
Evolution and genomic signatures of spontaneous somatic mutation in Drosophila intestinal stem cells
Abstract
Spontaneous mutations can alter tissue dynamics and lead to cancer initiation. Although large-scale sequencing projects have illuminated processes that influence somatic mutation and subsequent tumor evolution, the mutational dynamics operating in the very early stages of cancer development are currently not well understood. To explore mutational processes in the early stages of cancer evolution, we exploited neoplasia arising spontaneously in the Drosophila intestine. Analysing whole-genome sequencing data with a dedicated bioinformatic pipeline, we found neoplasia formation to be driven largely through the inactivation of Notch by structural variants, many of which involve highly complex genomic rearrangements. The genome-wide mutational burden in neoplasia was found to be similar to that of several human cancers. Finally, we identified genomic features associated with spontaneous mutation, and defined the evolutionary dynamics and mutational landscape operating within intestinal neoplasia over the short lifespan of the adult fly. Our findings provide unique insight into mutational dynamics operating over a short timescale in the genetic model system, Drosophila melanogaster.
The accumulation of mutations in somatic tissues plays a major role in cancer and is proposed to contribute to aging (Al Zouabi and Bardin 2020). Although the majority of mutations acquired throughout life are harmless, some alter cellular fitness and become subject to the selective forces operative in cells and tissues. Mutations that confer a selective advantage can lead to the formation of a clonal population of mutant cells under positive selection. Such events, termed driver mutations, underscore cancer formation and, as such, have been the subject of extensive investigation (Bailey et al. 2018; Alexandrov et al. 2020; Rheinbay et al. 2020; The ICGC/TCGA Pan-Cancer Analysis of Whole Genomes Consortium 2020). These initiating mutations are thought to arise in normal cells and can therefore provide key insights into the mutational processes at play in precancerous states. Large-scale sequencing projects have detailed the mutational burdens of human cancer genomes and have revealed the repertoire of somatic mutations driving cancer formation, illuminating the biological processes underlying somatic mutation. Cancer genomes, however, represent the end-point of a long evolutionary process that shapes the mutational landscape of tumors. Similarly, the mutations recently described to arise in aged normal cells and early-stage cancers represent the result of many years of selective pressure and mutational dynamics (Martincorena et al. 2015, 2018; Lee-Six et al. 2019; Moore et al. 2020; Yokoyama et al. 2019). Knowledge of mutational processes operative in the very earliest stages of cancer is therefore currently incomplete.
Our previous work has established the Drosophila midgut as an excellent model system for understanding somatic mutation in an adult tissue-specific stem cell population (Siudeja et al. 2015). In this tissue, intestinal stem cells (ISCs) self-renew and divide to give rise to two differentiated cell types: absorptive enterocytes (ECs) and secretory enteroendocrine cells (EEs) (Micchelli and Perrimon 2006; Ohlstein and Spradling 2006). We have previously shown that during aging, 12% of wild-type male flies harbor spontaneous mutations that inactivate the X-linked tumor-suppressor gene Notch, driving hyperproliferation of ISCs and EEs and resulting in neoplasm formation (Siudeja et al. 2015).
Here, we take advantage of the spontaneous formation of neoplasia in the intestine of the fruit fly to investigate the processes underlying early somatic mutation and evolution within a clonal cell population.
Results
A comprehensive pipeline to detect somatic structural variation in Drosophila ISCs
We have previously shown that ISCs of the Drosophila midgut spontaneously acquire structural variants during aging that disrupt tissue homeostasis via the inactivation of the X-linked tumor-suppressor gene Notch (Siudeja et al. 2015). Inactivation of the single copy of Notch in ISCs of male flies therefore leads to neoplasm formation, comprising a highly proliferative and rapidly expanding clonal population of Notch-mutant ISCs and EEs. Here, we leverage this system to dissect the mechanisms underlying Notch inactivation and characterize the landscape of somatic mutations in aging stem cell genomes via the development of a robust bioinformatic pipeline.
To define spontaneously arising somatic mutations, we analyzed whole-genome sequencing data generated from 35 intestinal neoplasia. As previously established (Siudeja et al. 2015), neoplasia were detected as GFP+ masses of cells in F1 progeny of flies (for details, see Methods) harboring ProsperoGal4 and UAS-2XGFP or DeltaGal4 and UAS-nlsGFP. To enable us to discern somatic events, we compared DNA from neoplasia to DNA from the head of the same fly as a direct matched control, and consistent with human cancer studies, we will refer to sequenced neoplasia and heads as “tumor” and “normal” samples, respectively (Supplemental Table S1). Three samples were reanalyzed from a previously published data set (Siudeja et al. 2015), and the remaining samples are also described in Siudeja et al. (2021). Using this approach, we can exploit the clonal nature of the tumors to identify somatic mutations in ISCs that are difficult to detect in genetically mosaic adult tissues (Fig. 1A). To accurately characterize structural variants, we developed a pipeline that combines multiple best-practice approaches and applies stringent filters with several novel annotation methods (Supplemental Code; Supplemental Table S2). This pipeline incorporates read-depth-based approaches for detecting copy number variants (CNVs), as well as those using read-mapping signatures. We created several novel tools to filter and annotate structural variant calls, and in cases in which multiple breakpoints were found within small (5-kb) windows, individual calls were collapsed into unified “complex” events (Methods) (Fig. 1B; Supplemental Fig. S1A; Supplemental Methods). We also attempted to categorize rearrangements by putative mechanism using criteria largely previously adapted (Supplemental Fig. S1B; Kidd et al. 2010; Yang et al. 2013).
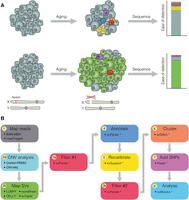
Clonal expansion of ISCs can be exploited to detect somatic mutations. (A) During aging, normal cells (gray) acquire somatic mutations (colored), typically restricted to small populations of cells. Bulk DNA-sequencing of such tissues fails to detect somatic mutations, as they are present in such small numbers. Somatic mutations occurring in an ISC (green) are inherited by the cell's progeny and, in the context of a neoplasm (e.g., as a result of loss of the X-linked tumor-suppressor gene Notch), are present in many cells within the tissue. As a result, sequencing of neoplasia increases the ability to detect somatic mutations in wild-type tissue. (B) A comprehensive bioinformatic pipeline was created in order to accurately detect and characterize structural variants from sequenced neoplasia. We have developed multiple packages to enable us to tag reads that map to multiple genomes (Siudeja et al. 2021), and filter and annotate structural variant breakpoints (svParser, svSupport, freqIn; Methods; Supplemental Methods). Our pipeline uses multiple approaches to detect structural variants and applies stringent filtering steps before annotating variants. Steps marked by an asterisk indicate bioinformatic tools developed for this study.
To ensure that only somatic variants were considered, we constructed a panel of normals (PON) by combining all normal samples and used this to filter out germline variants. We further verified the pipeline's ability to detect variants with a high success rate by using it to detect known variants of simulated data and a de novo assembly of a nonreference Drosophila melanogaster strain (for details, see Supplemental Fig. S2; Supplemental Tables S3–S6; Supplemental Methods; strain “A4” in Chakraborty et al. 2019). In addition, we used a tool that we recently developed, “readtagger,” to tag paired-end reads that partially map to, or have mates that map to, nonreference DNA sequences (Supplemental Methods; Siudeja et al. 2021). Here, we tagged reads associated with transposable elements (TEs) as well as enteric bacterial and viral species. In doing so, we were able to filter out microbial genomic sequences prevalent in the gut samples that artificially map to the D. melanogaster genome. The bioinformatic pipeline that we have developed therefore enables the comprehensive detection of multiple types of somatic mutation.
Diverse mutational events inactivate Notch in normal ISCs
We initially focused on mutations affecting Notch, and in the 35 tumor samples analyzed, we found Notch to be inactivated via multiple different classes of structural variants with lengths ranging from 2000 bp to 550 kb (Fig. 2A; Supplemental Table S7), several of which we verified by PCR (Fig. 2H; Supplemental Fig. S3A). These included deletions (20/35, 57.1%), complex rearrangements (8/35, 22.8%), an inversion (1/35, 2.9%), and one translocation (1/35, 2.9%). In five samples (P15, P37, P47, P51, D5), we found no evidence for inactivation of Notch by a structural variant. In sample P37, we detected multiple structural variants spanning 46 kb, which we hypothesize resulted in the biallelic inactivation of the Notch pathway component kuzbanian and thus was likely responsible for tumor formation. In the remaining four samples for which we did not find support for a structural variant in Notch (P15, P47, P51, D5), we detected evidence supporting de novo transposable element insertion in Notch, likely causing its inactivation. A further investigation of somatic TE insertions in this system is described elsewhere (Siudeja et al. 2021).
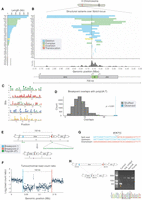
Notch is inactivated by multiple different mutational events. (A) Structural variants affecting Notch in each sample vary in size and class. Across all samples, we found Notch to be inactivated by deletions (20/35; blue), complex rearrangements (8/35; green), an inversion (1/35, sample P59; yellow), and one translocation (1/35, sample P35; red). In five samples (marked as “X”), we did not detect a structural variant in Notch. (B) Breakpoints were plotted over the Notch locus, and we observed a clustering around the TSS, indicated by a black vertical line. (C) Position-weight matrices showing highly repetitive motifs found enriched ±500 bp of Notch breakpoint. (D) Permutation tests showed that breakpoint flanking sequences were significantly enriched (P < 0.001) for poly(dA:dT) sequences. We observed 25 overlaps between breakpoint flanking sequences and poly(dA:dT) sequences (blue dashed line), and in 10,000 permutations, we detected a median of seven overlaps (black dashed line). (E) A schematic for the 102-kb Notch-inactivating deletion in sample P41, showing genomic regions before (top) and after (bottom) the rearrangement. Colored boxes represent breakpoints, with the resulting genomic adjacencies shown below. We detected poly(dA:dT) sequences within 250 bp of both breakpoints. (F) Read-depth ratio plot over the deleted region. Each point represents the Log2 ratio of read counts in 500-bp windows between the tumor and normal sample. Breakpoints are additionally indicated by dotted lines in both E and F. (G) Nucleotide sequences of breakpoint junctions detected in split-reads, with the upstream and downstream genomic sequences shown. Colors correspond to the breakpoints shown in E and F, and a short insertion is shown above in gray. (H) PCR validation of the Notch variant shown in E and F. Primers upstream of and downstream from the deletion were used to amplify the breakpoint. The orange arrowhead indicates the expected 470-bp amplicon detected in the tumor DNA but not in the controls isolated from the same fly: adjacent midgut, head (normal), or thorax DNA. The amplicon was sequenced (chromatogram). Black dashed lines indicate breakpoints, and underlined bases constitute a 7-bp breakpoint insertion.
In human cancer genomes, structural variant breakpoints are distributed nonuniformly and are commonly found to be located in regions of the genome that are inherently prone to double-strand break (DSB) damage (Glodzik et al. 2017). To establish whether hotspot regions existed in the Notch locus, we examined the distribution of breakpoints (Fig. 2B; Supplemental Fig. S4). Although no two breakpoints had the same genomic position, we observed clusters of breakpoints in close proximity (±5 kb) (Fig. 2B), including close to the transcription start site (TSS) of Notch (breakpoints in 7/35 samples within ±2 kb of the TSS) (Supplemental Fig. S4).
We next investigated whether breakpoints in Notch shared underlying sequence similarity that could provide insight into the mechanisms involved in their formation. In particular, we searched for sequences with the potential to form alternative DNA conformations (non-B-form DNA), including cruciform DNA, short inverted repeats (SIRs), and G-quadruplexes, all of which can promote genome instability (Kurahashi et al. 2004; Paeschke et al. 2011; Lu et al. 2015). We extracted the sequence ±500 bp from each breakpoint in Notch and performed permutation tests on the overlap between repeats and these breakpoint-flanking regions. We did not find significant enrichment of inverted repeats and G-quadruplexes around breakpoints. To determine whether other sequences might be associated with breakpoints, we used MEME (Bailey and Elkan 1994) to perform de novo motif discovery on breakpoint regions. All of the motifs recovered were highly repetitive, comprising mono- or di-nucleotide repeats, which resembled microsatellites: tandem repeats of 1–6 bp, sequences that have been previously shown to be prone to mutation owing to replication slippage, mismatch repair, or fork-stalling during DNA replication (Fig. 2C; Gadgil et al. 2017).
Of note, the two most highly overrepresented motifs we found—mononucleotide A/T repeats—were similar to the poly(dA:dT) tracts recently identified as preferential sites of replication fork collapse upon induction of replication stress by hydroxyurea (Tubbs et al. 2018). In light of this association between poly(dA:dT) tracts and replication fork collapse, we then performed permutation tests on the overlap between motif occurrences and the regions flanking Notch breakpoints. This analysis revealed that breakpoint regions were significantly enriched for poly(dA:dT) tracts compared with the genomic region surrounding Notch (Fig. 2D), with 26/42 (62%) of breakpoint regions containing one or more poly(dA:dT) tracts. For example, in one sample (P41), Notch was inactivated via a large deletion (102 kb) (Fig. 2E,F). At both breakpoints, we detected poly(dA:dT) sequences that could explain structural variant formation at this locus. This variant was supported by read-depth changes, split-reads, and PCR validation (Fig. 2F–H). The high enrichment of poly(dA:dT) tracts at breakpoints supports the hypothesis that replication fork collapse may promote many of the structural variants observed in Notch.
Transposable elements and viral inserts at structural variant breakpoints in Notch
In addition to finding an association with poly(dA:dT) sequences, we frequently observed evidence of TE sequences at breakpoints within the Notch locus (11/30 Notch-inactivating structural variants). Of these, 8/11 were at deletion breakpoints, and 3/11 were at breakpoints of complex rearrangements affecting Notch. One of the samples that we validated by PCR contained an I-element fragment at the breakpoint (Supplemental Fig. S3B).
One sample showing a complex rearrangement with TE involvement (P63) had a 23-kb deletion with breakpoints in the last exon of Notch and the second intron of dunce followed by a 1-kb duplication, a 4-kb inverted triplication, and a 1-kb quadruplication (Fig. 3A). On inspecting breakpoint junctions, we detected reads mapping to an I-element TE at both the 5′ breakpoint of the deletion and the 5′ breakpoint of the quadruplication. In another sample (P35), Notch was inactivated by a translocation with breakpoints located in the first intron of Notch and 2 kb upstream of Sox100B on 3R (Fig. 3B). Here, we observed multiple inverted copy number changes and found I-element mapping reads at multiple breakpoint junctions. One explanation for such a configuration is that the entire 2.05-Mb region upstream of Notch was incorporated on Chromosome 3R, which was then subsequently duplicated as part of a complex rearrangement (Fig. 3B).
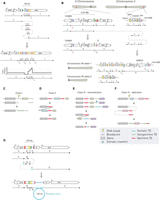
(See figure on preceding page.) Transposable element sequences and viral insertions in Notch-inactivating structural variants. (A,B) Two complex genomic rearrangements inactivating Notch based on read support and CNV calls. Schematics show genomic regions before (top) and after (bottom) each rearrangement. Colored boxes represent breakpoints, with the resulting genomic adjacencies shown below. Arrows indicate the order and orientation of genomic regions. Transposable elements are shown as genomic regions, with nongermline sequences (Tng) shown in green and germline sequences (Tg) shown in red. (A) In sample P63, a complex event generated a deletion in region B, followed by an inverted quadruplication of downstream sequence (regions C, D, and E), flanked by TE sequences. We detected a 12-bp locally templated insertion (indicated by an asterisk) at the breakpoint junction between regions C and D. A schematic of the resulting copy number profile is shown below. (B) In sample P35, a translocation from Notch to Chromosome 3R occurred. A 2.05-Mb region upstream of Notch (region B) was incorporated onto Chromosome 3R, and the entire region was duplicated. The region immediately upstream of the translocation breakpoint on the X Chromosome (region C) was deleted, and we detected TE sequence at the breakpoint, as well as at the 5′ breakpoint of region J, and the junction between regions H and I. In this model, one copy of Chromosome 3R contains the rearranged region from the X Chromosome (labeled allele 1), whereas the other is unaltered (allele 2). We note that other potential configurations may exist for such rearrangements. (C–F) Schematics show putative mechanisms of rearrangement that could explain the signatures of TE involvement detected in Notch-inactivating structural variants. In each class, the uppermost schematic shows a hypothetical genomic region, with genes indicated by colored boxes to help visualize the resulting rearrangement (shown at the bottom). (C) In class I events, read evidence supported a TE or TE fragment integrated at the breakpoint junction. We hypothesize that the TE sequence was integrated during DNA repair. (D) In class II events, two germline TE sequences were found at breakpoint junctions. We hypothesize that these sequences underwent nonallelic homologous recombination, deleting the central region. (E,F) Class III events had evidence for a nongermline TE sequence at one breakpoint junction and germline TE sequence at another. Two interpretations for the breakpoint signatures present in class III events. (E) In the first, a recombinative explanation posits that a de novo TE sequence was inserted (blue arrow) downstream from a germline TE belonging to the same family. Recombination between the two TEs deleted the central region. (F) A second possible explanation of class III breakpoint signatures, wherein DNA damage is repaired by a replicative polymerase that erroneously copies TE sequence into one or several of the breakpoint junctions. This results in a de novo TE signature. (G) A schematic showing genomic regions before (top) and after (bottom) the integration of a fragment of a viral genome in the context of a complex rearrangement detected in sample P31. Although our sequencing data support this configuration, it is possible that alternative explanations exist.
To further explore TE involvement in structural variant formation, we characterized TEs at breakpoints according to their family and somatic status (somatic or germline) and identified four classes of event, suggestive of distinct mechanisms (Fig. 3C–F). In Class I events (constituting 3/11 TE-associated Notch variants), TE-mapping reads were detected at both breakpoints, with no supporting evidence for TE sequences in the corresponding normal tissue. We believe that this represents the integration of either a full-length TE or a TE fragment at breakpoint junctions (Fig. 3C). In Class II events (3/11), we observed that both breakpoints were located within germline TEs. Here, we suspect that variants were generated via nonallelic homologous recombination (NAHR) between two germline TEs with high sequence similarity (Fig. 3D) as previously reported (Robberecht et al. 2013).
In Class III events (5/11), one breakpoint originated in a germline TE, whereas the other breakpoint was mapped to a nongermline TE sequence. Here, we classify TE sequences as being “nongermline” to distinguish them from putative somatic insertions. We observed that both germline and nongermline TE sequences belonged to the same TE family. We consider two explanations for such breakpoint signatures: First, given the sequence similarity, it is possible that structural variants were generated as result of a homologous recombination event between a somatic TE and the germline element (Fig. 3E). However, an alternative possibility is that a germline TE acted as a substrate for template switching during DNA replication, copying TE sequence into a novel locus. This would explain the association of this class with the breakpoints of complex rearrangements in Notch that likely arose via replicative mechanisms (Fig. 3F). That we detect the involvement of TEs in so many of the structural variants in Notch highlights the role that TE sequences may play in influencing somatic mutation, as well as underscoring the complexity of mutations inactivating a model tumor-suppressor locus.
In addition to detecting TE presence at breakpoints, we identified breakpoints at reads whose mates mapped to the double-stranded DNA (dsDNA) nudivirus Tomelloso in one sample (P31) (Supplemental Methods; Palmer et al. 2018). The breakpoint read orientation was consistent with a ∼100-kb fragment of viral DNA integrated into the Drosophila genome as part of a complex rearrangement (Fig. 3G), and to our knowledge, this is the first known example of a somatic dsDNA viral insertion in Drosophila. We did not detect virus-associated variants in other samples or genomic loci and found no correlation between the number of mutations detected and the viral load per sample (Supplemental Fig. S5A,B). Overall, around a quarter of the structural variants inactivating Notch comprised complex rearrangements (8/30, 26.6%) (Supplemental Table S7), many of which involve inserted DNA such as TE elements.
The mutational burden of ISCs
To further interrogate somatic mutation in adult stem cell genomes, we then extended our structural variant analysis to consider the genome-wide distribution and characteristics of all instances of somatic structural variation (Supplemental Table S8). Overall, we detected multiple classes of structural variants distributed throughout the mappable genome, with no breakpoint clustering apparent outside of the Notch locus (Fig. 4A). In total, we found 618 structural variants across all samples (median: six per sample), 36% of which were translocations in which the fraction of supporting reads was low, and were likely to be highly subclonal to the original mutation in Notch (Fig. 4B,C; Supplemental Fig. S6A). The relative frequency of structural variant classes genome-wide was quite distinct from those observed in Notch. We found translocations to be enriched genome-wide, whereas both deletions and complex rearrangements were considerably more frequently observed in Notch than genome-wide (Fig. 4B). It is likely that this difference is inherent to our assay, which selects for Notch-inactivating events. Owing to the greater disruptive potential of variants involving deletion, it is perhaps not surprising that this class of event is more frequently observed in Notch relative to genome-wide variants.
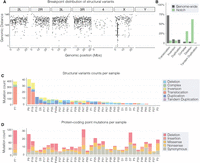
Multiple classes of somatic mutations were detected genome-wide. (A) A rainfall plot showing the distances between structural variant breakpoints across the genome. The y-axis shows the Log10 distance between consecutive breakpoints, with lower numbers representing smaller distances between breakpoints. (B) The percentage contribution of different structural variant classes to the total number of mutations identified genome-wide (gray bars) and in Notch-inactivating variants (green bars). (C,D) The number of each class of structural variant (C) and protein-coding point mutation (D; SNVs and indels) observed across samples. In both C and D, sample P7 is plotted on a separate axis to aid visualization.
Next, to characterize the full spectrum of somatic mutation in ISC genomes, we extended our analysis to include point mutations (SNVs and indels; Supplemental Table S9). As with the structural variant analysis, SNVs and indels were detected using multiple best-practice approaches, genotyped against a PON, and stringently filtered to ensure a high-quality call set of somatic events (see Methods; Supplemental Methods). As well as extensive manual inspection of calls, we also assessed the quality of SNVs by calculating the transition/transversion (Ti/Tv) ratio across samples. Considering that there are more possible transversions (A↔C, A↔T, C↔G, G↔T) than transitions (A↔G or C↔T), the Ti/Tv ratio is often used as a quality control to discriminate nonrandom substitution rates, generally indicated by Ti/Tv values in excess of 0.5. We calculated a genome-wide Ti/Tv ratio of 0.9, which, although substantially lower than values reported in mammalian data sets (e.g., Bainbridge et al. 2011), is broadly consistent with comparable Drosophila data sets (Petrov and Hartl 1999; Keller et al. 2007). This observed difference is largely explained by the lack, or very low levels, of DNA methylation in Drosophila and the associated absence of CpG hypermutability (Raddatz et al. 2013). We performed additional analyses and validation to rule out the possibility that events detected as somatic mutations could instead be owing to (1) miscalled germline SNPs (see Supplemental Methods; Supplemental Fig. S7; Supplemental Table S10), (2) contaminating adjacent ECs (Supplemental Fig. S8), and (3) other errors associated with real sequencing data (Supplemental Methods; Supplemental Fig. S9; Supplemental Tables S5, S6). Altogether these data argue against germline SNPs or contaminating EC SNVs being miscalled as tumor SNVs. Overall, through analyzing whole-gut sequencing data compared with normal heads, we show a low false-positive rate of mutation detection in our pipeline. In combining multiple detection strategies with stringent filtering steps, including manual inspection of calls, we are confident that our final call set comprises true somatic mutations in ISCs.
Genome-wide, we found approximately 1.4 somatic mutations per megabase with a median of 44 and 123 SNVs and indels per sample, respectively (Supplemental Fig. S6B,C). This mutation prevalence of 1.4 per megabase is broadly similar to those typically found in several human cancers such as ovarian (1.85 per megabase) and breast (1.29 per megabase) (Greenman et al. 2007; Alexandrov et al. 2013a; Angus et al. 2019), as well as a Drosophila induced brain tumor model (Rossi et al. 2018). Considering that tumors were dissected from 6-wk-old files, this suggests an overall high mutation rate in flies relative to human cancer genomes. We found no evidence of hypermutation in localized genomic regions, which is sometimes observed in cancer genomes (Alexandrov et al. 2013a).
Next, to focus on mutations in protein-coding regions, we combined SNV and indel calls and annotated mutations with their functional impact (Supplemental Methods) and found a median of 12 protein-coding mutations per sample (Fig. 4D; Supplemental Table S11). Of note, our analysis did not uncover any protein-coding mutations in Notch or in components of the Notch signaling pathway. Although we detected several genes with multiple protein-coding mutations in different samples, we did not find statistical support for any of these being potential drivers.
Human cancer genomes frequently show patterns of mutation—mutational signatures—that are often associated with exposure to distinct underlying mutational processes (Alexandrov et al. 2013a,b, 2020; Nik-Zainal et al. 2012) and have been categorized in the COSMIC database (https://cancer.sanger.ac.uk/cosmic/signatures_v2). To investigate underlying mutational processes, we first extended our SNV analysis to consider mutations within a trinucleotide sequence context. Combining data from all samples, we observed that T > C and C > T transitions were marginally more frequent than C > A, C > G, T > A, and T > G transversions (Supplemental Fig. S6D). Next, to examine mutational patterns operative within individual samples, we calculated the per-sample cosine similarity between mutational profiles and COSMIC signatures (Blokzijl et al. 2018). Although we did not find any one signature contributing to large numbers of mutations across samples, we identified several signatures that contributed heavily in several samples (Supplemental Fig. S6E). Signature 3, often associated with failure of DSB-repair (Nik-Zainal et al. 2016), was observed to contribute strongly in five samples. The liver-cancer-associated signature 16 was also detected in five different samples. Because these signatures are found in flies as well as humans, it is likely the biological processes that generate such signatures are also operative in the fly gut.
Overall, these analyses show the genome-wide mutational spectrum in ISC-derived neoplasia. This included multiple different classes of structural variants, as well as point mutations highlighting the range of mutational processes operative in the fly gut.
Association of mutations with genomic features
In human cancer genomes, several features contribute to the nonrandom distribution of somatic mutations, including local base composition, chromatin structure, and gene expression (Schuster-Böckler and Lehner 2012). To determine the extent to which mutations are enriched or depleted in a given genome feature, we compared the number of mutations observed in the feature to the number of mutations expected in the feature by chance considering its total length. As one particular sample (P7; Fig. 4C; Supplemental Fig. S6B,C) contributed heavily to the total mutational burden across all samples, we excluded this sample from all subsequent aggregate analyses to avoid sample-specific bias.
First, we concentrated on the mutations in both coding (CDS) and noncoding (UTR, introns) gene features. Overall, we found all classes of mutation to be weakly depleted in genic regions of the genome (Fig. 5A), consistent with the observation that euchromatic regions are depleted for mutations (Pleasance et al. 2010; Schuster-Böckler and Lehner 2012; Woo and Li 2012). However, we found CDS to be strongly depleted for both SNVs and indels (Fig. 5A), suggesting that such regions may be maintained under negative selection. To investigate this further, we annotated our data to include expression levels using recently published ISC-specific RNA-seq data (Dutta et al. 2015). We found that the coding sequences of ISC-expressed genes were more strongly depleted for indels, but not SNVs, than those of nonexpressed genes (Supplemental Fig. S10A). We also observed that both 3′ and 5′ UTRs were approximately threefold more strongly enriched for mutations in expressed versus nonexpressed genes. Considering that mutations in UTRs have been traditionally overlooked by studies focusing on protein-coding regions of the genome and that a subset of highly expressed oncogenes are frequently mutated at their 3′ UTR in human cancer genomes (Supek et al. 2014), this finding highlights that mutations in UTRs may be an important, but underinvestigated, class of mutation.
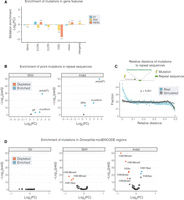
Distribution of somatic mutations in genome features. (A,B,D) The Log2 FC enrichment of mutations in genomic features. In each case, we compared the number of mutations in a given feature with the number expected given the feature's size. (A) Point mutations (SNVs and indels) were both strongly depleted in CDS regions. (B,D) Volcano plots showing enrichment or depletion of mutations in repeat regions (B) and chromatin features from the Drosophila modENCODE data set (D). Highlighted features represent those that with an E-score (−Log10(p) × Log2(FC); Methods) >5. (C) The distribution of relative distances between combined somatic mutations (breakpoints, SNVs, and indels; blue points) and the closest instance of repeat sequences in the genome. Simulated data are shown for comparison in gray. The y-axis of B and D are restricted to a maximum −Log10(padj) value of 50. Asterisks denote significance: (***) P < 0.001, (*) P < 0.5. All P-values shown have been generated from a two-sided binomial test and adjusted for multiple comparisons using a Benjamini–Hochberg adjustment.
Repeat sequences have previously been associated with increased mutability and have been shown to be enriched around structural variant breakpoints (Lu et al. 2015) and for point mutations in human cancers (Zou et al. 2017). Consistent with such reports, we observed a strong enrichment for point mutations in inverted repeats, which was particularly notable in cruciform DNA (Fig. 5B), suggesting that similar mutational dynamics are operative in the fly genome. We also detected an extreme genome-wide enrichment of point mutations in the poly(dA:dT) tracts (Fig. 5B). In comparing the relative distance of mutations to poly(dA:T) tracts (Supplemental Methods), we found that not only are mutations enriched in poly(dA:dT) tracts, but they are also found closer to such repeats more frequently than in randomly distributed data (Fig. 5C), suggesting that such repeats are both inherently mutable and play a role in determining the mutation rate of flanking DNA.
Considering that chromatin organization has also been shown to influence mutation in cancer genomes (Schuster-Böckler and Lehner 2012), we next investigated the distribution of mutations in the publicly available Drosophila modENCODE adult fly data sets, including chromatin landscape and transcription factor binding sites (The modENCODE Consortium et al. 2010). There was a strong depletion for both SNVs and indels in chromatin regions enriched for several marks, including H3K36me2/3. We also observed an association with H3K9me2/3, which is associated with transcriptional repression (Fig. 5D). In contrast, SNVs in cancer genomes are enriched in H3K9me2/3 (Schuster-Böckler and Lehner 2012), suggesting that these marks may influence mutational processes differently in Drosophila. Conversely, we found an enrichment of indels in additional marks (H4K8ac, H3K18ac) and several transcription factor binding sites, all of which belonged to either the C2H2 family of zinc-finger proteins (Br, Trl, Cf2, Odd, Hb) or HMG proteins (D, Pan) (Supplemental Fig. S10B).
Finally, to establish whether a similar distribution of mutations was observable in ISC-specific chromatin profiles, we repeated enrichment analyses using our recently published DamID profiles of chromatin binding factors in ISCs (Gervais et al. 2019). Mutations were found to be depleted in regions associated with silent chromatin, marked by Heterochromatin Protein 1 (HP1). In contrast, mutations were enriched in regions bound by Trithorax-related (Trr), RNA polymerase II (Pol II), and Kismet (Supplemental Fig. S10C), all of which have been previously shown to map transcriptionally active chromatin (Gervais et al. 2019).
Thus, somatic mutations in ISCs are distributed nonrandomly across the Drosophila genome and are found associated with features that influence mutation distribution in cancer genomes, as well as with features with no known associations. Taken together, these findings show the necessity of exploring mutation distribution across whole genomes and the value of performing such analyses in Drosophila.
Mutational timing and tumor evolution
Each tumor genome bears the cumulative damage acquired over its evolutionary history, which can be partially reconstructed using whole-genome sequencing. Mutations arising early in adult life will be propagated throughout the ISC lineage, whereas those arising after tumor formation will be subclonal to the driving mutation and will be present in a smaller fraction of cells (Fig. 6A,B). Considering the time frame of our experiment, we estimate that the mutations driving Notch inactivation arise at ∼3 wk post eclosion and that tumors then develop for another 3 wk before dissection (Siudeja et al. 2015). To reconstruct the evolutionary history of each tumor, we treated the variant allele frequency (VAF) of each variant as a proxy for mutational time. Although we use this to estimate mutational timing, it must be noted that this is an approximation, as the confidence interval around such estimates can be large (Slatkin and Rannala 2000). To normalize between mutations on sex chromosomes and autosomes, we multiplied the VAF on autosomes by a factor of two. As the origin of each tumor we have sequenced can be explained by a mutational event affecting the Notch signaling pathway, we approximated mutational timing relative to Notch-inactivating events.
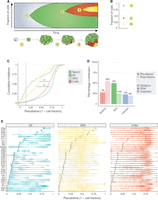
The evolution of somatic mutational in ISC genomes. (A) A schematic illustrating the accumulation of mutations in a stem cell and clonal neoplasm over time. At time point 1, an ISC acquires a somatic mutation that inactivates Notch, driving hyperproliferation of Notch− cells (in green). At time points 2 and 3, subsequent mutations are acquired (shown in red and yellow) that are present in smaller numbers of cells. (B) A modified VAF (Methods) can be used to estimate the fraction of cells carrying each mutation that we use as a proxy for time (pseudotime). (C) The cumulative distribution of mutations (aggregated over all samples) over pseudotime shows that mutations in Notch occurred before other classes of mutation. SNVs arose before other mutations and had a significantly different distribution to both indels and non-Notch SVs (P < 0.001; Kolmogorov–Smirnov test). The median pseudotime value for Notch-inactivating events is shown as a dotted vertical line. (D) For each sample, VAF values of Notch-inactivating mutations were used to divide point mutations observed genome-wide as occurring before Notch (darker shaded bars) and post Notch (lighter shaded bars). Numbers on top of each bar show the number of mutations observed in each category. (E) Per-sample estimates of tumor evolution. Notch-inactivating events for each sample are shown as vertical black bars. Each dot represents a single mutation, and violin plots ease the visualization of mutation distribution over pseudotime. Asterisks in C denote significance: (***) P < 0.001.
Consistent with the notion that Notch mutations occur early in tumor evolution, we found the majority of all mutations types across samples to be subclonal to the mutation in Notch (84%; 4664/5546) (Fig. 6C,E). This would also suggest that cells in the tumor experience a higher mutational burden than those in pretumor ISCs. However, ∼8.8% of point mutations, of which half were SNVs, had a VAF consistent with their likely origin during development or in young-adult ISCs, before the mutation in Notch (467/5267) (Fig. 6D,E).
Together, our findings indicate that in this model system of spontaneous neoplasia formation in wild-type flies, the major driver events are loss of Notch activity through deletions and complex rearrangements. Subsequent genome diversity then arises via the accumulation of SNVs, indels, and additional structural variants. Our data show a rapid accumulation of mutations over a short evolutionary time span during the adult life of D. melanogaster.
Discussion
We have applied whole-genome sequencing to interrogate how spontaneous tumors arise from stem cells in the Drosophila intestine. Our in-depth analysis of the causes of driver inactivation and subsequent tumor evolution provides insight into spontaneous somatic mutation over short timescales.
A potential role for replication stress in promoting Notch inactivation
Our investigation of the underlying causes of spontaneous inactivation of the Notch locus suggests that replication stress may be a contributing factor. Replication fork collapse has been proposed to generate complex structural variants through a mechanism known as microhomology-mediated break-induced replication (MMBIR), related to the previously proposed fork stalling and template switching (FoSTeS) (Lee et al. 2007; Hastings et al. 2009; Carvalho and Lupski 2016; Li et al. 2020). Consistent with this, we found that ∼25% of the Notch-inactivating events were classified as complex rearrangements, involving multiple connected breakpoint junctions, many of which harbored inserted sequences frequently observed in MMBIR. In addition, sequences flanking the breakpoints of Notch-inactivating structural variants were found to be highly enriched for poly(dA:dT) repeats that have previously been implicated as preferential loci for replication fork-stalling and collapse after induction of replication stress (Tubbs et al. 2018). We detected a strong enrichment for point mutations in both poly(dA:dT) sequences and inverted repeats, and mutations were found to occur closer to repeat sequences than expected by chance. One explanation for this association is that these sequences can cause replication fork collapse or lead to stalling replication forks, exposing highly mutable single-stranded DNA (Kurahashi et al. 2004; Lu et al. 2015; Tubbs et al. 2018).
Genome-wide distribution and mutational burden in ISCs
By exploiting the clonal nature of tumors, we were also able to characterize somatic mutations genome-wide. Overall, highly subclonal translocations were the most frequent type of structural variant detected across the genome, although these largely originated from one sample (P7). It is important to note that the read-depth-based approach to detecting CNVs is inherently less sensitive than read-mapping-based approaches, and as such, we expect to detect copy-number-neutral variants (such as translocations and inversions) at lower VAFs than CNVs. In addition, considering that our pipeline integrates both read-depth and frequency changes of heterozygous SNPs to filter CNVs, we have less information with which to discern false-positive events for copy-number-neutral variants. However, we found both the Notch variants and those detected genome-wide to be highly deficient for tandem duplications, a class of structural variant commonly found in both cancer (Li et al. 2020), and normal somatic genomes (Lee-Six et al. 2019; Moore et al. 2020). Unlike the structural variants in Notch, this difference observed in genome-wide variants cannot be explained by an influence of our experimental setup (which selects for Notch-inactivating events), suggesting the possibility of alternative DNA repair strategies in Drosophila that would explain this difference.
Tumor evolution
Using a tumor-purity-adjusted VAF, we attempted to reconstruct the evolutionary history of somatic mutation in ISC genomes. Although similar approaches have been used recently and applied to human cancer data (Gerstung et al. 2020), it is important to note the potential limitations of this strategy. First of all, explicitly using VAF as a proxy for mutational timing assumes a linear propagation of mutations within clones, which will almost certainly fail to capture the dynamics of clone contraction/expansion operating within a tumor. Here, a mutation that occurs very early in tumor development may be selected against in the tumor and be present in few cells of the dissected tissue. The approach we take is therefore unable to distinguish early-occurring, negatively selected mutations from those that are late-occurring and positively selected. In addition, although it has little bearing on our interpretation, it is unable to distinguish mutations in separate cell populations from those occurring within the same clone. Nonetheless, by estimating the timing of mutations in this fashion, we were able to establish the Notch events as founding mutations and divide other somatic mutations into those occurring before and after Notch inactivation. We have shown that the majority of mutations occur post Notch inactivation, consistent with an accelerated rate of mutation in tumors. This could be owing to an increased number of cell divisions following neoplasia formation or owing to changes in the mutation rate arising during aging or tumor development.
Mutational dynamics in somatic tissues
The adult intestine is the most mitotically active tissue in the adult fly, although other tissues such as those associated with the germline are actively renewing throughout adult life. Further studies will be required to determine whether the mutation rate of the adult gut is higher than that of developing tissues or is influenced by environmental exposure, alteration of the microbiome, or dietary changes. On extending our analysis to include point mutations across the whole genome, we found a relatively high frequency of mutation (1.4 per Mb), a mutation prevalence comparable to several human cancers (Greenman et al. 2007; Alexandrov et al. 2013a; Angus et al. 2019). Considering that we dissect tumors from flies at ∼6 wk post eclosion and that the tumor itself has only been developing for ∼3 wk, this would imply that in a matter of weeks, Drosophila ISCs reach a mutational burden equivalent to several decades’ worth of mutation in human cancers.
Methods
Sequencing of Drosophila neoplasia and controls
A detailed methodology can be found in Siudeja et al. (2021). In brief, for selection of neoplastic tissue, clusters of EEs or ISC cells were selected by expression of Prospero-Gal4-driven (Pros > 2XGFP) or Dl-Gal4-driven (Dl > nlsGFP) UAS-nlsGFP. Pros > 2XGFP adult flies (genotype: w; P{w[ + mC] = UAS-2xEGFP}AH2)/+; ProsVoila1GAL4/+) were obtained by crossing w; ProsVoila1GAL4/TM6BTbSb females (gift from J. de Navascués) with w; UAS-2XGFP; males (Bloomington Stock Center: Bl 6874 w[*]; P{w[ + mC] = UAS-2xEGFP}AH2). Dl > nlsGFP flies (genotype: w;; DlGal4 / P{w[ + mC] = UAS-GFP.nls}8) were obtained by crossing w; DlGal4/TM6TbHu (gift from S. Hou) females with w; UAS-nlsGFP males (Bloomington Stock Center: Bl 4776 w[1118]; P{w[ + mC] = UAS-GFP.nls}8). Six- to seven-week-old Pros > 2XGFP or Dl > nlsGFP males were used to visually identify midguts containing neoplasia based on clonal accumulation of GFP-positive cells. To isolate neoplasia, the midgut region containing an estimated 40%–80% neoplastic cells (GFP+), which represents >80% of DNA from neoplastic cells (Supplemental Fig. S8A), was manually dissected together with the head as a direct comparison. Genomic DNA for short-read Illumina sequencing was isolated with the QIAamp DNA microkit (Qiagen) according to the manufacturer's protocol dedicated to processing laser-microdissected tissues. DNA quantity was measured with Qubit dsDNA high-sensitivity assay kit. For three tumor normal pairs (samples P1, P3, and P5), data were reanalyzed from our previous work (Siudeja et al. 2015), and sequencing reads are available under the ArrayExpress (https://www.ebi.ac.uk/arrayexpress/) accession number E-MTAB-3917.
DNA sequencing
Genomic DNA libraries were prepared with the Nextera XT protocol (Illumina). Whole-genome 2 × 100-bp or 2 × 150-bp paired-end sequencing was performed on HiSeq 2500 or NovaSeq (Illumina) on a total of 37 samples and their respective head controls (Supplemental Table S1). Two samples (P13, P21) were excluded from further analysis owing to low coverage.
Structural variant calling and filtering
First, CNVs were called using two different approaches: CNV-seq (Xie and Tammi 2009) and Control-FREEC v11.0 (Boeva et al. 2012). Next, we used three different read-based approaches to precisely identify breakpoints: novoBreak (Chong et al. 2017), LUMPY (Layer et al. 2014), and DELLY (Rausch et al. 2012). We created a PON by genotyping all normal samples using SVTyper (Chiang et al. 2015) and used these to remove germline calls. We then used the tool svParse v0.3.1 developed within the svParser suite of tools (https://github.com/bardin-lab/svParser) to filter variants, selecting for those in the mappable genome, supported by at least three reads, with a coverage of at least 10 in both the tumor and normal, as well as a ratio of coverage to supporting reads >0.05. We did not generally consider somatic events of transposable element insertions, which is the subject of a companion paper (Siudeja et al. 2021), although we did annotate those found associated with structural variants. In addition, we excluded several events that appeared to be false-positive duplications called owing to inconsistent coverage between the tumor and normal samples (Supplemental Table S2). We then categorized structural variants according to putative underlying mechanism using criteria largely adapted from previous studies (Supplemental Fig. S1B; Supplemental Tables S7, S8; Kidd et al. 2010; Yang et al. 2013).
VAF calculation
We extracted reads that supported and directly opposed each variant, and used these read counts to calculate VAF by dividing the number of supporting reads by the number of supporting reads + number of opposing reads. Tumor purity values obtained from Control-FREEC were then used to calculate a tumor-purity-adjusted value by adjusting the number of reads expected to oppose each variant, given a per-sample purity value (Supplemental Methods).
Manual inspection of Notch variants
For each sample in Notch, we manually inspected breakpoints using the Integrative Genomics Viewer (IGV; Robinson et al. 2011). In several samples, we manually added CNVs that were below the detection threshold of our pipeline, and in cases in which breakpoints were unresolved, we adjusted these in line with split-read evidence where possible. In one sample (P59), we manually reclassified a complex event as two distinct inversions, as we could not find any read evidence linking the breakpoints. We then used IGV to identify TE-tagged reads at breakpoints, and manually characterized the signature of TE-tagged reads for each variant in Notch. Here, we classified TE presence at breakpoints as either somatic or germline and recorded the TE family best supported by clusters of TE-tagged reads.
Association of mutations with genomic regions
To detect the enrichment or depletion of genomic regions for mutations, we counted the number of mutations in a given region and compared this to the expected number considering the region's size. The association was tested by performing a two-sided binomial test, adjusting for multiple comparisons using Benjamini–Hochberg adjustment.
Point mutation calling and filtering
Reads were aligned to the Drosophila genome release 6.12 using BWA-MEM v0.7.15, and duplicate reads were marked using Picard MarkDuplicates (v2.7.1; http://broadinstitute.github.io/picard/). A PON was constructed by running Mutect2 (v4.1.2) (Cibulskis et al. 2013) on all normal samples, and we called somatic point mutations using multiple different tools (Mutect2 v4.1.2, Varscan2 v2.4 [ Koboldt et al. 2012]; Strelka v2.9.10 [Kim et al. 2018]; SomaticSniper v1.0.5.0 [Larson et al. 2012]; FreeBayes v1.2.0-dirty [Garrison and Marth 2012]), as described in Supplemental Methods. These calls were then filtered against the PON to remove germline variants, and we selected for variants called at regions with coverage ≥ 20 in both the tumor and normal sample (Supplemental Methods).
Mutational signature analysis
We used MutationalPatterns (Blokzijl et al. 2018) to detect relative contributions made by mutational signatures in the COSMIC database (https://cancer.sanger.ac.uk/cosmic/signatures_v2). Filtered VCF files were used to generate a matrix of mutational counts at trinucleotide positions relative to that observed in the Drosophila genome. The fit_to_signatures function was used to assign the optimal linear combination of mutational signatures that most closely explained the per-sample mutational spectrum, and contributions were plotted using the plot_contribution_heatmap function.
Data access
The whole-genome sequencing data generated in this study have been submitted to the NCBI BioProject database (https://www.ncbi.nlm.nih.gov/bioproject/) under accession number PRJNA641572 (intestinal neoplasia and heads) and the European Nucleotide Archive (ENA; https://www.ebi.ac.uk/ena/browser/home) under accession number PRJEB44312 (whole-gut).
Competing interest statement
The authors declare no competing interests.
Acknowledgments
We thank P.-A. Defossez, N. Servant, J. Waterfall, and members of the Bardin laboratory for critiques and comments on the manuscript and J. de Navascués, S. Hou, and the Bloomington stock center for Drosophila stocks. This work was supported by grants from the Fondation ARC pour la Recherche sur le Cancer (N.R.; postdoctoral fellowship award PDF20161205270), Fondation pour la Recherche Médicale (A.J.B.; DEQ20160334928), as well as funding from the program “Investissements d'Avenir” launched by the French Government and implemented by ANR (references: ANR SoMuSeq-STEM [A.J.B.], Labex DEEP [ANR-11-LBX-0044], and IDEX PSL [ANR-10-IDEX-0001-02 PSL]). We thank the NGS platform of the Institut Curie. High-throughput sequencing has been performed by the ICGex NGS platform of the Institut Curie supported by the grants ANR-10-EQPX-03 (Equipex) and ANR-10-INBS-09-08 (France Génomique Consortium) from the Agence Nationale de la Recherche (“Investissements d'Avenir” program), by the Canceropole Ile-de-France, and by the SiRIC-Curie program–SiRIC (Institut National Du Cancer) grant INCa-DGOS-4654.
Author contributions: N.R. developed software, analyzed and interpreted the data, and wrote the manuscript. K.S. prepared samples and helped interpret data. M.v.d.B. developed readtagger and helped interpret data. B.B. helped prepare samples and helped interpret data. A.J.B. conceived and supervised the study, interpreted the data, and wrote the manuscript.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.268441.120.
-
Freely available online through the Genome Research Open Access option.
- Received July 9, 2020.
- Accepted June 15, 2021.
This article, published in Genome Research, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








