
Chromosome-specific telomere lengths and the minimal functional telomere revealed by nanopore sequencing
- Samantha L. Sholes1,2,
- Kayarash Karimian1,2,
- Ariel Gershman1,2,
- Thomas J. Kelly3,
- Winston Timp1,4 and
- Carol W. Greider1,5
- 1Department of Molecular Biology and Genetics, Johns Hopkins University School of Medicine, Baltimore, Maryland 21205, USA;
- 2Biochemistry, Cellular and Molecular Biology Graduate Program, Johns Hopkins University School of Medicine, Baltimore, Maryland 21205, USA;
- 3Program in Molecular Biology, Sloan Kettering Institute, Memorial Sloan Kettering Cancer Center, New York, New York 10065, USA;
- 4Department of Biomedical Engineering, Johns Hopkins University School of Medicine, Baltimore, Maryland 21205, USA;
- 5Department of Molecular Cell and Developmental Biology, University of California, Santa Cruz, California 95064, USA
Abstract
We developed a method to tag telomeres and measure telomere length by nanopore sequencing in the yeast S. cerevisiae. Nanopore allows long-read sequencing through the telomere, through the subtelomere, and into unique chromosomal sequence, enabling assignment of telomere length to a specific chromosome end. We observed chromosome end–specific telomere lengths that were stable over 120 cell divisions. These stable chromosome-specific telomere lengths may be explained by slow clonal variation or may represent a new biological mechanism that maintains equilibrium unique to each chromosome end. We examined the role of RIF1 and TEL1 in telomere length regulation and found that TEL1 is epistatic to RIF1 at most telomeres, consistent with the literature. However, at telomeres that lack subtelomeric Y′ sequences, tel1Δ rif1Δ double mutants had a very small, but significant, increase in telomere length compared with the tel1Δ single mutant, suggesting an influence of Y′ elements on telomere length regulation. We sequenced telomeres in a telomerase-null mutant (est2Δ) and found the minimal telomere length to be ∼75 bp. In these est2Δ mutants, there were apparent telomere recombination events at individual telomeres before the generation of survivors, and these events were significantly reduced in est2Δ rad52Δ double mutants. The rate of telomere shortening in the absence of telomerase was similar across all chromosome ends at ∼5 bp per generation. This new method gives quantitative, high-resolution telomere length measurement at each individual chromosome end and suggests possible new biological mechanisms regulating telomere length.
Maintenance of the telomere length equilibrium is critical for cell survival. Telomeres shorten with each cell division, whereas a few are stochastically elongated by the enzyme telomerase (Greider and Blackburn 1985; Teixeira et al. 2004). This balance of shortening and lengthening is exquisitely regulated to generate a normal distribution of lengths that is established around a mean or “set point.” Different species have very different telomere length set points; for example, Oxytricha has 20–30 bp, yeasts have 300 bp, humans have 10 kb, and some mouse strains have up to 80 kb mean telomere lengths (Greider 1996). How this distribution is established and maintained is not fully understood. When telomerase levels are low or absent, the distribution shifts toward shorter lengths, and the shortest telomeres signal a DNA damage response. These cells undergo a permanent cell cycle arrest, termed senescence or apoptosis, depending on the cell type (Enomoto et al. 2002; d'Adda di Fagagna et al. 2003; IJpma and Greider 2003).
Loss of telomere length maintenance in humans leads to disease. Individuals with inherited mutations in telomerase, or other telomere regulatory genes, have short telomere syndrome, which includes bone marrow failure, immunodeficiency, pulmonary fibrosis, and other diseases (Armanios 2013). In contrast, activation of telomerase (Counter et al. 1992; Greider 1998) or inheritance of long telomeres allows cell immortalization, which can predispose to cancer (Stanley and Armanios 2015; McNally et al. 2019). Thus, deviation from the mean telomere length in either direction affects cellular lifespan and plays a critical role in disease.
The yeast Saccharomyces cerevisiae serves as an excellent model for probing mechanisms of telomere length regulation. Many genes and regulatory mechanisms that affect human telomere length were first discovered in yeast (Wellinger and Zakian 2012). Yeast telomere sequences are more variable than the consistent TTAGGG repeats found in vertebrates (Moyzis et al. 1988). They are composed of a mixture of GT, GGT, or GGGT sequence motifs, usually abbreviated, G1-3T. The subtelomeric region in yeast contains specific families of repeated DNA.
Deletion of any component of telomerase in yeast leads to progressive telomere shortening, termed ever-shorter telomere (EST) phenotype (Lundblad and Szostak 1989; Lendvay et al. 1996). Previous work suggests that telomeres shorten by ∼3–5 bp per cell division in the absence of telomerase and that this value is constant and independent of telomere length (Marcand et al. 1999; Wellinger and Zakian 2012; Xu et al. 2013). There is initially no effect of telomere shortening on cell growth and survival, but when telomeres become very short after passaging in culture for several days, most cells arrest at G2/M (Enomoto et al. 2002; IJpma and Greider 2003). A few cells can rescue their short telomeres through recombination and will generate “survivors” that grow in the absence of telomerase (Lundblad and Blackburn 1993). Survivors are generated through a break-induced replication (BIR) pathway (Bosco and Haber 1998; McEachern and Haber 2006) that can initiate within the telomere repeats (Type II survivors) or in the subtelomere repeated elements (Type I survivors) (Teng and Zakian 1999). Deletion of the recombination gene RAD52 significantly reduces production of both types of survivors (Lundblad and Blackburn 1993), although very rare survivors have been found in est2Δ rad52Δ double mutants (Lebel et al. 2009).
The normal distribution of the telomere length equilibrium is due primarily to sequence loss through replication and sequence addition by telomerase. Shampay and Blackburn (1988) showed that independent chromosome ends can slightly shift their mean length owing to clonal variability. If a cell that happens to have a short telomere at Chromosome 8L, for example, seeds a new colony on a plate, the descendants of that cell will initially have a new, shorter telomere length distribution until enough divisions occur in the population to homogenize all ends toward a population mean (Shampay and Blackburn 1988).
The “protein counting” model for telomere length regulation established in S. cerevisiae (Marcand et al. 1997, 1999) proposes that proteins bound to the telomeric repeats negatively regulate telomere elongation by telomerase, so that the probability of telomere elongation decreases with telomere length. Mean telomere length is stable when the average rate of length-dependent elongation is equal to the average rate of length-independent shortening. (Bianchi and Shore 2008). This model has received significant attention and is now the accepted model for telomere length regulation in organisms as distantly related as mammals, plants, and fission yeast (Smogorzewska and de Lange 2004; Watson and Riha 2010; Wellinger and Zakian 2012; Armstrong and Tomita 2017; Maciejowski and de Lange 2017). Sequence analysis of individual telomeres showed that telomerase added between 3 bp and 179 bp (average, 44 bp) onto chromosome ends in one cell cycle; however, only a few telomeres were elongated at each cycle. Further, shorter telomere repeat tracts were preferentially elongated compared with those with longer repeat tracts (Marcand et al. 1999; Teixeira et al. 2004). This work further supported the protein counting model by providing evidence that telomerase preferentially elongates short telomere repeat tracts. How preferential elongation of short telomeres allows the robust maintenance of the telomere length equilibrium over many cell divisions is not yet clear.
The standard technique for visualizing telomere length in yeast is by Southern blot. Genomic DNA from yeast cells is cut with a restriction enzyme near the telomere and hybridized with a probe to the telomere repeats or to the subtelomere, generating smeared bands visible on the autoradiogram (Shampay and Blackburn 1988). The smears represent the heterogenous distribution of telomere lengths across both the different chromosomes and between cells. Because restriction sites on different chromosomes are at different locations, the absolute size of each smear differs. Frequently, the restriction enzyme XhoI is used to cut genomic DNA because many telomeres have a conserved XhoI restriction site in the Y′ subtelomere element. Multiple telomeres are then visualized in a heterogeneous band centered at ∼1.2 kb. Measuring median length of this 1.2-kb-sized band can allow large changes in bulk telomere length to be measured in various mutant backgrounds, but it is not possible to deconvolute the length of individual telomeres from each other.
To provide a more quantitative analysis of telomere length variation, we developed a nanopore sequencing method to measure telomere length. Nanopore sequencing allowed us to capture a full telomere sequence as well as the subtelomere and unique chromosomal sequence in one long read. Using this quantitative method for measuring telomere length, we re-evaluated fundamental questions about telomere length distribution and changes in mutant backgrounds that alter telomere length.
Results
Telomere tagging and bioinformatic analysis
To accurately measure telomere length and retain the telomeric 3′ overhangs, we modified a method (Teixeira et al. 2004) to tag the molecular end of the chromosome. We prepared high-molecular-weight (HMW) DNA (Denis et al. 2018) and added poly(A) to the 3′ ends with terminal transferase. We next annealed an oligo(dT) primer that also contained a unique adapter sequence, termed TeloTag. The addition of Sulfolobus DNA polymerase IV, which lacks strand displacement and exonuclease activity, was used to generate a double-stranded blunt end, as well as T4 DNA ligase to seal the nicks. Nanopore library adaptors were added to allow sequencing (Fig. 1A).
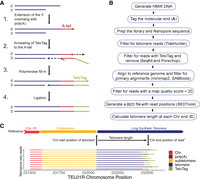
Telomere length measurement by nanopore sequencing. (A) Schematic of method to tag the molecular end of the chromosome. (1) Terminal transferase adds poly(A) sequence to the 3′ overhang of the telomere. (2) A complementary oligo(dT) sequence on the TeloTag oligonucleotide end containing a unique TeloTag sequence is annealed to the poly(A) sequence. (3) Polymerase fill-in generates double-stranded blunt ends. (4) Ligase is used to seal the nick. Nanopore adaptors are then added using the manufacturer's protocol. (B) Bioinformatic pipeline to generate telomere length information (see Methods). (C) Telomere length is calculated using the chromosomal position of each telomere sequence start and calculating the distance to the TeloTag sequence at the chromosome end. A random sample of 10 sequence reads aligned at TEL01R is presented as an example.
Tagged genomic DNA was run on nanopore MinION or Flongle flowcells to generate whole-genome sequencing (WGS) data (Fig. 1B). For optimal use of a nanopore MinION flowcell, samples were multiplexed, and only reads with barcodes on both ends were retained. Raw reads with telomeric sequence were extracted using TideHunter (Gao et al. 2019). To ensure that reads had sequenced to the end of the telomere, we retained reads only if they contained the TeloTag. Adapter sequences were then bioinformatically removed using Porechop to ensure that they are not calculated as part of the telomere length (https://github.com/rrwick/Porechop). These filters were used on the raw reads before alignment. The resulting reads were aligned using minimap2 to a S. cerevisiae reference genome, sacCer3 (Foury et al. 1998). To ensure that the reads mapped only to the correct chromosome end, we removed all reads that aligned in multiple places in the genome using SAMtools to select only reads with a map quality score greater than 20 and to remove all secondary and supplemental alignments. Finally, the position where the subtelomere ends and telomere repeats begins was identified for each chromosome end, and the telomere sequence length was calculated using the aligned reads (see Methods) (Fig. 1C).
Telomere length by nanopore sequencing recapitulates length measurement by Southern blot
To test the accuracy of our nanopore method for telomere length measurement, we compared telomere fragment length analysis by nanopore and Southern blot. We first looked at strains with expected clear differences in telomere length profiles, wild type and rif1Δ. Deletion of the RIF1 gene results in very long telomeres on Southern blots (Hardy et al. 1992), allowing us to probe a greater distribution of telomere length profiles and test whether these length differences are captured with nanopore sequencing. We measured telomere fragment length after XhoI digest using a probe against the common Y′ subtelomere element; this will measure telomere lengths in bulk, probing 17 of 32 subtelomeres (Fig. 2A; Supplemental Fig. S1C). We ran all samples in duplicate on a Southern blot and included an additional probe to the unique sequence at CEN4 that runs at 1.4 kb as an internal molecular marker for densitometry plots (Fig. 2A; Supplemental Fig. S1C). We used densitometry scanning to quantify the signal on the Southern blot (see Methods).
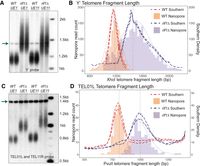
Telomere fragment length measured by nanopore sequencing accurately reflects length measurements by Southern blot. (A) Southern blot probed with Y′ subtelomeric probe and with CEN4 probe (arrow) as a marker at 1.4 kb to calibrate for densitometry. Two replicates of wild-type (WT) cells and two replicates of rif1Δ cells are shown for each strain, UE1 and UE11. (B) Comparison of densitometry (dotted lines) of Southern blot shown in A and nanopore sequencing (histogram fill bars) for UE1 strain. The light orange bars represent nanopore read counts for WT cells; the light purple bars represent nanopore read counts for rif1Δ cells. The spike in densitometry data at 1400 bp is owing to the CEN4 internal control signal. (C) Southern blot probed with the unique single telomere TEL01L probe and CEN4 probe (arrow). (D) Comparison of densitometry (dotted lines) of Southern blot in C and nanopore sequencing (bars) of UE1 TEL01L. The light orange bars represent nanopore read counts for WT cells; the light purple bars represent nanopore read counts for rif1Δ cells.
On the same samples, we performed our nanopore sequencing method as described (Methods) (Fig. 1). Nanopore sequencing data statistics are summarized in Supplemental Table S1. To directly compare to Southern blot telomere length, we included the internal chromosomal sequence up to the XhoI site for Y′ on the nanopore telomere read file. The histogram of the nanopore telomere sequence read lengths aligned well with the Southern blot densitometry for both wild-type telomeres and the much longer rif1Δ telomeres in two different strains (Fig. 2B; Supplemental Fig. S2A). The differences at the edges of the telomere length distribution between the nanopore and noisier Southern densitometry data highlight the increased ability to detect subpopulations in nanopore that are not detected by Southern blotting densitometry scans (Fig. 2D; Supplemental Fig. S2B). We used WALTER, a tool for Southern blot densitometry that generates summary length statistics (Lyčka et al. 2021), to compare Southern blot and nanopore telomere length profiles, and again saw comparable results (Supplemental Fig. S3A). We then generated Q–Q plots to examine the distribution of nanopore and Southern density data against a normal distribution (Supplemental Fig. S4A,B) and against each other (Supplemental Fig. S4C). The linearity of the Q–Q plots of both the Southern and nanopore data confirms that these profiles resemble a normal distribution (for details, see Supplemental Fig. S4), and we assumed a normal distribution for our statistical analysis throughout this work, including calculating correlation coefficients between data sets, performing t-tests, and performing analysis of the mean (ANOM) testing. The sample standard deviations of nanopore and Southern density distributions were similar, and there was no significant difference in the means by Welch's t-test (Supplemental Fig. S3A).
Because the Y′ probe measures 17 telomere length distributions simultaneously, it is challenging to detect potential small effects and chromosome-specific telomere lengths with this probe on a Southern blot. To assess the length distribution of single chromosome ends, we generated strains in which the subtelomere was replaced by a unique sequence. We used two different strains (Shubin et al. 2021) with unique sequence at either chromosome end TEL01L unique end 1 (strain UE1) or at chromosome end TEL11R unique end 11 (strain UE11) (Supplemental Fig. S1A,B). We examined the telomere lengths of these single TEL01L and TEL11R chromosome ends in both wild-type and rif1Δ backgrounds, using a probe to the unique ends (Fig. 2C; Supplemental Fig. S1D). Again, we saw good agreement between the single telomere length distributions determined by nanopore sequencing and Southern blotting (Fig. 2D; Supplemental Fig. 2B). In both genetic backgrounds, we observed no significant differences between the sample means (Supplemental Fig. S3B). We conclude that nanopore sequencing can accurately measure telomere fragment length in a bulk population or single telomere population across different telomere length profiles.
To determine the most effective flowcell type to achieve the minimum number of reads required for telomere length measurement using nanopore sequencing, we compared the MinION flowcells to the lower-cost, but lower-yield, Flongle flowcells. Multiplexing two samples per Flongle and MinION flowcell yielded many fewer reads from the Flongle (about 150–250 per barcode) versus the MinION (about 2000 reads per barcode), but we found no statistically significant differences between the mean and distributions of bulk telomere lengths across technical replicates and flowcell types (Supplemental Fig. S5). Thus, although the lower-cost Flongle flowcells yield reliable bulk telomere length data, the low number of reads does not allow analysis of individual chromosome ends (see below) and thus may not provide as useful a platform for investigating telomere biology.
Each chromosome end shows reproducible chromosome-specific telomere length distributions
Probing telomere lengths on unique chromosome ends is challenging with Southern blots, but nanopore sequence reads are long enough to span the subtelomeric elements and extend into unique chromosomal sequence. To examine individual chromosome ends, we sequenced three independent wild-type clones and three rif1Δ clones (Supplemental Fig. S6B). For the wild type, we used two independent clones of UE1 and one clone of UE11. Each clone was isolated by picking an independent single colony from a freshly streaked plate of cells (Supplemental Fig. 6B). We analyzed both the bulk telomere length of all telomeres and individual chromosome ends (Fig. 3A).
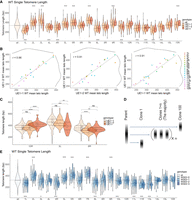
Nanopore sequencing mapping of the telomere length distribution of each individual chromosome end. (A) Bulk telomere length (all) and telomere length for each chromosome end were mapped by aligning to internal unique sequences. Three independent clones were sequenced: two from UE1 (UE1-1 and UE1-2) and one from UE11 (see Supplemental Fig. S6). Telomere lengths are reported as violin plots with a line at the mean. A dotted line shows the mean of the total population at 343.3 for WT cells. Analysis of the mean (ANOM) multiple contrast test of each telomere against the grand mean of all telomere length profiles was performed (see Supplemental Fig. S8A), and P-values adjusted by the Bonferroni method are reported as (*) <0.05, (**) <0.01, (***) <0.001, (****) <0.0001, or (n.s.) not significant. To note, TEL01L is engineered (as described in the text) in the UE1 strains, and TEL11R is engineered in the UE11 strains. (B) Pairwise correlations of mean telomere length across each of the three replicates for WT. A linear model was fit to the data (dotted line) to calculate the correlation constant, reported as the r-value. Marginal density of each data set is plotted as solid black lines across the top and right of the plots to show the spread of telomere lengths across each population. (C) Clonal variation was observed at TEL10R, TEL03L, and TEL08R in WT cells. Individual telomere length reads are represented as single points within the violin plots. The significance of the difference between two samples was tested using a multiple t-test (Methods) and P-values adjusted by the Bonferroni method are reported as above. (D) Model of clonal variation in which the telomere length of the majority of clones remains at the parental average, but occasional length outliers may seed new distributions represented by Clone 1 and Clone 100. (E) Three independent clones from the W303-0 parent were passages for 120 population doublings in independent cultures. The resulting clones W303-1, W303-2, and W303-3 were sequenced along with the parent W303-0 (see Supplemental Fig. S9A). Telomeres for each chromosome end were mapped by aligning to internal unique sequence. ANOM multiple contrast test of each telomere against the grand mean of all telomere length profiles was performed (see Supplemental Fig. S8B) and P-values adjusted by the Bonferroni method are reported as (*) <0.05, (**) <0.01, (***) <0.001, (****) <0.0001, or (n.s.) not significant.
We tested alignment to the S288C (Foury et al. 1998) as well as the W303 (Berlin et al. 2015; Matheson et al. 2017) reference genomes (Supplemental Fig. S7) and found the genome assembly in the subtelomeres was more complete for S288C and allowed reads to correctly map to more specific telomeres, so we used S288C as the reference. We were able to individually map 20 of 32 total chromosome ends with unique subtelomere and upstream genic sequences in our UE1 and UE11 strains. However, we were not able to map 12 chromosome ends uniquely (TEL01R, TEL02R, TEL03R, TEL06L, TEL07L, TEL09L, TEL12L, TEL12R, TEL13R, TEL14R, TEL16L, and TEL16R) owing to variation in subtelomeric structure from the reference genome. Subtelomeric elements are known to recombine and generate end diversity (Louis and Haber 1990, 1992; Maxwell et al. 2004), and we therefore excluded these 12 difficult to map telomeres in our analysis.
We were surprised to find that some chromosome ends showed telomere length distributions that were consistent across three independent clones but distinctly longer or shorter from the population mean of all telomeres (Fig. 3A). To quantify the differences, we applied ANOM (Pallmann and Hothorn 2016) to compare the mean length of individual chromosome ends to the population mean length of all the telomeres and found that eight chromosome ends (TEL04L, TEL04R, TEL08L, TEL10L, TEL11L, TEL11R, TEL13L, and TEL14L) had significantly shorter telomeres than the population mean, whereas six chromosome ends (TEL02L, TEL03L, TEL05L, TEL05R, TEL07R, and TEL09R) had significantly longer telomeres than the population mean (Supplemental Fig. S8A). A pairwise comparison of the three data sets suggests that telomere length of individual chromosome ends was consistent across replicates with correlation coefficients (r) of 0.96, 0.91, and 0.91 (Fig. 3B). We note that for chromosomes for which both ends could be analyzed, there was a trend toward concordance with both ends being long like TEL05L/5R and TEL015L/15R or with both ends being short like TEL04L/4R, TEL08L/8R, and TEL11L/11R, but there was no correlation between the length of chromosomes and the mean length of telomeres.
To determine whether chromosome end–specific lengths were maintained in the absence of Rif1, we analyzed two rif1Δ clones from UE1-1 and UE11 (Supplemental Fig. S6). Each clone showed telomeres whose mean lengths were significantly different from the clonal population mean (Supplemental Fig. S10A–C). However, in contrast to the wild-type cells, we did not see consistent length differences at individual chromosome ends in the two clones, evidenced by the correlation coefficient of 0.039 (Supplemental Fig. S10D). Because of the lack of correlation, we performed ANOM separately for the two clones and found significant deviations from the mean as expected from the broader length distribution seen in Figure 2 (Supplemental Fig. S10B,C). These results suggest that deletion of RIF1 may disrupt the mechanism responsible for maintenance of telomere-specific lengths or that the very broad length distribution obscures the differences between specific telomeres.
The apparent chromosome end–specific telomere lengths in wild-type cells could be due to stochastic clonal variation as previously proposed (Shampay and Blackburn 1988). That is, when a cell with a short telomere on Chromosome 4L forms a new colony, it will seed a new telomere distribution that will center around a shorter midpoint (Fig. 3D). We found a few cases of statistically significant variation among the three clones analyzed. In particular, TEL03L was significantly shorter in UE11 than in UE1-1 and UE1-2, and TEL08R was significantly longer in UE1-1 than in UE1-2 and UE11, and TEL10R was significantly longer in UE1-1 and UE1-2 than in UE11 (Fig. 3C). This rate of clonal variation was infrequent compared with the rate of clonal variation in the previous study (Shampay and Blackburn 1988), although this may be due to different number of cell divisions in propagating clones.
Shampay and Blackburn (1988) introduced the concept of the telomere length “set point,” in which all telomeres will gradually move back toward a population mean length distribution and suggested that differences in mean telomere length between clones might be erased during further propagation with all telomeres evolving toward a common mean length. Therefore, given enough cell divisions in the presence of telomerase, we might expect all chromosome ends to approach a mean value. In fact, Shampay and Blackburn (1988) showed that continued growth in liquid culture led to a broadening of the length distribution, which they interpreted as homogenization of all end lengths. The “protein counting model” for telomere length regulation (Marcand et al. 1997) suggests that telomere length is actively regulated around a mean based on the number of Rap1 proteins bound and, thus, the length of telomere sequence. These length regulation models are supported by the observation that telomerase apparently preferentially elongates short telomeres while ignoring long ones (Marcand et al. 1999; Teixeira et al. 2004). To test whether the telomere lengths would homogenize with more cell divisions, we isolated a clone of W303 cells (W303-0) and propagated it in liquid culture for 120 generations. Strain W303-0 is the parent of the clones UE1-1, UE1-2, and UE11 analyzed in Figure 3A (Supplemental Fig. S6A) and has a similar pattern of end-specific telomere lengths with some exceptions, such as that at TEL03L, which could be due to clonal variation. After 120 generations in liquid culture, three independent clones were isolated on plates and sequenced (W303-1, W303-2, and W303-3) (Supplemental Fig. S9A). We expected, based on previous models, that the telomere length distributions at individual chromosome ends would revert toward a grand population mean with more cell divisions. However, we saw that most chromosome-specific telomere lengths were maintained across all W303 clones, for example TEL02L, TEL04L, TEL04R, TEL05L, TEL05R, TEL07R, TEL08L, TEL08R, TEL09R, TEL10L, TEL10R, TEL11L, TEL11R, TEL13L, and TEL14L (Fig. 3E; Supplemental Fig. S8B). Although there were some examples of significant deviations from the initial length, for example, at TEL03L, TEL06R, TEL15L, and TEL15R, the major trend was reproducibility of the parents’ chromosome end–specific lengths (Supplemental Fig. S8A,B). To quantify the variation between different clones, we examined pairwise correlations between the W303-0 parent and the W303-1, W303-2, and W303-3 subclones and found correlation coefficients (r) of 0.92, 0.82, 0.89, 0.82, 0.86, and 0.75 (Supplemental Fig. S9B). Our data indicate that after 120 doublings, the chromosome end–specific telomere lengths are largely maintained. This implies a very slow rate of reversion to a common mean length distribution, if it occurs at all, and implies new models for telomere length regulation are needed to explain these data.
Y′ elements have a small effect on telomere length regulation by TEL1 and RIF1
The possibility of chromosome end–specific telomere lengths raised the idea that subtelomeric sequence might influence telomere length. A similar suggestion was made by Craven and Petes (1999), who proposed that the subtelomeric X and Y′ elements influenced the effects of TEL1 and RIF1 on telomere length. Specifically, they suggested that TEL1 is epistatic to RIF1 only at telomeres with Y′ elements but not at telomeres with only X elements. That is, telomeres are long in rif1Δ mutants, short in tel1Δ mutants, and also short in the tel1Δ rif1Δ double mutant at Y′ telomeres.
We reexamined this question using both Southern blots and nanopore sequencing. We generated tel1Δ, rif1Δ, and tel1Δ rif1Δ mutants in the in UE1 background (see Methods). As expected, in the Southern blot probed with Y′ sequence, rif1Δ mutants had long telomeres and tel1Δ mutants had short telomeres. The double mutant, tel1Δ rif1Δ, had short telomeres very similar to the single tel1Δ mutant (Fig. 4A), indicating that TEL1 is epistatic to RIF1 at telomeres with Y′ elements. Nanopore sequencing showed the same result at chromosomes with Y′ elements; tel1Δ rif11Δ double mutants had short telomeres indistinguishable from tel1Δ telomere lengths, supporting the previous finding (Fig. 4B). However, for chromosome ends that lacked Y′ elements and thus had only X elements, telomere length in tel11Δ rif11Δ was slightly longer (∼65 bp on average) and statistically different from tel1Δ (Fig. 4D). This trend also was seen at the unique chromosome TEL01L, where the subtelomeric sequence was removed entirely. Both nanopore sequencing (Fig. 4D, TEL01L) and Southern blot (Fig. 4C) showed that telomeres in the tel1Δ rif1Δ double mutant were longer than the tel1Δ alone. Although the difference in length was not as large as seen by Craven and Petes (1999), it is reproducible and suggests the need for further study.
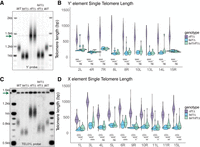
Y′ element subtelomeric sequences have a small effect on telomere length regulation by TEL1 and RIF1. (A) Southern blot of WT, tel1Δ, rif1Δ, and tel1Δ rif1Δ cells with bulk telomere length hybridized with the Y′ and CEN4 probes (arrow). (B) Individual telomere lengths determined by nanopore for telomere ends containing Y′ subtelomeric elements reported for tel1Δ, rif1Δ, and tel1Δ rif1Δ cells. (C) Southern blot of WT, tel1Δ, rif1Δ, and tel1Δ rif1Δ samples hybridized with unique probe to TEL01L and CEN4 probe (arrow) to examine unique TEL01L with no subtelomeric elements. (D) Individual telomere lengths determined by nanopore for telomere ends containing only X subtelomeric elements or no telomeric elements (TEL01L) reported for tel1Δ, rif1Δ, and tel1Δ rif1Δ cells. A multiple t-test was used to assess the significance of telomere length differences between genotypes, and P-values are reported as (*) <0.05, (**) <0.01, (***) <0.001, (****) <0.0001, or (n.s.) not significant.
Telomere shortening in the absence of telomerase reveals the minimal functional telomere
To examine the dynamics of telomere loss in cells without telomerase, we examined telomerase-null cells. We sporulated an EST2/est2Δ RAD52/rad52Δ heterozygous diploid and selected individual haploid colonies of est2Δ, est2Δ rad52Δ, and wild type (EST2). We grew these genotypes from a single colony and passaged each three times in liquid culture (Supplemental Fig. S11). To determine population doubling numbers, we estimated growth from a single cell to colony formation to be about 20 cell doublings. Both est2Δ and est2Δ rad52Δ cells grew slowly after 55 population doublings, and est2Δ rad52Δ cells showed significantly slower growth than est2Δ. Telomere shortening in the absence of telomerase leads to cell cycle arrest (Enomoto et al. 2002; IJpma and Greider 2003), as well as to telomeric elongation by BIR (Bosco and Haber 1998) that ultimately allows the outgrowth of “survivors” (Lundblad and Blackburn 1993). This implies there must be a competition between arrest and BIR, and presumably, all chromosomes must be elongated for phenotypic survivors to emerge.
The ability to measure the length distributions of individual telomeres using nanopore sequencing suggested that it might be possible to detect possible BIR events in the progeny of the est2Δ cells. Such events would not be visible by traditional Southern blotting of telomeres in bulk (Fig. 5A,B). In fact, we observed some telomere length distributions that were highly skewed, unlike any in the wild-type populations that we studied (see, e.g., TEL03L and TEL08R in Fig. 5C). To quantify this effect, we calculated the sample standard deviation of each telomere distribution in wild-type cells (P0; see Methods) and in est2Δ cells. For most telomeres we observed, as expected, that the sample standard deviation decreased relative to that of the parental wild-type telomere during passaging of the est2Δ cells. This observation is consistent with the fact that in the absence of Est2, telomeres progressively shorten, but their lengths are bounded by the minimum telomere length consistent with cell survival (see below). We found seven telomeres for which the sample standard deviation actually increased relative to that of the wild-type parent: TEL02R, TEL03L, TEL04L, TEL07R, TEL08R, TEL11R, and TEL15R (Fig. 5C). These telomere samples contain subpopulations with unexpectedly long telomeres, which we refer to below as outliers. Qualitative inspection of the distributions confirmed that these telomere distributions are highly skewed (Fig. 5C). Although we did not passage est2Δ cells for enough generations to observe outgrowth of survivors, our data suggest that telomere elongation events due to BIR may be occurring in these cells at an early stage, albeit at a relatively low frequency.
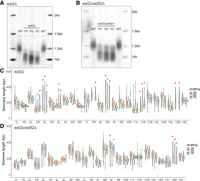
Telomere shortening and outlier elongation events in a telomerase-null, est2Δ background at individual telomeres over time. Freshly generated est2Δ and est2Δ rad52Δ haploids were grown up (P1, 35 doublings) and passaged twice (P2, 55 doublings; P3, 75 doublings) to allow for telomere shortening (see Methods). Telomere length was measured by Southern blot hybridized with the Y′ probe and by nanopore sequencing on the same samples. (A) Southern blot of WT, P1, P2, and P3 est2Δ hybridized with the Y′ probe. (B) Southern blot of WT, P1, P2, and P3 est2Δ rad52Δ hybridized with the Y′ probe. (C,D) Individual telomere length of est2Δ (C) and est2Δ rad52Δ (D) cells determined by nanopore sequencing of P1, P2, and P3. We calculated the sample standard deviations of (σ) of each telomere at every passage and in the wild-type parental strain. Telomeres for which the standard deviation was greater than that of the wild type are considered outliers and are marked with a red caret. At P2, 55 doublings, in the est2Δ sample, not enough data were collected at TEL07R and TEL15R to plot, and at P3, 75 doublings, in the est2Δ rad52Δ sample, not enough data were collected at TEL04L and TEL05L to plot.
To further examine the role of BIR in the generation of outlier telomere lengths, we examined the est2Δ rad52Δ cells, which are recombination deficient (Fig. 5B/D). If the long telomere outliers in est2Δ cells are owing to BIR, we would expect to see fewer of these outlier length increases in est2Δ rad52Δ cells. Indeed, there was a trend toward fewer outliers as we saw only five examples of outlier telomeres at TEL09L and TEL15R in the est2Δ rad52Δ cells (Fig. 5D). These length increases may be owing to rare events that occur even in est2Δ rad52Δ cells that allow telomere elongation (Lebel et al. 2009; Claussin and Chang 2016). Nanopore sequencing will allow detailed mechanistic insight into telomere elongation by BIR and the generation of survivors.
To determine the rate of telomere shortening in the absence of telomerase before cell division arrest, we compared the rate of shortening at individual chromosome ends in est2Δ and est2Δ rad52Δ cells over three outgrowths or passages (P1, P2, P3) (Supplemental Fig. S11). We exclude data where the telomere length increases likely owing to BIR over the 75 population doublings at TEL02R and TEL11L in est2Δ cells, as it would skew the analysis. To establish the initial rate of shortening, we need to establish the initial telomere length before EST2 deletion. Because this was not possible in the newly dissected haploid est2Δ strain, we instead used the wild-type telomere length for each chromosome as a proxy for the initial length and designated this P0 (Supplemental Fig. S11). The rate of shortening for each chromosome end was very similar from P0 to P1; there was a loss of 5.33 bp/population doubling in est2Δ and 5.13 bp/population doubling in est2Δ rad52Δ cells (Fig. 6A,B). This telomere shortening rate is similar to other estimates of 3-4 bp/population doubling (Marcand et al. 1999; Wellinger and Zakian 2012; Xu et al. 2013). If we include the later passages P2 and P3 in the rate, there is a continuous decline in the apparent rate because cells with very short telomeres do not divide and thus are lost from the calculated rate. To compare individual ends, we separately examined the rate of shortening from P1 to P3 and found all ends had a similar loss rate of 0.76 bp/population doubling in est2Δ cells and 0.39 bp/population doubling in est2Δ rad52Δ cells (Fig. 6A,B). The loss rates of the P1 to P3 est2Δ cells were higher than those of est2Δ rad52Δ cells on average perhaps owing to the more severe growth defects we observed in est2Δ rad52Δ compared with est2Δ cells. The rates of telomere shortening at each individual chromosome end were very similar in both est2Δ and est2Δ rad52Δ. Our quantitative single telomere data support previous analysis (Marcand et al. 1999) that telomere shortening rates are similar across chromosome ends.
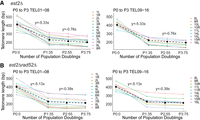
Telomere shortening rate in the absence of telomerase. (A) The progression of mean telomere length at individual chromosome ends in est2Δ cells over three passages. TEL01-08 and TEL09-16 are shown on separate graphs for clarity. The initial telomere length, P0, is estimated from the wild-type parental strain for each chromosome, and so it is not the exact genotype (see Results). The line of best fit to the average of the shortening rate is shown as a black dotted line. The rate of shortening for P0 to P1 and P1 to P3 are plotted separately because the two rates differ and each rate is reported above the line. (B) The rate of telomere shortening for the est2Δ rad52Δ strain was measured as described above in A.
To determine the minimum telomere length in telomerase-null cells, we examined telomeres at early passages before survivor events were predicted to occur (Lundblad and Blackburn 1993). We calculated the bottom 99th length percentile at each chromosome end and identified critically short telomeres in both the est2Δ and est2Δ rad52Δ ranging from 70 to 80 bp. The shortest three values are highlighted in each genotype at each passage (Table 1). This minimal telomere length is also consistent with the minimal length we found in telomerase-positive wild-type and tel1Δ cells of ∼75 bp (Table 1). Previous work has suggested that telomeres <100 bp could be considered “critically short” (Abdallah et al. 2009). Our sequence analysis now more precisely identified a minimum of ∼75 bp at individual chromosome ends.
The bottom 99th percentile of telomere length
Discussion
Nanopore telomere sequencing uncovered the unexpected finding of chromosome end–specific telomere length differences that were preserved across multiple independent clones over 120 population doublings. The stable maintenance of telomere-specific length differences, with mean values differing by as much as 200 bp, implies that current models for telomere length homeostasis require some revision (Marcand et al. 1997, 1999; Teixeira et al. 2004; Greider 2016). Both the protein counting and the replication fork models suggest that the average rate of telomere elongation by telomerase decreases with telomere length because of increased binding of inhibitory proteins to the telomeric repeats. In the context of this model, our new data indicate that the functional relationship between the number of bound proteins and the decrease in the average elongation rate is different for different telomeres. Thus, the balance point between length-dependent elongation and length-independent shortening occurs at different lengths for different telomeres. This finding implies the existence of as-yet-undiscovered telomere-specific influences on length homeostasis. It also suggests that the observed preferential elongation of short telomeres (Marcand et al. 1999; Teixeira et al. 2004) must be applied in a telomeric-specific sense, that is, preferential elongation of telomeres that are short relative to their specific equilibrium length. In addition, our new data indicate that the rate of shortening in the absence of telomerase is similar for all telomeres, confirming and extending previous studies (Marcand et al. 1999; Wellinger and Zakian 2012; Xu et al. 2013). It will be of interest to determine whether in the absence of telomerase, telomeres with a shorter equilibrium length may have a higher probability of becoming critically short and triggering cell cycle arrest.
Our work adds to other evidence showing that counting Rap1 binding proteins does not fully explain telomere length regulation. Alteration of the telomerase RNA template to generate TTAGGG repeats that do not bind Rap1 has little effect on telomere length (Henning et al. 1998; Alexander and Zakian 2003; Brevet et al. 2003; Bah et al. 2011). In addition, recent work suggests the apparent preferential elongation of short telomeres may be due to replication fork collapse in telomere repeats with subsequent elongation by telomerase (Paschini et al. 2020). Current models of length regulation need to be modified to account for end-specific equilibrium distributions and for Rap1-independent length maintenance.
What mechanism might regulate these end-specific telomere lengths? One obvious hypothesis is that sequences adjacent to the telomere repeats might affect length regulation, through either the sequence itself, chromatin modifications or transcription, or the noncoding RNA TERRA (Feuerhahn et al. 2010). In fact, there were small differences in response to RIF1 length regulation at chromosomes that had Y′ elements compared with those that did not (Fig. 4). Subtelomeric DNA-binding proteins such as Tbf1 and Reb1 that have binding sites in X and Y′ are thought to play some role in length regulation (Berthiau et al. 2006). However, the small effect of these proteins cannot explain the large length differences of up to 200 bp on different chromosome ends observed in wild-type cells. The hint that there may be concordance between telomeres on the same chromosome (Fig. 3A,D) suggests that perhaps chromosome territories within the nucleus (Noma 2017) have an effect on length regulation. Alternatively, topologically associated domains (TADs) (Noma 2017) or localization to the nuclear periphery (Sobecki et al. 2018) may play a role in chromosome end–specific telomere length differences. The fact that deletion of RIF1 disrupted telomere-specific lengths might provide a hint at the mechanism; however, it might be that that deletion of RIF1 simply broadens the length distribution, obscuring any differences between telomeres. Nanopore sequencing of telomeres provides a method to further interrogate these interesting new questions.
In the absence of telomerase, all telomeres shortened at a similar rate as predicted by the end replication problem. A shortening rate of ∼5 bp/population doubling is similar to what has been seen previously with other methods (Marcand et al. 1999; Abdallah et al. 2009; Wellinger and Zakian 2012; Xu et al. 2013). The shortest telomeres in telomerase-null cells, as well as in wild-type and tel1Δ cells, were ∼75 bp. This is shorter than previously suggested (Abdallah et al. 2009) and allows specific hypotheses to be made for what might cause loss of telomere function. For instance, Rap1 binds telomeric DNA approximately every 20 bp (Gilson et al. 1993; Wahlin and Cohn 2000), so telomere dysfunction might be owing to loss of Rap1 binding or perhaps loss of the terminal nucleosome binding. However, it was observed previously that a yeast chromosome with human telomere repeats can be elongated by telomerase, even though Rap1 cannot bind human telomere repeats (Henning et al. 1998; Alexander and Zakian 2003; Brevet et al. 2003). This suggests that there may be distinct factors, independent of Rap1, that negatively regulate telomere length. We note that even at a minimal length of 75 bp, Ku should be able to bind DNA through its end binding function (Gravel et al. 1998; Chen et al. 2018). Given these new data on end-specific telomere lengths, it will be of interest to re-evaluate the role of specific protein binding in determining length regulation.
This novel nanopore sequencing method allows for the analysis of single molecules and provides the ability to assign telomere length measurement to individual chromosome ends. Leveraging this, we were able to uncover new aspects of telomere biology. Nanopore sequencing can be performed in any laboratory and requires relatively little capital investment. Even a smaller, less expensive Flongle flowcell provides a suitable number of independent reads for bulk telomere length measurement. With further improvements and the use of targeted enrichment (Gilpatrick et al. 2020; Kovaka et al. 2021), the cost and yield of this method can be even further improved. Telomere length measurement by nanopore sequencing will allow the telomere field to study new questions and revisit past unanswered questions in telomere biology. A different method of long-read sequencing, Pacific Biosciences (PacBio) sequencing, has recently been applied to human telomere sequences, (Grigorev et al. 2021) indicating long-read sequencing technologies are widely useful in telomere research. It will be of interest to determine whether chromosome end–specific telomere length differences are generalizable to other organisms, as well as perhaps even humans, and understanding how they are established and maintained will be a fascinating new area of telomere biology to explore.
Methods
Biological resources
Yeast strains and plasmid vectors used in this study are listed in Supplemental Tables S4 and S6. Yeast strains are available upon request and plasmid vector (pCS105) is available from Addgene (plasmid 174830).
Molecular cloning
Yeast molecular cloning, culturing, sporulation, and transformation were performed as previously described (Green and Sambrook 2012; Keener et al. 2019; Shubin et al. 2021).
The UE1 (previous named 1L 5×UAS landing pad) and UE11 (previously named 11R 5×UAS landing pad) constructs were designed in silico using SnapGene software and constructed as previously described (Shubin et al. 2021). UE11 was generated in the same manner as UE1, but with homology at chromosome arm 11R, rather than chromosome arm 1L. The subtelomere was removed in these strains using homology with the unique gene region directly upstream of the telomere providing a 3′ overhang that is endogenously elongated by telomerase.
Yeast strain culturing and transformation
CVy61 (W303-0 WT haploids), UE1 (previously CALy117, 1L::leu2), UE11 (previously CALy119, 11R::leu2), UE1 rif1Δ (previously CALy202, 1L::leu2, rif1Δ::kanMX), UE11 rif1Δ (previously CALy204, 11R::leu2, rif1Δ::kanMX), and UE1 tel1Δ (previously CALy472, 1L::leu2, tel1Δ:: hphMX4) strains were generated as previously described (Shubin et al. 2021). These strains were renamed UE1 and UE11 to highlight the function of a unique end, rather than a 5×UAS landing pad, which was important to this study. A list of all yeast strains and genotypes is presented in Supplemental Table S4.
The UE1 tel1Δrif1Δ was generated by transformation. Briefly, transformation was performed by treating 50 mL of logarithmically growing UE1 rif1Δ cells with 0.1 M lithium acetate (Sigma-Aldrich L6883) and adding 1 µg of linear DNA to replace TEL1 with tel1Δ::hphMX4 cassette for one-step integration. Cells were incubated for 10 min at 30°C before the addition of 0.5 mL 40% polyethylene glycol (PEG4000; Sigma-Aldrich P4338), 0.1 M lithium acetate. Cells were then incubated for 30 min at 30°C followed by a 45-min heat shock at 42°C. Cells were plated after a water wash onto YPD + Hygromycin B (Corning; at 200 µg/mL). Integration at the TEL1 locus was confirmed by junction polymerase chain reaction (PCR). A list of primers and plasmids used in this study is presented in Supplemental Tables S5 and S6, respectively.
To perform the W303-0,1,2,3 experiment, CVy61 was streaked to single colony on YPD media. One colony was selected (W303-0) and grown in 5 mL YPD to 0.5 OD. This culture was split into two subcultures: (1) growth to 1.0 OD in 100 mL YPD broth for HMW genomic DNA extraction (see below) and called W303-0 for nanopore sequencing and (2) growth in 10 mL YPD broth to saturation for 24 h and passaged as 5 µL of saturated culture in 10 mL fresh YPD. This was repeated a total of five times before cells were plated onto YPD media. Three independent colonies were selected (W303-1, W303-2, and W303-3) to inoculate three cultures of 100 mL YPD broth for DNA extraction as described for subculture 1. We estimate 120 population doublings between W303-0 and W303-1,2,3 (for illustration, see Supplemental Figure S6A).
To perform the UE1 and UE11 experiments in wild-type and rif1Δ backgrounds, frozen stocks of UE1 wild type, UE11 wild type, UE1 rif1Δ, and UE11 rif1Δ were streaked onto YPD media to single colony. One colony each was selected from UE1 wild type, UE11 wild type, UE1 rif1Δ, and UE11 rif1Δ, grown to 0.5 OD in 5 mL YPD broth, followed by growth to 1 OD in 100 mL YPD for DNA extraction and nanopore sequencing as described above. An additional colony was selected from UE1 wild type and UE1 rif1Δ and grown in 10 mL YPD to saturation for 24 h and passaged as 5 µL of saturated culture in 10 mL fresh YPD. This was repeated a total of three times in liquid culture before cells were grown to 0.5 OD in 5 mL YPD broth and then to 1.0 OD in 100 mL YPD broth for DNA extraction and sequencing of UE1-2 wild type and rif1Δ. We estimate 60 population doublings between UE1-1 and UE1-2 (for illustration, see Supplemental Figure S6B).
To generate telomerase-null est2Δ and est2Δ rad52Δ cells, the heterozygous diploid strain yAY139 (IJpma and Greider 2003) was sporulated and dissected to yield sister spores for analysis.
Because these mutants will senesce with passaging, we attempted to keep cell division to a minimum, allowing telomeres to shorten but not form survivors (Chen et al. 2001). For both est2Δ and est2Δ rad52Δ, cells were taken directly from the original dissection plate and grown to saturation in 10 mL cultures of YPD broth. These mutant cultures were (1) grown to 1 OD in 100 mL for DNA extraction and sequencing, called passage 1 (P1) or (2) diluted to 105 cells/mL in YPD broth every 24 h before being grown to 1 OD in 100 mL for DNA extraction and sequencing. Dilution and growth for these mutants were continued for two more rounds, collecting cells after each passage called P2 and P3 (for illustration, see Supplemental Figure S10). A wild-type haploid spore was also selected from the original dissection plate of yAY139, grown to saturation in 10 mL YPD, and then grown to 1 OD in 100 mL YPD for DNA extraction and sequencing. This sample was called passage 0 (P0). The sample standard deviation of the telomeres in the P0 wild-type sample was compared with the telomerase-null telomere sample standard deviations as a threshold for characterizing potential recombinant telomere length outliers in the mutants. We additionally confirmed this outlier measurement with a qualitative examination of comparisons of mutant and wild-type telomere length distributions. This P0 wild-type sample was used a 0-population doubling initial length in examining the rate of telomere shortening.
HMW genomic DNA extraction
We modified a DNA isolation protocol (Denis et al. 2018) to collect HMW genomic DNA from yeast. Cultures were grown at 30°C to 1.0 OD in 100 mL YPD broth, collected in a Sorvall Legend XTR centrifuge with swinging bucket rotor for 5 min at 1500g and washed once with 20 mL TE buffer (10 mM Tris, 1 mM EDTA at pH 8.0).
To generate spheroplasts, cell pellets in a 50-mL conical centrifuge tube (Falcon) were resuspended in 2 mL 1 M sorbitol with 0.5 mL of 2.5 mg/mL zymolyase 100T (Amsbio 120493-1) and shaken gently (75 rpm) for 1 h at 30°C. Spheroplasts were collected at 300g for 4 min in the swinging bucket centrifuge, and the supernatant was carefully discarded.
Pelleted spheroplasts were lysed by resuspension in 1.7 mL lysis buffer (100 mM Tris at pH 7.5, 100 mM EDTA at pH 8.0, 500 mM NaCl, 1% PVP40) and 250 µL 10% SDS. After inverting the tube gently10 times, 20 µL of 100 mg/mL RNase A (Sigma-Aldrich R6513) was added, and the mixture was shaken at 75 rpm for 30 min at 37°C. To digest protein, 25 µL of 20 mg/mL Proteinase K (Invitrogen 25530-015) was added, and the mixture was incubated for 2 h at 50°C with occasional inversion until there was a uniform cloudy suspension.
To precipitate and remove protein, 5 mL TE and 2.5 mL 5M potassium acetate (pH 7.5) were added to the suspension, and the tube was inverted 10 times to mix and then placed on ice for 5 min. The tube was inverted again and placed on ice for an additional 5 min before centrifugation for 15 min at 4°C and 4000g in the swinging bucket centrifuge. The supernatant was decanted into a clean tube, and the centrifugation was repeated. The supernatant was again decanted into a clean tube.
DNA was precipitated by adding an equal volume of 100% isopropanol and swirling the tube until the DNA strands were observed. Precipitated DNA was collected from the bottom of the tube using a p1000 wide-bore pipette tip and placed in a clean Eppendorf microcentrifuge tube. The DNA was pelleted in a minicentrifuge for 15 sec and the supernatant carefully decanted. Then 500 µL 75% ethanol was added to rinse the DNA pellet. After decanting the wash, the residual ethanol was removed with a pipette tip. The DNA pellet was resuspended in 50–200 µL of EB buffer (Qiagen; 10 mM Tris-Cl at pH 8.5), depending on the size of the pellet. The DNA was allowed to resuspend on an inverting rotator overnight at 4°C. The final concentration of resuspended DNA was measured on a Qubit 3.0 fluorometer (Thermo Fisher Scientific).
DNA molecular end tagging
To tag the molecular end, HMW genomic DNA (gDNA, see above) was first A-tailed using terminal transferase (NEB M0315) in a reaction by incubating 2 µg of gDNA, 1× terminal transferase buffer, 1× CoCl2, 5 mM dATP, and 20 U of terminal transferase enzyme for 1 h at 37°C, followed by heating for 10 min at 70°C to stop the reaction.
Five primers were designed to make a mixture of TeloTags (see Supplemental Table S6).
To the A-tailed gDNA reaction, the following were added to complete the TeloTag addition: 1× ThermoPol reaction buffer pack (NEB B9004S), 2.5 mM dNTP mix, 1 mM ATP, 2.5 mM TeloTag primer mix, and 4 U Sulfolobus DNA Polymerase IV(NEB M0327S). The TeloTag reaction was incubated in a Veriti 96-well thermal cycler (Applied Biosystems) for 1 min at 56°C and 10 min at 72°C. Four hundred units of T4 ligase (NEB M0202) was then added to the reaction and allowed to continue incubating for 20 min at 12°C. Ampere XP beads (Beckman Coulter) were used to purify the tagged gDNA from the reaction mixture.
Size selection of the tagged gDNA was performed using the short-read eliminator XS kit (Circulomics SS-100-121-01), which retains DNA molecules >10 kb. Once this step is completed, the tagged gDNA is ready for nanopore library preparation (see below).
Southern blotting and densitometry
HMW genomic DNA was extracted and quantitated as described above. Telomere length analysis by Southern blot was performed on the same strains used for nanopore sequencing according to a protocol previously described (Kaizer et al. 2015). Briefly, 250 ng of gDNA was digested with PvuII and XhoI for 2–4 h at 37°C and resolved on a 1% agarose gel overnight. Two concentrations of two-log ladders (NEB) were included as reference for analysis, 10 ng for single telomere assays and for bulk telomere assays. After transfer, the membrane was hybridized with 32P-radiolabeled two-log ladder and PCR fragments unique to either TEL01L, TEL11R (a purified PCR product of the LEU2 gene and CYC1 terminator as described by Shubin et al. 2021), Y′ subtelomeric elements described by Kaizer et al. (2015), or CEN4 (Laterreur et al. 2018). After washing, the membrane was exposed on a storage phosphor screen (GE Healthcare) for 1 d for bulk assays and 4–5 d for single telomere assays. Southern images were captured on a STORM using ImageQuant (GE Healthcare). ImageQuant GEL files were downloaded into Adobe PhotoShop CS6 and saved as TIF files.
Southern blot densitometry was measured to allow for comparison to nanopore data as it shows both the normal distribution and spread of telomere length for each sample. However, these plots are often inconsistent and are not a direct measure of telomere length as they are generated from the Southern blot image. To somewhat counter this, we measured densitometry using two programs. We first used ImageQuant to generate a distribution fully across each sample lane to be overlaid with nanopore telomere fragment length data, so that all molecular markers and the resolution cut-off would be visible (Fig. 2C,D,F,G). In ImageQuant, we used the two-log ladder and the CEN4 band at 1.4 kb as molecular markers to determine a linear range, and the resulting CSV files were loaded into RStudio to generate telomere length plots. Second, PhotoShop TIF files were inverted and loaded into the WALTER ScanToItensity and IntensityAnalyzer to remove background and generate intensity profiles of the telomere signal alone (Lyčka et al. 2021). The reported summary statistics were loaded into RStudio to produce boxplots of telomere length.
Nanopore library preparation and sequencing
Library preparation of 1 µg input DNA with a molecular tag was performed using the native barcoding genomic DNA kit (Oxford Nanopore Technologies EXP-NBD104 and SQK-LSK109). Samples were run on a MinION flowcell (v.9.4.1) or Flongle flowcell (v.9.4.1 pore) using the MK1B or GridION for 24–72 h and were operated using MinKNOW software (v.19.2.2). Only reads with barcodes on both ends were selected to pass. The read counts and characteristics are shown in Supplemental Tables S1, S2, and S3. We note that this WGS sequencing from a MinION flowcell provides enough coverage (about 50–200 reads per telomere) to analyze specific single chromosome ends, but Flongle flowcells produce only about 10 reads at a single chromosome end, which, at present, are not enough to analyze specific single chromosome ends. On average, we found that a Flongle flowcell costs $33 per 100 telomere reads and 35 cents per 100 total reads (including flowcell and reagent costs), whereas a MinION flowcell costs $6.9 per 100 telomere reads and 4 cents per 100 total reads. We conclude that it may be more efficient to highly multiplex samples on a MinION flowcell to generate bulk telomere data than to use a Flongle flowcell to examine individual telomeres.
Nanopore analysis, alignment, and telomere length calculation
Base calling to generate FASTQ sequencing files from the electrical signal data (FAST5 files) was performed with GUPPY (v.4.2.3). FASTQ reads containing telomere sequence were selected using TideHunter (v1.4.4) (Gao et al. 2019). Raw base called reads containing the TeloTag were selected using SeqKit (v0.16.1) before alignment (Shen et al. 2016). Nanopore adapters, barcodes, and the TeloTag were removed from reads using Porechop (v0.2.4) before alignment as to not skew length measurement (https://github.com/rrwick/Porechop). FASTQ sequence reads were aligned to a custom reference genome modified from sacCer3 (Foury et al. 1998) (where 2 kb of the S. cerevisiae consensus 13-mer telomere repeat sequence [Wahlin and Cohn 2000; Wellinger and Zakian 2012; Li 2018] was added to each chromosome end) using the map-ont preset of minimap2 (v2.18) for alignment of noisy long-read sequences. Custom references for WT W303-0, UE1, UE11, and S288C are available below in GitHub and Supplemental Code. Reads with a map quality score of less than 20, as well as secondary and supplemental reads were filtered out using SAMtools (v1.12), so that only high-confidence alignment primary reads were selected to prevent measuring telomere length of any reads that multimapped to multiple telomeres (Li et al. 2009). The resulting BAM files were examined in the Integrative Genomics Viewer (IGV) (Robinson et al. 2011; Thorvaldsdottir et al. 2013) to ensure that telomere reads are not clipped during alignment, that there are no large indels in the telomere alignment to skew length measurement, and that the telomere alignment begins at the expected “telomere start” position in the reference. BEDTools (v2.30.0) was used to generate files with the start and end chromosomal position of each read from the aligned reads (Quinlan and Hall 2010). These BED files and a BED file with the start position of each telomere end (available for WT W303, UE1, and UE11 references at below) were read into a custom script in RStudio (Supplemental Code; R Core Team 2020) to calculate telomere length for each read and generate plots using the R package ggplot2 (v3.3.3) (Wichman et al. 2016). This R script contains an additional filter to remove any read name duplications within a BED file to again ensure that any multimapping reads are removed. Statistics were calculated using the R package rstatix (v0.7.0) and ANOM (v0.2) (https://cran.r-project.org/web/packages/rstatix/index.html).
Data access
The whole-genome raw sequencing reads data generated in this study have been submitted to the NCBI BioProject database (https://www.ncbi.nlm.nih.gov/bioproject/) under accession number PRJNA730563. The reference genomes, telomere start position files, and code for the above analysis and plots are hosted on GitHub (https://github.com/timplab/Telomere_Length) and are available as Supplemental Code.
Competing interest statement
W.T. has two patents (8,748,091 and 8,394,584) licensed to Oxford Nanopore Technologies.
Acknowledgments
We thank Carla Connelly for strain generation and Southern blotting. We thank Dr. Brendan Cormack, Dr. Rebecca Keener, and Dr. Calla Shubin for thoughtful discussions about experimental design and conclusions. We thank Dr. Rebecca Keener also for assistance in generating analysis scripts. We thank Carla Connelly, Dr. Brendan Cormack, Dr. Rebecca Keener, Dr. Paul Hook, Dr. Akshi Jasani, and Thea Egelhofer for critical reading and editing of the manuscript. This study was supported by the National Institutes of Health, R35CA209974 (to C.W.G.) and U01CA253481 (to W.T.), National Institute of General Medical Sciences, T32 GM007445 (to the BCMB graduate training program), and a Turock Scholar award (to S.L.S.).
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.275868.121.
- Received June 6, 2021.
- Accepted October 20, 2021.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see https://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








