzed the impact of frequent SHMs on antibody repertoires.
To capture both germline variations and frequent somatic hypermutations (further referred to as germline or somatic variations or GSVs) for each subject, we generated a combined data set from all four time points by collapsing identical nucleotide sequences and analyzed sequences aligned to the same V gene. Given a position in a germline V gene, we analyzed all reads aligning to this gene in a single combined data set and computed a vector (fA, fC, fG, fT), where fN is the fraction of reads that have the nucleotide N aligned at this position. We collected such vectors from all subjects and define N1 and N2 as nucleotides with the highest and the second-highest total fractions. Note the same N1 and N2 nucleotides are defined for all individuals.
For most positions, fN1 is close to 1, indicating that these positions do not exhibit variations and frequent SHMs. We were interested in positions where fN1 falls below a frequency threshold freq (the default value freq = 0.55), as such positions likely reflect one of the following situations:
-
If a subject is homozygous by N2 (i.e., N1 is substituted by N2 in the germline), we expect that fN1∼0 and fN2∼1.
-
If a subject is heterozygous by N1/N2, we expect that fN1∼0.5 and fN2∼0.5.
-
If the germline nucleotide N1 is replaced by a frequent SHM represented by N2 (with frequency at least 50%), we expect that fN1 ≤ freq.
We classified a position P in a gene G as a GSV if fN1 ≤ freq for at least one subject.
Each GSV (represented by a position P in a gene G, and nucleotides N1 and N2) was encoded as (P, G, N1/N2). Figure 3B illustrates the procedure for identifying GSVs using examples of a GSV (126, IGHV1-7, A/G) that represented a known germline variation, a GSV (167, IGHV1-10, G/A) that represents a likely frequent SHM, as well as a non-GSV (180, IGHV1-27, C/T).
Fractions fN1 for position 126 in IGHV1-7 vary from 0.01 to 0.98 and form a trimodal distribution. Because GSV (126, IGHV1-7, A/G) is a known germline variant, the three modes correspond to the homozygous states AA and GG, and a heterozygous state AG. (126, IGHV1-7, A/G) is classified as a GSV because the minimum of fractions fN1 across all individuals (0.01) does not exceed the default frequency threshold freq = 0.55. (167, IGHV1-10, G/A) was classified as a GSV because fractions fN1 for position 167 of IGHV1-10 vary from 0.54 to 0.86 (likely frequent SHMs). In contrast, (180, IGHV1-27, C/T) was classified as non-GSV because fractions fN1 for position 180 in IGHV1-27 vary from 0.78 to 0.95 and did not fall below the frequency threshold.
In total, we classified 52 GSVs in seven V genes: IGHV1-7 (8 GSVs), IGHV1-10 (10), IGHV1-14 (3), IGHV1-17 (7), IGHV1-20 (8), IGHV1-21 (8), and IGHV1-27 (8). Seventeen out of 52 GSVs represent known germline variations described by Sinclair et al. (1997), Ma et al. (2016), and Rosen et al. (2020) (Supplemental Fig. S2).
Generating the genotype matrix
For each GSV (P, G, N1/N2) in each animal, IgQTL computes the R-ratio as R = fN1/(fN1 + fN2). The R-ratio represents a more flexible and expressive alternative to the conventional binary description of SNP states (e.g., AA or AC) because it enables description of SHMs and their relative abundances. The R-ratios also distinguish subjects that are heterozygous by the same pair of alleles but have different expression profiles for these alleles (e.g., 20%–80% vs. 50%–50%).
We refer to a 52-mer vector of all R-ratios for a given animal (across all GSVs) as its IGenotype. We further analyzed IGenotypes across all 204 animals for finding their correlations with various phenotypes. The IGenotypes of 204 animals across 52 GSVs form a 52 × 204 IGenotype matrix, an analog of a genotype matrix that describes both genomic SNPs and SHMs (Fig. 4A).
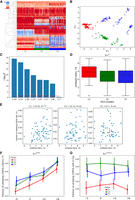
GSVs of V genes are associated with gene usages and antibody titers. (A) The 52 × 204 IGenotype matrix for 52 GSVs of V genes across 204 calves. Rows represent GSVs, columns represent animals. Rows are ordered using the hierarchical clustering, and columns are ordered according to the three clusters found using PCA, followed by k-means clustering. Three clusters C1, C2, and C3 are shown in red, green, and blue in the lower horizontal panel, respectively. The order of animals within a cluster is chosen arbitrarily. R-ratios vary from 0 (blue) to 1 (red). (B) Principal components 1 and 2 of the IGenotype matrix shown in A. Three identified clusters are shown in red, green, and blue. (C) Likelihoods of association P-values between three PCA clusters and usages of V genes. Usages are computed in the combined data sets. Likelihood is computed as the negative logarithm of the P-value to the base of 10. Genes are shown in the descending order of likelihoods. The red line corresponds to P = 0.05. (D) Antibody titers at time point “+6” for three PCA clusters. (E) Antibody titers at time point “+6” versus fractions of ultralong CDR3s in all CDR3s at time point “+3” across clusters C1–C3. The Pearson's correlations (r) and P-values (P) are shown at the top of the panel. (F) Fractions of ultralong CDR3s among all CDR3s derived from IGHV1-7 in clusters C1, C2, and C3 at four time points. (G) Fractions of ultralong CDR3s with six cysteines among all ultralong CDR3s in clusters C1, C2, and C3 at four time points. Vertical lines in F and G show 95% confidence intervals.
Clustering animals with similar IGenotypes
We clustered animals into groups with similar IGenotypes (these groups represent analogs of common genotypes) by applying the principal component analysis (PCA) to the IGenotype matrix. Iterative k-means clustering of the first two principal components with k from 2 to 10 followed by the elbow method (Thorndike 1953) reveals that k = 3 provides the optimal decomposition of 204 animals (Fig. 4B; Supplemental Fig. S3). Although decompositions into more clusters resulted in similar values of inertia (Supplemental Fig. S3), we focused our analysis on k = 3 because it simplified further statistical analysis and allowed us to apply popular statistical methods such as the Kruskal–Wallis test (Kruskal and Wallis 1952).
We say that the computed clusters are associated with the R-ratios (or usages/titers/fractions of ultralong CDR3s) if the differences between distributions of R-ratios (or usages/titers/fractions of ultralong CDR3s) across these clusters are statistically significant. The computed clusters C1, C2, and C3 are associated with the R-ratios for 47 out of 52 GSVs, including 16 out of 17 known germline variations (Supplemental Fig. S4). We thus conclude that the decomposition of 204 calves into clusters C1–C3 is driven by multiple linked GSVs (GSVs with correlated R-ratios) that represent common genotypes of V genes.
To better understand the nature of 47 GSVs associated with clusters C1–C3, we applied IgQTL to sequences from the time point “–3” only. We detected 46 out of 47 previously detected GSVs, except for a variation at position 288 in IGHV1-7 that likely represents a frequent somatic hypermutation. We assume that this observation indicates that GSVs associated with clusters C1–C3 are largely driven by genomic variations and pre-existing immunity rather than BRD vaccinations.
GSVs explain variance in usages of V genes and antibody titers
Clusters C1–C3 are associated with usages of all highly used V genes except for IGHV1-7 (Fig. 4C; Supplemental Fig. S5). Figure 4D shows that the clusters are also associated with antibody titers collected at time point “+6”: cluster C1 has higher antibody titers compared with clusters C2 and C3, with P = 0.017 according to the Kruskal–Wallis test. This observation suggests that IGenotypes of V genes are associated with the response to the BRD vaccination. Antibody titers collected at three other time points do not have statistically significant associations with clusters C1–C3.
Because the fractions of ultralong antibodies are not associated with clusters C1–C3 (Supplemental Fig. S6), we hypothesize that generation of ultralong antibodies is not specific to genotypes described by the revealed clusters but rather is a general feature of cattle antibody repertoires. However, our analysis revealed subtle correlations between genotypes and some features of ultralong CDR3s. Figure 4E shows that clusters C1–C3 partially explain the variance in Figure 2D: The fraction of ultralong CDR3s among all CDR3s at the time point “+3” positively correlates with titers at the time point “+6” only for animals from cluster C1 (r = 0.32, P = 0.0072). Similar correlations do not exist and are not statistically significant for clusters C2 and C3 (r = 0.12 and r = 0.10, respectively). We thus assume that ultralong CDR3s from the cluster C1 work better in response to the BRD vaccine.
Figure 4, F and G, shows that animals from cluster C1 are characterized by a lower initial fraction of ultralong CDR3s (in all CDR3s derived from IGHV1-7) and a lower fraction of ultralong CDR3s with six cysteines (important for knob formation) as compared with animals from clusters C2 and C3 (P-values for the cluster variable are 3.96 × 10–4 and 2.63 × 10–5, respectively). Because the initial number of cysteines in ultralong CDR3s is four (Wang et al. 2013), a higher number of cysteines suggests that ultralong CDR3s of animals from clusters C2 and C3 underwent more extensive affinity maturation before the BRD vaccination compared with animals from cluster C1. Because cluster C1 is associated with higher titers after the second BRD vaccination, we extend the hypothesis about pre-existing immunity and suggest that it might partially consist of mature ultralong CDR3s (with six cysteines) generated before the vaccinations in animals from clusters C2 and C3. However, because titers at time points “–3” and “+6” are anticorrelated in all clusters (Supplemental Fig. S7), we also suggest that mature ultralong antibodies might not be the only component of the pre-existing immunity. Further exploration of cattle antibody repertoires would help to understand the origin of the pre-existing immunity (e.g., maternal antibodies or microbiota) and its impact on the BRD vaccination.
A GSV at position 148 in IGHV1-7 is associated with the fraction of ultralong CDR3s
The fraction of ultralong CDR3s among all antibodies varies between 0.0033 and 0.0543 in the combined data sets (Supplemental Fig. S8A). The fraction of ultralong CDR3s limited to CDR3s derived from IGHV1-7 varies from 0.09 to 0.64 in the combined data sets (Supplemental Fig. S8B). Because clusters C1–C3 are not associated with fractions of ultralong CDR3 antibodies (Supplemental Fig. S6), we also examined potential associations of individual GSVs with fractions of ultralong CDR3s.
The GSV (148, IGHV1-7, A/G) has the most significant association with the fraction of ultralong CDR3s whether computed with all antibodies or limited to IGHV1-7 containing antibodies: P = 3.03 × 10–28 and P = 1.49 × 10–47, respectively (Fig. 5A,B). P-values were computed using the linear regression model. The GSV with the next most significant association was GSV (144, IGHV1-17, C/T), but this significance was many orders of magnitude lower (P = 0.0016) (Supplemental Fig. S9A). The closest IGHV1-7 containing GSV in terms of significance was GSV (71, IGHV1-7, C/T), which also has an association that is orders of magnitude lower (P = 0.0002) (Supplemental Fig. S9B). Thus, GSV (148, IGHV1-7, A/G) is unique because its association P-values are many orders of magnitude lower than association P-values of all other GSVs.
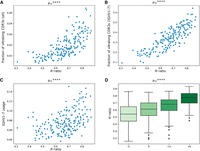
GSV at position 148 of IGHV1-7 is associated with production of ultralong antibodies. (A) Fractions of ultralong CDR3s in all CDR3s versus R-ratios of the GSV G148A in IGHV1-7. (B) Fractions of ultralong CDR3s in all CDR3s derived from IGHV1-7 versus R-ratios of the GSV G148A in IGHV1-7. (C) Usages of IGHV1-7 in the combined data set versus R-ratios of the GSV G148A at position 148 in IGHV1-7. (D) Distributions of R-ratios for position 148 in IGHV1-7 at four time points.
This GSV (that we refer to as G148A for brevity) also has the most significant association with the usage of IGHV1-7 in the combined data set (P = 6.11 × 10–17, the linear regression model): the higher the fraction of nucleotide A at position 148 of IGHV1-7, the higher the usage of IGHV1-7 (Fig. 5C). Figure 5D shows that R-ratios of the GSV G148A grow after both vaccinations (P = 3.36 × 10–204). This position was not previously identified as a germline variation (Fig. 3B); the known alleles of IGHV1-7 have a nucleotide G at position 148 classified as the second popular nucleotide N2 by our analysis (Supplemental Table S2). The R-ratios of GSV G148A are not associated with clusters C1–C3, suggesting that this GSV is not linked with 47 GSVs for which R-ratios are associated with clusters C1–C3 (Supplemental Fig. S4). The R-ratios for this GSV vary from 0.31 to 0.87, indicating that both nucleotides A and G are always present in Rep-Seq reads in each animal (Supplemental Fig. S10A). Supplemental Figure S10B also shows that R-ratios of the GSV grow similarly for clusters C1–C3. We thus assume that GSV G148A is a frequent SHM that is often selected after vaccinations and is important for generating ultralong antibodies.
The role of the GSV G148A in ultralong CDR3s
The most abundant nucleotide N1 = A of the GSV G148A replaces the germline amino acid Gly (encoded by the codon GGT) with the amino acid Ser (encoded by codon AGT) at amino acid position 50. To analyze frequencies of amino acids Gly and Ser at this position, we translated antibody sequences derived from IGHV1-7 into amino acids. The amino acid position 50 represents the last amino acid of the second framework region (FR2) according to the IMGT notation (Lefranc et al. 2003) but is classified as a CDR2 position according to Kabat et al. (1979) and Paratome (Kunik et al. 2012) notations. Wang et al. (2013) showed that, unlike Gly at position 50 (referred to as Gly50), Ser at position 50 (referred to as Ser50) can form hydrogen bonds with the conserved Gln at position 97 in the stalk part of an ultralong CDR3. On average, 24.8% and 3.8% of ultralong antibodies derived from IGHV1-7 in the combined data sets contain Ser50 and Gly50, respectively (Fig. 6A). In contrast to ultralong antibodies, where Ser50 is six times more frequent than Gly50, Ser50 and Gly50 appear in similar proportions (30.4% and 24.8%, respectively) in nonultralong CDR3s derived from IGHV1-7 (Fig. 6A).
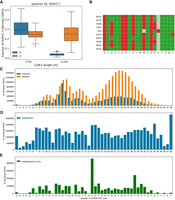
The anatomy of ultralong CDR3s. (A) Fractions of nonultralong CDR3s (lengths <150 nt) and ultralong CDR3s derived from IGHV1-7 with amino acids Gly (blue) and Ser (orange) at amino acid position 50. Fractions are computed in the combined data sets. (B) Impact of substitutions at position 50 in 10 crystallized antibodies predicted by the I-Mutant2.0 tool. Accession IDs of antibody structures are shown on the left. Gray cells show amino acids at position 50 present in structures (N or S). A green (red) cell (Ab, AA) indicates that mutation to amino acid AA is predicted to increase (decrease) stability of antibody structure Ab. (C–E) IGHD8-2 is a template for generating cysteines through SHMs. The germline sequence of IGHD8-2*01 is shown at the bottom with four cysteines shown in red. (C) The bar plot shows the number of ultralong CDR3s that have insertions (blue) and deletions (orange) at a given position of IGHD8-2*01. The position of insertion is defined as the position in the germline sequence that precedes the insertion. The average lengths of insertions and deletions in IGHD8-2 are 1.6 and 2.4 aa, respectively. (D) The bar plot shows the number of ultralong CDR3s that have a substitution at a given position of IGHD8-2*01. (E) The bar plot shows the number of ultralong CDR3s that have a substitution into cysteine at a given position of IGHD8-2*01.
We also analyzed 13 3D structures of crystallized bovine antibodies (reported by Wang et al. 2013; Stanfield et al. 2016; Dong et al. 2019) available in the Protein Data Bank (Berman et al. 2000). None of the known ultralong antibodies have the germline Gly at position 50: All but one of them have Ser at position 50 (Supplemental Fig. S11). We further applied the I-Mutant2.0 tool (Capriotti et al. 2005) to analyze the effect of substitutions at this position on the stability of the analyzed antibodies. I-Mutant2.0 generated prediction for only 10 out of 13 analyzed antibodies (three structures were processed with errors), and it turned out that substitution of Ser by the germline amino acid Gly decreases antibody stability for all 10 of them (Fig. 6B). We thus assume that amino acid Ser at position 50 is critically important for maintaining the structure of ultralong antibodies.
Notable features of ultralong CDR3s
All ultralong CDR3s are generated through recombination of a 148-nt-long D gene IGHD8-2 that encodes four cysteines in its third open reading frame and contains 39 codons (in the same frame) differing from the cysteine-encoding codons by a single nucleotide, providing multiple opportunities for generating novel disulfide bonds in an ultralong antibody by somatic mutations of noncysteines into cysteine. During VDJ recombination, all short IGHD genes undergo intensive exonuclease removals that contribute to the overall diversity of an antibody repertoire (Ralph and Matsen 2016). To understand the recombination properties of the long IGHD8-2 gene, we collected 2,855,428 distinct amino acid sequences of ultralong CDR3s across all individuals, aligned them to the IGHD8-2*01 gene, and identified positions corresponding to substitutions, insertions, and deletions. In contrast to the short D genes, the first six and the last three amino acid positions of IGHD8-2 do not accumulate insertions and deletions (Fig. 6C) and only the first and last positions undergo substantial numbers of substitutions (Fig. 6D). Therefore, in contrast to the short D genes, IGHD8-2 does not undergo extensive truncations from both sides.
The distribution of observed indels in the IGHD8-2*01 segment shows an uneven distribution throughout the middle portion, between positions 6 and 45 (Fig. 6C). Relatively increased numbers of indels can be detected in the region between positions 14 and 17 and downstream from position 25 to 40, with deletions much more common than insertions in the latter. We note that the positions prone to indels do not include those encoding four germline cysteines (positions 2, 12, 18, and 23). Furthermore, the germline cysteine codons accumulate about five times fewer substitutions compared with other positions of IGHD8-2 (Fig. 6D). Thus, most ultralong CDR3s preserve germline cysteines of IGHD8-2. The introduction of additional cysteines in the ultralong CDR3 by SHMs are most commonly the result of substitutions at position 24 (about four times more common than the average at other positions; Fig. 6E).
Discussion
Longitudinal study of cattle antibody repertoires developed in response to the BRD vaccine
We conducted a personalized immunogenomics study of 204 calves to analyze the efficacy of the BRD vaccine. Our analysis showed that the BRD vaccinations increase both the usage of IGHV1-7 and the fraction of ultralong antibodies, suggesting that ultralong antibodies play an important role in immune responses against antigens of the BRD vaccine. It also showed that antibody titers measured after the booster vaccination are weakly correlated with the usage of IGHV1-7 and the fraction of ultralong antibodies. Usages of other cattle IGHV genes are not associated with antibody titers before and after the BRD vaccination. We also showed that antibody titers before the initial vaccination anticorrelate with titers after the booster vaccination. This suggests that pre-existing immunity to BRD may prevent successful development of the immune response to the BRD vaccine.
The IgQTL analysis of antibody repertoires
Although the analysis of eQTLs in genomic studies is well developed, there are still no tools for analyzing IgQTLs in immunogenomics data sets. We developed an IgQTL tool for detecting important germline and somatic variations or GSVs (based on analyzing Rep-Seq data), applied it to identify GSVs in cattle IGHV genes, and found their associations with various phenotypes (gene usages, fractions of ultralong antibodies, and antibody titers). Our analysis demonstrates that IgQTL can be used for analyzing antigen-specific antibody responses in a population. Although it has only been tested on cattle Rep-Seq data sets, it can be applied to any vertebrate species, including humans, and thus improve our understanding of the specifics of adaptive immune responses associated with various antigens.
SHM G148A in IGHV1-7 is important for generating ultralong antibodies
Analysis of the identified GSVs revealed that a GSV G148A in IGHV1-7 is strongly associated with both the usage of IGHV1-7 and the fraction of ultralong antibodies. This GSV results in a substitution of the germline amino acid Gly into Ser that is specific to ultralong antibodies. Whereas nonultralong antibodies derived from IGHV1-7 have similar fractions of Gly and Ser, the ultralong antibodies have a highly elevated fraction of Ser as compared with the fraction of Gly. Wang et al. (2013) showed that Ser encoded by this GSV forms a hydrogen bond with the conservative Gln at position 95 in the stalk region of an ultralong CDR3. We thus assume that the GSV G148A is not specific to responses induced by the BRD vaccine but rather is a general feature of ultralong antibodies. Different patterns of the GSV G148A in IGHV1-7 in ultralong and nonultralong antibodies suggest that it represents a frequent SHM rather than a novel germline variation. Further investigation of the origin and the role of this GSV will likely require paired WGS and Rep-Seq data sets, as well as analyzing the 3D structures of ultralong antibodies.
Germline variations and SHMs explain variance in titers
Further analysis of GSVs revealed three clusters (C1, C2, and C3) representing common genotypes of IGHV genes. The detected clusters are associated with usages of all highly used IGHV genes except for IGHV1-7. Cluster C1 is associated with higher titers after the booster vaccination and a higher correlation between the final titers and the fraction ultralong CDR3s compared with clusters C2 and C3. Cluster C1 is also characterized by a significantly lower fraction of ultralong CDR3s before the vaccination and a lower fraction of ultralong CDR3s with six cysteines. Because the initial number of cysteines in ultralong CDR3s is four (Wang et al. 2013), a higher number of cysteines might indicate that ultralong CDR3s of animals from clusters C2 and C3 underwent more extensive affinity maturation compared with animals from cluster C1. One possible explanation suggests that the pre-existing immunity in animals from clusters C2 and C3 partially consists of “mature” ultralong CDR3s (with six cysteines) detected before the vaccinations. However, further exploration of cattle antibody repertoires is needed for understanding the origin of the pre-existing immunity, its impact on the BRD vaccination, and associations with its components. Also, similarly to the G148A variation, further investigation of the origin of GSVs associated with clusters C1–C3 will require paired WGS and Rep-Seq data sets. Such data sets will also help to identify additional types of genomic variations (e.g., copy number variations of V genes, structural variations of IGH locus, mutations in RSSs) that can shape expressed antibody repertoires (Sasso et al. 1996; Avnir et al. 2016) but are difficult to detect using Rep-Seq data alone.
Limitations of the study
The study describes a large-scale vaccination effort against the BRD caused by multiple pathogens. The live attenuated virus vaccine used in the experiment contains antigens against four distinct pathogens, which might complicate downstream analysis in conventionally reared animals exposed to numerous antigen challenges. Although we made sure that the analyzed calves were vaccinated in the same environment and that their ages are not associated with the vaccine outcomes, we still cannot completely rule out the impact of multiple pathogens and pre-existing immunity on the variance in antibody responses.
Further analysis of antibody responses to BRD
The GSVs detected by IgQTL might be used as the first markers for further exploration of efficient responses to the BRD vaccine and identification of animals that can benefit from the vaccine most. Further studies examining GSVs and their relationship with developing antibody repertoires in response to vaccination have a potential to identify animals with successful responses to the BRD vaccine and thus contribute to the ongoing selection strategies by including not only genomic but also immunogenomic traits.
Our study has revealed that ultralong antibodies play an important role in the antibody response against BRD antigens. Although our study has already combined experimental (Rep-Seq) and computational approaches, these approaches would further benefit from functional experiments aimed at identification of ultralong antibodies that bind BRD antigens. Such experiments (and further antibody engineering analysis) represent the topic of a follow-up paper.
Methods
Sample preparation
The Iowa State University Animal Care and Use Committee (IACUC) approved all animal work before the study was conducted. Purebred American Angus calves (n = 204) were vaccinated with a modified live vaccine (Bovi-Shield Gold 5; Zoetis, Inc.) containing antigens of four viruses associated with bovine respiratory disease as described in Kramer et al. (2017). A second booster vaccination was applied 3 wk later. Bovine whole blood was collected and stored in Tempus blood RNA tubes (Applied Biosystems). Collections occurred at four time points; 3 wk prior to vaccination, at vaccination, 3 wk postvaccination (at booster vaccination), and 3 wk after booster vaccination. RNA was isolated using the Tempus spin RNA isolation kit as recommended by the manufacturer (Applied Biosystems).
Repertoire sequencing
The RNA was converted to cDNA using the Clontech SMARTer kit (Takara Bio USA) as directed by the manufacturer, with minor modification and oligonucleotides listed in Supplemental Table S3. Double-strand cDNA synthesis was performed using the SMARTer IIA oligonucleotide that comes with the kit and anneals to the poly(A) tail of mRNA by mixing 2 µg of RNA in 11-µL volume with 1 µL of 12 µM SMARTer IIA Oligonucleotide and annealing for 30 min at 72°C and then for 2 min at 42°C, followed by addition of 9 µL of a mixture of IGHG-specific primer 87747, 5× first-strand buffer, 0.1 M DTT, 10 mM dNTP mix, Recombinant RNasin Ribonuclease inhibitor (Promega), and SuperScript II reverse transcriptase (Invitrogen). The first-strand synthesis was not targeted to IGHG transcripts by the poly(T) primer, but second-strand synthesis was primed with a targeted oligonucleotide that anneals to the 5′ leader sequence of cattle IGHG first-strand cDNA, providing initial selection for target transcripts in the double-strand cDNA products (Supplemental Fig. S12).
The target region of IGHG transcripts spanning the regions FR1–4 and CDR1–3 was amplified from the cDNA as previously described (Larsen and Smith 2012). Briefly, primers targeting the 3′ end of the leader region (primer 87934) (Supplemental Table S3) and 5′ end of the CH1 constant region (primer 87935) (Supplemental Table S3) were used to amplify the cDNA (Supplemental Fig. S12). These primers were designed to include adapter sequences that directly prepared the amplification products for sequencing on Illumina platform instruments (Supplemental Fig. S12). PCR amplification was performed using AccuPrime Tag DNA polymerase high-fidelity enzyme (Invitrogen) with initial denaturation of the cDNA for 2 min at 94°C, followed by 33 cycles of 15 sec at 94°C, 15 sec at 64°C, and 1 min at 68°C. A final extension for 5 min at 68°C was included after all cycles completed. Amplified DNA was purified using AMPure XP beads as recommended by the manufacturer (Beckman-Coulter). Library concentration was determined by quantitative PCR using a NEBNext Library Quant kit for Illumina (New England Biolabs). Amplicon fragment size distribution was determined using a Fragment Analyzer System (Agilent Technologies). Sequencing was performed on a MiSeq instrument using 600 cycle v3 Reagent kit (Illumina, Inc.) with 2 × 300 base paired-end reads.
Antibody titers
Viral neutralization assays were performed against four viruses: BVDV1, BVDV2, BRSV, and BHV1. Serum samples from each animal were tested for the four viruses, with five replicates for BVDV1 and BVDV2, three replicates for BVDV2, and two replicates for BRSV and BHV1 samples. Samples were diluted from 1:4 to 1:2048, with BRSV and BHV1 starting at 1:8 dilution. Neutralization antibodies were detected for each dilution and the greatest dilution was recorded as the log2 reciprocal for each calf sample using the Spearman–Kärber method (Finney 1978). An average across replicates was used for each calf to obtain a final antibody titer score, with a 0 being given if the first dilution showed a cytopathic effect.
Preprocessing Rep-Seq data
Each paired-end read was merged into a single sequence using the PairedReadMerger tool (Safonova et al. 2015). For each resulting sequence, the V gene, the J gene, and the CDR3 contributing to this sequence were inferred using the DiversityAnalyzer tool (Shlemov et al. 2017; https://immunotools.github.io/immunotools/) based on the cattle germline immunoglobulin genes listed in the IMGT database (Lefranc et al. 2009). To make sure that cattle V genes do not represent a special case of highly similar sequences so that DiversityAnalyzer can perform accurate V gene assignments, we computed alignments between all pairs of eight highly used cattle V genes and, for each V gene, found the closest V gene. The percent identities of the closest cattle V genes vary from 92% to 98%. In humans, 69 (24) out of 106 V genes have the closest V genes with the percent identity exceeding 92% (98%). Thus, we expect that accuracy of DiversityAnalyzer for cattle Rep-Seq data is comparable with human Rep-Seq data. To simplify the downstream analysis of gene variations, we kept only the first allele of each V gene and ignored its allele variants. The germline and somatic variations were computed using alignments against the closest V genes reported by the DiversityAnalyzer.
Statistical analysis
Statistical analysis was performed using Python (version 3.8.5). The Kruskal–Wallis test and the Pearson's correlation were computed using the SciPy package (version 1.6.0). The linear regression model and the linear mixed effect model were called from the statmodels package (version 0.12.2).
Data access
Sequencing data sets generated in this study have been submitted to the NCBI BioProject database (https://www.ncbi.nlm.nih.gov/bioproject) under accession number PRJNA607961. MiAIRR-compliant metadata, antibody titers, animal ages, IgQTL code, and results for 204 analyzed animals are available as Supplemental Materials, Supplemental Code, and at GitHub (https://github.com/yana-safonova/great_cattle_ab_repertoire).
Competing interest statement
The authors declare no competing interests.
Acknowledgments
We thank Vaughn Smider for fruitful discussions and thoughtful comments. We also thank Kristen Kuhn, Jacky Carnahan, and William Thompson for technical support. Mention of trade names or commercial products in this publication is solely for the purpose of providing specific information and does not imply recommendation or endorsement by the U.S. Department of Agriculture. USDA is an equal opportunity provider and employer. Y.S. is supported by the UCSD Data Science Fellowship 2017 and the American Association of Immunologists, AAI Intersect Fellowship 2019. S.B.S. and T.P.L.S. are supported by National Institute of Food and Agriculture (NIFA) Award No-2017-67011-26043 and USDA CRIS 3040-31000-100-00-D. C.T.W. is supported in part by the National Institutes of Health (NIH), National Institute of Allergy and Infectious Diseases award number R21AI142590. P.A.P. is supported by the NIH 2-P41-GM103484PP grant.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.276027.121.
- Received July 25, 2021.
- Accepted February 28, 2022.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see https://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








