
Systematic evaluation of the effect of polyadenylation signal variants on the expression of disease-associated genes
- 1State Key Laboratory of Genetic Engineering, Collaborative Innovation Center of Genetics and Development, Human Phenome Institute, School of Life Sciences and Eye & ENT Hospital, Fudan University, Shanghai, 200438, China;
- 2Eye Institute, Eye & ENT Hospital, Shanghai Medical College, Fudan University, Shanghai, 200031, China;
- 3NHC Key Laboratory of Myopia (Fudan University), Key Laboratory of Myopia, Chinese Academy of Medical Sciences, and Shanghai Key Laboratory of Visual Impairment and Restoration (Fudan University), Shanghai, 200031, China;
- 4State Key Laboratory of Genetic Engineering, Collaborative Innovation Center of Genetics and Development, Human Phenome Institute, School of Life Sciences and Huashan Hospital, Fudan University, Shanghai, 200438, China;
- 5Department of Pathology, Fudan University Shanghai Cancer Center, Department of Oncology, Shanghai Medical College, Fudan University, Shanghai, 200032, China;
- 6MOE Key Laboratory of Contemporary Anthropology, School of Life Sciences, Fudan University, Shanghai, 200438, China;
- 7Bio-X Institutes, Key Laboratory for the Genetics of Developmental and Neuropsychiatric Disorders (Ministry of Education), Collaborative Innovation Center of Genetics and Development, Shanghai Jiao Tong University, Shanghai, 200030, China;
- 8Singlera Genomics (Shanghai) Limited, Shanghai, 201318, China;
- 9Shanghai Engineering Research Center of Industrial Microorganisms, School of Life Sciences, Fudan University, Shanghai, 200438, China
-
↵10 These authors contributed equally to this work.
Abstract
Single nucleotide variants (SNVs) within polyadenylation signals (PASs), a specific six-nucleotide sequence required for mRNA maturation, can impair RNA-level gene expression and cause human diseases. However, there is a lack of genome-wide investigation and systematic confirmation tools for identifying PAS variants. Here, we present a computational strategy to integrate the most reliable resources for discovering distinct genomic features of PAS variants and also develop a credible and convenient experimental tool to validate the effect of PAS variants on expression of disease-associated genes. This approach will greatly accelerate the deciphering of PAS variation-related human diseases.
Genome-wide association studies (GWASs) have reported that a large proportion of human disease-associated single nucleotide variants (SNVs) are located in noncoding regions, including enhancer, promoter, 5′ untranslated region (5′ UTR), intron, 3′ untranslated region (3′ UTR), and intergenic regions (Lawrenson et al. 2016; Zhu et al. 2017; Shao et al. 2019). Although candidate gene-based approaches have identified a few disease-causal genetic variants (Lawrenson et al. 2016), the majority remains unexplored. Distinct mechanisms may underlie the disease causality of variants located in different noncoding regions. SNVs located in enhancer or promoter regions may affect the transcription efficiency of target genes (Westra et al. 2018; Shao et al. 2019; Tian et al. 2019). Those variants inside 5′ UTRs and 3′ UTRs are most likely to impact the stability and/or translation efficiency of their corresponding mRNAs (Lawrenson et al. 2016; Zhu et al. 2017; Gu et al. 2019). Intronic variants may function through affecting alternative splicing of corresponding genes (Hsiao et al. 2016; Pasutto et al. 2017). Besides these functional genomic regions, the polyadenylation signal (PAS), a specific six-nucleotide sequence motif typically located 10–40 nucleotides (nt) upstream of the poly(A) tail and necessary for mRNA maturation (Elkon et al. 2013), may impair gene expression and cause human diseases. However, few PAS variants have been identified to contribute to RNA maturation and disease development (Higgs et al. 1983; Orkin et al. 1985). Most studies merely investigated the correlation between clinical phenotypes and PAS variants, lacking evidence of causality (Jankovic et al. 1990; Harteveld et al. 2010). Therefore, a comprehensive and genome-wide investigation of this type of variant and an easy-to-use method for evaluating the impact of a variant on gene expression level are urgently needed, which will improve our understanding of the effect of PAS variants on diverse diseases. Meanwhile, three interesting and important questions in this field remain unclear: (1) Why are some PAS variants pathogenic whereas others are benign (neutral)? (2) Do variants in each PAS position have equal contribution to gene expression and to the corresponding disease? (3) Why is a systematic validation tool for PAS variants on gene expression lacking?
To address these questions, we performed an integrative analysis by combing available public databases/data sets and discovered that pathogenic variants inside PASs had distinct genomic features including PAS type and position preferences. Moreover, we developed a reliable and convenient tool named mpCHECK2 to experimentally evaluate the effect of PAS variants on expression of disease-associated genes.
Results
Genomic feature differences between pathogenic and benign variants in PASs
We analyzed all pathogenic variants recorded in HGMD (Stenson et al. 2014) and ClinVar (Landrum et al. 2014) databases, the most widely used and reliable resources for disease-associated variants, and identified overall 107,306 pathogenic variants covering 4540 genes, 4049 of which were protein coding. Exons had the highest number of pathogenic variants, followed by introns, 5′ UTRs, 3′ UTRs, promoters, and PASs (Fig. 1A, left). Of note, each region had different lengths and the PAS had only six nt, and we thus normalized the variant numbers to the average length of each region. Exons still had the highest frequency of pathogenic variants (Fig. 1A, right), which is in line with the idea that the variants in coding regions had the highest possibility to affect gene function. However, PASs had the second highest density of pathogenic variants, followed by introns, 3′ UTRs, promoters, and 5′ UTRs (Fig. 1A, right). This finding emphasizes the importance of investigating the pathogenic variants in PASs at a genome-wide scale.
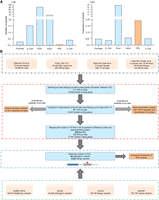
Genomic distribution of pathogenic variants and pipeline for obtaining variants in PAS. (A) Number (left) and frequency (right) of pathogenic variants in different genomic locations. The frequency is normalized to the average length of each genomic region. (B) Flowchart for identifying potential PAS locations in the human genome.
To systematically understand the relationship between PAS variants and human diseases, we first performed a comprehensive analysis by including all publicly available data sets/databases of human polyadenylation (pA) sites (covering 28 tissues, 25 cell lines, and 11 primary cell types) (Fig. 1B; see details in Methods). To be rigorous, we only included 18 known PAS types supported by at least two independent studies for analysis (Fig. 2A; Beaudoing et al. 2000; Tian et al. 2005; Gruber et al. 2016; Ha et al. 2018). We searched 40 nt upstream of each pA site for the 18 PAS types and identified 229,014 unique PASs belonging to 17,947 protein-coding genes. The base composition around these pA sites was consistent with previously identified polyadenylation features (Fig. 2B; Chen et al. 1995). The traditional PAS “AA/UUAAA” was significantly enriched in the 40 nt upstream of pA sites (Fig. 2C; Bailey and Elkan 1994). “AAUAAA” (34.6%) and “AUUAAA” (14.6%) ranked as the top two of the 18 PAS types (Fig. 2D), consistent with previous results (Ni et al. 2013). These findings indicate that the identified pA sites and their corresponding PASs are of satisfactory quality for further analyses.
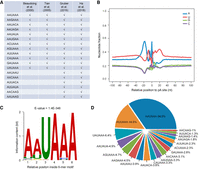
Comprehensive identification of human pA sites and their corresponding PASs in the human genome. (A) The 18 PASs curated from four literature sources. (B) Base composition analysis around the identified pA sites. (C) Motif analysis within 40 nt upstream of the identified pA sites by MEME. (D) The relative distribution of 18 PASs identified in human pA sites.
To discover the features of disease-associated variants in PASs, we collected 107,306 pathogenic (deleterious) and 837,753 benign (neutral) variants from three databases that are widely used for variant annotation and have strong credibility for studying human diseases (Fig. 1B; Peterson et al. 2013; Sarkar et al. 2020). It is worth noting that a variant within a PAS is annotated as pathogenic or benign by these databases based on multiple lines of evidence, and the mechanism through which a variant has an effect may be complicated and may not always depend on the function of polyadenylation; therefore, interpretation of these variants in PASs should be cautious. Using integrative analysis of PASs and database-annotated variants narrated above, we identified a total of 68 pathogenic (Supplemental Table S1) and 4522 benign variants inside PASs, which covered 14 and 18 PAS types, respectively (Fig. 3A). In the 14 shared PAS types, pathogenic variants were significantly enriched in the PAS “AAUAAA” compared to benign ones (Fig. 3B), implying that variants in this strongest signal were likely to affect gene expression and therefore might be related to disease-associated phenotypes. Moreover, pathogenic variants occurred more frequently at the third position, whereas benign variants were more likely to be found at the second position of the PAS (Fig. 3C). One possible explanation is that variants at the third position may disrupt the corresponding PAS, whereas variants at the second position may not. For example, “AAUAAA” to “AUUAAA” or conversely, a variant occurring at the second position, is unlikely to change the effect of polyadenylation, thus making this variant benign. These results suggest pathogenic variants in PASs tend to disrupt mRNA maturation.
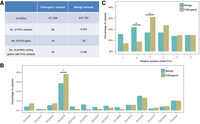
Distinct features of PAS-located pathogenic variants and their associations with human diseases. (A) Classification of disease-associated variants located in the PASs. (B) The distribution difference between pathogenic and benign variants in the 13 shared PASs. (C) The position preferences between pathogenic and benign variants in the PAS. The x-axis denotes six positions (1–6) of the PAS. (*) P < 0.05, (**) P < 0.01, Fisher's exact test.
We noticed that most of the 68 pathogenic variants in PASs are related to monogenic disorders with Mendelian inheritance (Supplemental Table S1). To examine whether PAS variants also appear in other independent disease databases, we analyzed variants located in PASs using public GWAS data of various common diseases (https://www.ebi.ac.uk/gwas/). From 94,471 variants of high confidence (P value ≤ 5 × 10−8), we identified 35 variants located in PASs. Based on the annotated phenotypes associated with these 35 variants, we found that those SNVs were linked with various common diseases (e.g., cardiovascular disease, asthma, and rheumatoid arthritis) and multiple cancers (e.g., glioblastoma, uterine fibroids, and melanoma), as well as other phenotypes (e.g., intraocular pressure, triglycerides, and heel bone mineral density) (Supplemental Table S2). These results further support the importance in studying those variants located in PASs.
Meanwhile, a literature search showed that variants in the PAS of HBA2 and HBB could cause thalassemia, a well-known Mendelian disease, which was supported by the result that cells derived from patients carrying PAS variants exhibited lower HBA2 or HBB expression compared to cells from normal individuals (Higgs et al. 1983; Orkin et al. 1985). Therefore, we chose HBA2 and HBB as candidate genes for developing a universal tool for evaluating the impact of PAS variants on expression of disease-associated genes.
Establishment of an mpCHECK2 system for evaluating the effect of PAS variants
A straightforward and stringent strategy to study the function of PAS variants is to introduce the mutated and wild-type PAS to the same cell types. Although the CRISPR-Cas9 gene-editing system is a powerful genetic tool to modify endogenous genes (Behan et al. 2019), it is difficult to generate the exact PAS variants through the current gene-editing methods, due to the lack of proper guide RNAs targeting this A-T rich region. Carol Lutz and collaborators developed a luciferase assay to assess the effect of PAS variants on luciferase protein production (Hague et al. 2008). However, they used Renilla activity as the internal control in a separate vector and thus the ratio of firefly to Renilla could be affected by the cotransfection efficiency of two plasmids into a single cell. In addition, by simply removing the SV40 pA signal in the test vector in their strategy, the inserted fragment with mutated PAS may cause unexpected transcriptional read-through near the potential polyadenylation site and introduce possible noise, which may affect the reliability of the results. Therefore, we developed an improved approach to evaluate the impact of PAS variants on poly(A)+ RNA and protein production by modifying the dual luciferase vector psiCHECK2, which has been applied for evaluating the impact of 3′ UTR lengths on gene expression efficiencies (Chen et al. 2018; Shen et al. 2019). The original psiCHECK2 vector is not suitable for studying the consequence of PAS variants because it contains a highly efficient synthetic PAS (Supplemental Fig. S1; Levitt et al. 1989) downstream from the multiple cloning sites (Fig. 4A–D), which will overwhelm the function of the weak PAS in the inserted (or tested) 3′ UTR. To overcome this barrier, we removed this strong PAS from the psiCHECK2 vector and subsequently added an artificial terminator with high efficiency (>95%) (Cambray et al. 2013) downstream from the multiple cloning sites to ensure the proper transcription termination (Supplemental Figs. S1, S2). This highly efficient artificial terminator is a palindromic sequence and was registered in the NCBI GenBank databse (https://www.ncbi.nlm.nih.gov/genbank/), named BBa_B1006; the detailed information is shown in Supplemental Figure S2. With these two modifications of the original psiCHECK2 plasmid, this new vector now has the ability to evaluate the impact of PAS variants by quantifying either poly(A)+ RNA or Renilla versus firefly luciferase activity (Fig. 4E–H). We named this modified vector mpCHECK2 (mutated PAS psiCHECK2) (see Supplemental Data 1 for the full sequence of mpCHECK2), which is suitable for studying the impact of PAS variants.
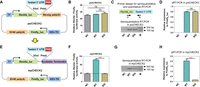
The establishment of an mpCHECK2 system to evaluate the effect of PAS variants. (A) The schematic diagram of the original psiCHECK2 vector with a highly efficient synthetic PAS downstream of the tested 3′ UTR (labeled strong polyA). (B) Renilla luciferase activity (normalized by firefly) in HUVEC transfected using psiCHECK2 without a tested 3′ UTR (NC), with the 3′ UTR of HBA2 containing a WT PAS (AATAAA) (WT), and a 3′ UTR of HBA2 containing a mutated PAS (GGATCC) (SIG), respectively. (C,D) Comparison of mRNA expression levels among NC, WT, and SIG (based on psiCHECK2, described in panel B, that was transfected into HUVEC cells using both semiquantitative PCR (C) and qRT-PCR (D). Primer design is illustrated on the top and M denotes molecular size markers. (E) The modified mpCHECK2 vector resulted from replacing the strong PAS in psiCHECK2 with a highly efficient synthetic terminator. (F) Comparison of normalized Renilla luciferase activity in HUVEC transfected with mpCHECK2 vector (panel E) of NC, WT, and SIG, as described above. Dashed line indicates the basal luciferase signal of NC. (G,H) Comparison of mRNA expression levels in HUVEC transfected with NC, WT, and SIG (based on mpCHECK2) using semiquantitative PCR (G) and qRT-PCR (H). All luciferase activity assays were performed with four replicates and all qRT-PCR reactions were carried out with three replicates. Data are presented as mean ± SEM. (***) P < 0.001, (ns) not significant; one-way ANOVA test.
To confirm that mpCHECK2 works properly, empty vector (negative control [NC], without a 3′ UTR insertion), vector with HBA2’s wild-type 3′ UTR (WT), and vector containing HBA2’s 3′ UTR with the PAS replaced by a palindromic sequence (SIG, AATAAA → GGATCC) were constructed and transfected into both human umbilical vein endothelial cells (HUVEC) and embryonic kidney (HEK) 293T cells. Renilla/firefly relative luciferase activity was used to reflect the polyadenylation efficiency of the tested PAS. In cells transfected with vectors based on the original psiCHECK2, Renilla/firefly relative luciferase activity did not show obvious differences among NC, WT, and SIG in either HUVEC (Fig. 4B) or 293T cells (Supplemental Fig. S3A). Quantitative reverse transcription polymerase chain reaction (qRT-PCR) and semiquantitative RT-PCR also showed no significant differences of poly(A)+ RNA level between WT and SIG (Fig. 4C,D; Supplemental Fig. S3B,C). NC exhibited no amplification signal due to its lack of the inserted 3′ UTR. These results suggested that, no matter what kind of 3′ UTR was inserted into psiCHECK2, the fusion Renilla gene will always use the vector's own strong poly(A) signal, thus overwhelming the effect of tested PAS variants. In contrast, in cells transfected with vectors based on mpCHECK2, SIG showed significantly reduced Renilla/firefly relative luciferase activity, similar to NC, compared to WT in both HUVEC (Fig. 4F) and 293T cells (Supplemental Fig. S3D), consistent with the lack of a PAS in the SIG vector and thus impaired mRNA maturation. The qRT-PCR and semiquantitative RT-PCR also showed a significant RNA level decrease in SIG compared to WT in both HUVEC (Fig. 4G,H) and 293T cells (Supplemental Fig. S3E,F). These findings demonstrated that the mpCHECK2 vector could serve as a robust and reliable system to evaluate the effect of PAS variants on gene expression.
Evaluation of PAS variants on gene expression using the mpCHECK2 system
After demonstrating the feasibility of mpCHECK2, we further examined whether known pathogenic PAS variants in HBA2 and HBB genes discovered in patients could affect polyadenylation. Two point mutations, one from HBA2 (AATAAA → AATAAG, Mut6G) and the other from HBB (AATAAA → AACAAA, Mut3C) (Fig. 5A–H; Supplemental Table S1), were cloned into mpCHECK2 and then transfected into HUVEC and 293T cells. As we suspected, Mut6G, the PAS variant in HBA2, led to an almost complete loss of Renilla luciferase activity (similar level to NC) in both HUVEC and 293T cells (Fig. 5B; Supplemental Fig. S4A). Consistent with the reduced protein abundance, Mut6G led to significantly decreased poly(A)+ RNA level compared with WT, as confirmed by both qRT-PCR and semiquantitative RT-PCR in the two tested cells (Fig. 5C,D; Supplemental Fig. S4B,C). Similar results were obtained for Mut3C (of HBB) at both protein and RNA levels (Fig. 5F–H; Supplemental Fig. S4D–F). Considering that HBB has multiple known pathogenic variants, we tested two more PAS variants (Mut4G and Mut6G) with the mpCHECK2 system and observed similar decreased protein activity and RNA level in both HUVEC and 293T cells (Supplemental Fig. S5). These results indicated that disease-associated variants in PASs (as exemplified by the two test genes HBA2 and HBB) could impair polyadenylation ability and thus reduce mRNA and protein levels of disease-associated genes.

Investigation on the effect of PAS variants occurring in patients using the mpCHECK2 system. (A) The sixth position of the PAS in HBA2 was changed from A (WT) to G (Mut6G) in mpCHECK2. (B) Comparison of normalized Renilla luciferase activity in HUVEC transfected with mpCHECK2 constructs of NC, WT, and Mut6G (panel A). (C,D) Comparison of mRNA expression levels in HUVEC transfected with vectors in panel A (NC, WT, and Mut6G) using both semiquantitative PCR (C) and qRT-PCR (D). (E) Mutations in the third PAS position of HBB (T → C) in mpCHECK2. (F) Comparison of normalized Renilla luciferase activity in HUVEC transfected with mpCHECK2 constructs of NC, WT, and Mut3C (panel E). (G,H) Comparison of mRNA expression levels in HUVEC transfected with NC, WT, and Mut3C (panel E) using semiquantitative PCR (G) and qRT-PCR (H). All luciferase activity assays were performed with four replicates and all qRT-PCR reactions were carried out with three replicates. Data were presented as mean ± SEM. (***) P < 0.001, (**) P < 0.01; one-way ANOVA test.
High correlation of the performance between mpCHECK2 and polyApredictor in evaluating the effect of PAS variants on gene expression
In a recent study, a polyadenylation efficiency prediction tool (polyApredictor) was developed based on a deep learning model (Vainberg Slutskin et al. 2019). We therefore conducted a comparison of the performance between our mpCHECK2 system and polyApredictor. As the polyApredictor method is based on the DNA sequence 250 nt downstream from the stop codon, it is limited to predicting the effect of variants located in the PASs within those 250 nt. We found that only 23 pathogenic variants and 822 benign variants in PASs occurred within 250 nt downstream from the stop codon in our data and thus used these variants for a fair comparison. We first calculated the RNA level changes using polyApredictor for variants within the PAS region and compared the effect between pathogenic and benign groups. The result showed that pathogenic variants in PASs had a higher effect on expression changes (reflected by absolute RNA level changes between mutant and wild type, abs [alt-ref]) than benign variants in PASs (P = 0.002, t-test) (Fig. 6A). We next explored whether our mpCHECK2-quantified expression changes between variants and wild-type PASs were in line with those reported by polyApredictor. For this purpose, we constructed 16 pathogenic and 15 benign variants in PASs and subsequently performed a dual-luciferase assay for systematic analysis. A positive correlation between mpCHECK2 experimental data and predicted values reported by polyApredictor was observed (Fig. 6B), suggesting that the performances of the two methods were consistent in evaluating the effect of PAS variants on gene expression.
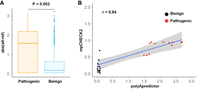
Comparison between our method and polyApredictor for evaluating the effect of PAS variants on RNA-level gene expression. (A) Box plot of predicted expression changes between database-annotated pathogenic (left) and benign (right) variants in the PAS using polyApredictor. Abs (alt-ref) denotes absolute value reported by polyApredictor when comparing mutated to reference sequence of the PAS. (B) Scatterplot for evaluating the correlation of the analytic results reported between mpCHECK2 and polyApredictor. x-axis denotes the difference value (reported by polyApredictor) between mutated and reference base inside PASs. y-axis denotes the degree of normalized luciferase activity changes (reported by mpCHECK2) between mutated and reference base inside PASs. Each dot denotes a PAS variant. Red and black dots denote pathogenic and benign variants in PASs, respectively. Blue line shows the fitting of linear regression trend, and gray region denotes 95% confidence interval (CI). Pearson correlation coefficient (r = 0.84) is shown on the top left.
Discussion
SNVs occurring in the PAS region may disturb the proper termination and polyadenylation of mRNAs, resulting in an aberrant mRNA expression, possibly leading to human disease. Such a concept is only supported by a few genes with PAS variants associated with aberrant RNA production and certain diseases (i.e., HBA2 in α-thalassemia, HBB in β-thalassemia, and TP53 in cutaneous basal cell carcinoma) (Higgs et al. 1983; Orkin et al. 1985; Stacey et al. 2011). With the rapid development of high-throughput sequencing technology, the correlation between PAS variants and clinical phenotypes has been expanded to more genes and diseases (Jankovic et al. 1990; Harteveld et al. 2010). However, whether these disease-associated variants in PASs have causal effects on gene expression along with clinical phenotypes, and whether a genetic variant in each PAS position has an equal contribution, remain unknown. The difficulty in acquiring disease-associated tissues also limits the causal test of PAS variants in disease-associated genes. Although systematically investigating the impact of PAS variants on disease phenotypes is challenging, we found evaluation of the effect of PAS variants on expression of disease-associated genes was feasible. The integration of four resources of human polyadenylation maps (containing 28 tissues, 25 cell lines, and 11 primary cell types) and two disease-related databases makes our study the most comprehensive one. By performing such analysis, we obtained 68 pathogenic (covering 45 genes) (Supplemental Table S1) and 4522 benign (covering 3346 genes) variants in PASs. Whereas pathogenic variants were significantly enriched in the third PAS position, the benign variants were enriched in the second position. Through transforming the original psiCHECK2 vector into an mpCHECK2 system, we demonstrated that mpCHECK2 can reliably evaluate the effect of PAS variants on expression of disease-associated genes. The consistency of the performance between mpCHECK2 and polyApredictor provided strong support for the reliability of this experimental validation tool (Fig. 6). Thus, the computational pipeline coupled with the experimental validation tool provided in our study can greatly facilitate the study of pathogenic variants located in PAS regions.
The first disease-causing variant in PAS was from the hemoglobin subunit alpha 2 coding gene HBA2, where AATAAA was mutated to AATAAG and likely disrupted the polyadenylation process. Three lines of evidence support the causal role of such a variant on affecting gene expression and α-thalassemia (Higgs et al. 1983): (1) This variant was identified in Saudi Arabian patients with α-thalassemia and has a relative frequency of 1.2% in the United Kingdom (Kountouris et al. 2014); (2) northern blot analysis showed that a homozygous mutation from a Saudi Arabian patient with α-thalassemia had a significantly lower level of RNA in peripheral blood reticulocytes than in unaffected individuals; (3) fragments with a mutant and a normal PAS were cloned into a pSVED expression vector, respectively, and the mutant HBA2 generated a longer transcript that possibly read through the AATAAG and presumably terminated in the vector sequence. Although such an expression system can detect the impact of PAS variants on gene expression, it has two limitations. One is the requirement for radiolabeling to sensitively detect the signal, and the other is the lack of internal control, which restrains the general usage of this methodology. The pA signal luciferase assay developed by Carol Lutz and collaborators using Renilla activity from a separate vector as the internal control (Hague et al. 2008) thus was affected by the cotransfection efficiency of two plasmid into the same cells. To solve these problems, we modified a dual luciferase assay vector to make it suitable for such a purpose. We used an internal control (firefly luciferase) to rule out the different transfection efficiencies of human cells between mutated and wild-type samples. The changes in relative luciferase activity (reflected by fluorescence signal) can quantitatively evaluate the effect of a PAS variant on gene expression. Consistent with the pathogenicity of the PAS variant, for example, Mut6G in HBA2, by previous results (Higgs et al. 1983), our mpCHECK2 system showed a significant decrease of both Renilla/firefly luciferase activity and poly(A)+ RNA expression. This result supported the reliability of our experimental validation tool.
Another well-investigated PAS variant was from the HBB gene, where AATAAA is changed to AACAAA (Mut3C). Both HGMD and ClinVar databases annotate this variant as pathogenic, with relative frequencies of 4.3% in Guadeloupe, 3.39% in Cuba, 1% in Morocco, 0.7% in Sri Lanka, and 0.4% in the UK. Northern blot and S1 nuclease mapping indicated a longer transcript that possibly escaped the polyadenylation site of HBB due to the PAS variant and terminated by using a cryptic downstream PAS (Orkin et al. 1985). Such read-through may largely reduce the normal transcript abundance and ultimately contribute to the phenotypes of β-thalassemia. In line with these results, our mpCHECK2 system demonstrated that AACAAA in the 3′ UTR of HBB reduced the transcript level and in turn generated less Renilla/firefly luciferase activity than the wild-type PAS (Fig. 5E–H). This again demonstrated the reliability of our generalized validation tool.
It is worth noting that the annotation of “pathogenic” of certain PAS variants in these databases is based on large-scale sequencing data, and most of them depend on prediction relying on previous knowledge. Although some variants have different allele frequencies in different populations and experimental validations, they are correlative validations rather than causality confirmations. Additionally, the “pathogenic” and “benign” annotations could be due to the affected gene rather than affected poly(A) signal and related polyadenylation efficiency. For example, some genes can be substantially affected by variants at their poly(A) signal without clinical consequences, and some genes can have clinical manifestation even when a subtle change takes place, regardless of poly(A) signal or elsewhere. Only by combing comprehensive analysis of all these pathogenic variants with experimental validation tools can we fully reveal the genome-wide features of these PAS variants and their real impacts on expression of disease-related genes. Additional work such as analyzing the change of mRNA decay and/or translational control associated with PAS variants and phenotype validation using animal models is still required for further confirmation.
To examine whether genes containing pathogenic variants in PAS have only this type of variants, we analyzed all 45 genes having 68 pathogenic variants in PAS and found all of them (45/45) had both pathogenic and benign variants outside the PAS. For example, there are 504 pathogenic variants and 38 benign variants outside the PAS region of the HBB gene. The TPM1 gene, associated with heart disease, oculopathy, and various cancers (Kubo et al. 2017; Wang et al. 2019; Hirono et al. 2020), has 35 pathogenic variants and 134 benign variants outside the PAS region. These results suggest that variants within the PAS are annotated as pathogenic in the databases not just because the corresponding genes are relevant to diseases—they also have variants annotated as benign.
Pathogenic variants can also appear in noncoding regions other than the PAS, and the analysis of noncoding variants outside the PAS hexamer is also valuable. Whereas benign variants showed a relatively even distribution along the region 200 nt upstream of and 50 nt downstream from the poly(A) site, pathogenic variants exhibited an increased distribution frequency upstream of the poly(A) site (Supplemental Fig. S6). One possible reason why the upstream region had a higher pathogenic variant frequency is that this region not only contains upstream sequence elements required for polyadenylation but also contains other key elements, including microRNA-binding motifs, sequences required for secondary structure formation in the 3′ UTR, and RNA-binding protein recognition motifs that are important for RNA stability, localization, and translation, all of which can affect gene expression. Another possibility for why the downstream region had a lower pathogenic variant frequency could be due to research bias and/or lack of easy-to-use validation methods. This result suggests that pathogenic variants outside the PAS are prevalent and deserve in-depth investigation in the future.
To estimate the sensitivity and specificity of our tool (mpCHECK2), we tested an additional 10 pathogenic and 10 benign PAS variants covering 18 genes. The results showed that eight out of 10 pathogenic variants in PASs had significantly decreased expression compared to the reference sequence (Supplemental Fig. S7), whereas none of the 10 benign variants in PASs had obvious expression changes compared to the reference sequence (Supplemental Fig. S8). In addition, we constructed eight more database-annotated pathogenic variants in the PAS of the HBB gene (Supplemental Fig. S9) and found that all of them led to reduced expression. Actually, we found one special case (from gene DCLRE1B) that was annotated as benign in the database but was confirmed to cause elevated expression using our method (Supplemental Fig. S10). The variant converts the weak PAS (TATAAA) to a stronger one (AATAAA), which may explain the elevated expression in the luciferase assay (Supplemental Fig. S10). Literature searching showed that abnormal expression of DCLRE1B was likely to cause disease-related phenotypes such as cell proliferation defects and developmental delay (Akhter et al. 2010; Michailidou et al. 2013). This special case highlights the sensitivity of our experimental validation tool in studying the impact of PAS variants on gene expression. We believe that this generalized method can be applied to any PAS variants associated with disease. For example, a GWAS study on 457 Icelanders discovered a new risk variant for cutaneous basal cell carcinoma, where AATAAA in the TP53 gene is mutated to AATACA, resulting in impaired 3′ end processing of TP53 mRNA, as demonstrated by RT-PCR and 3′ rapid amplification of complementary DNA ends (3′ RACE) (Stacey et al. 2011). Such variants can also be validated in our mpCHECK2 system to determine the effect at both the RNA and protein levels.
As RNA maturation is the last critical step for mRNA expression, our comprehensive computational analyses coupled with an experimental validation tool enable the possibility of fully understanding the genomic features regarding disease-associated variants located in polyadenylation signals. Compared to existing strategies, mpCHECK2 provides a quantitative, reliable, and easy-to-use method to examine the impact of a PAS variant on the expression level of corresponding genes. This study will extend our understanding of genetic mechanisms underlying disease-associated genes.
Methods
Polyadenylation site collection and polyadenylation signal acquisition
The goal of this study was to provide a comprehensive approach to investigating distinct features of disease-associated variants located in the PAS. We pooled together human pA sites collected from four resources: (1) PA-seq data published by Ni et al. (2013) (NCBI Sequence Read Archive [SRA; https://www.ncbi.nlm.nih.gov/sra] accession number SRA059064); (2) PolyA_DB (https://exon.apps.wistar.org/PolyA_DB/, version 3.2, NCBI Gene Expression Omnibus [GEO; https://www.ncbi.nlm.nih.gov/geo/] accession number GSE111134) (Wang et al. 2018); (3) PolyA-seq data published by Derti et al. (2012) (accession number: SRA039286); and (4) PolyASite (https://polyasite.unibas.ch/, accession number: SRP065825) (Gruber et al. 2016). Considering that those pA sites were from four different sources, the redundant PASs for the same gene were removed. We curated 18 robust PASs from four publications and utilized a series of analysis steps to retrieve reliable PASs for integrative analysis with SNVs data (Figs. 1B, 2A; Beaudoing et al. 2000; Tian et al. 2005; Gruber et al. 2016; Ha et al. 2018).
Data processing and evaluation
Considering the differences between data from different sources, we performed a series of data processing to ensure the reliability of collected pA sites (Fig. 1B). In brief, we annotated and obtained pA sites which were located in protein-coding gene regions using Ensembl Release 75 (ftp://ftp.ensembl.org/pub/release-75/gtf/homo_sapiens/). Then, we used BEDTools (Quinlan and Hall 2010) to extract 6 nt downstream from pA sites. To eliminate internal priming, we filtered out pA sites with downstream “AAAAAA.” To evaluate the reliability of those pA sites, we conducted both base composition and motif analyses near the acquired pA sites. For base composition analysis, we obtained 100 nt upstream of and downstream from pA sites defined in Figure 1B based on BEDTools (Quinlan and Hall 2010), and the base composition at each position was counted. For motif analysis, we used MEME (Bailey and Elkan 1994) to analyze 6-mer enrichment within the 40 nt upstream of the defined pA sites.
Identification of 18 PASs and their relative distribution
After confirming the reliability of those pA sites, we mapped the location of 18 PASs (Fig. 2A) in the 40 nt upstream of the pA sites (Fig. 1B; Ni et al. 2013). The relative percentage of the 18 PASs was then calculated (Fig. 2D).
Analysis of disease-associated variants located in PASs
To reveal the characteristic features of disease-associated variants inside PASs, we collected 107,306 pathogenic variants from HGMD public version (Human Gene Mutation Database, 2014) (Stenson et al. 2014) and ClinVar (downloaded on 11/20/2020) (Landrum et al. 2014). To improve the reliability of the data, we respectively performed a filter step in the two databases. In the HGMD database, we only retained the variants with an annotation of “DM,” which means disease mutation. Similarly, we only selected those variants annotated as “Pathogenic” in the ClinVar database. Meanwhile, 837,753 benign variants were pooled from VariSNP (Schaafsma and Vihinen 2015) and ClinVar (downloaded at 11/20/2020) (Landrum et al. 2014). Only variants that showed consistent pathogenic or benign annotation among all databases were used for further analysis. After mapping pathogenic and benign variants to the PASs, respectively, we compared the difference between pathogenic and benign variants that shared 14 PASs. We also calculated the percentage of pathogenic and benign variants at each position of the PAS to explore if PAS variants have location preference.
Cell cultivation and transfection
HUVEC and 293T cells were cultured in DMEM (1×; Gibco) with 10% fetal bovine serum at 37°C in 5% CO2. For transient transfection, seeded cells in six-well plates with 70% confluence were transfected with a mixture consisting of 6 μL Lipofectamine 2000 (Invitrogen) reagent and 2 μg vector in 500 μL serum-free medium.
Luciferase assay, semiquantitative PCR, and qRT-PCR
The relative Renilla/firefly luciferase activity was measured by the Dual-Luciferase Reporter 1000 Assay System (Promega) 24 h after transfection with either psiCHECK2 or mpCHECK2 (with or without a variant in the PAS) in HUVEC or 293T cells. For semiquantitative PCR, mRNA was reverse-transcribed into cDNA using oligo (dT)25 and then amplified by PCR with primer pairs listed in Supplemental Table S3. When performing semiquantitative RT-PCR, the PCR reaction was stopped at the exponential amplification cycle that was determined by qRT-PCR. Agarose gel electrophoresis was used to evaluate the relative abundance based on the intensity of the bands. For qRT-PCR analysis, we used the same aforementioned cDNA and primers listed in Supplemental Table S3. SYBR Green reagent (Vazyme) and a Bio-Rad qPCR machine were used for the qPCR reaction.
Data access
The full sequence of the mpCHECK2 vector is available in Supplemental Data 1. The mpCHECK2 plasmid is available from Addgene (https://www.addgene.org/; plasmid #167576). All the codes used in this study are available as Supplemental Code. The chromosome coordinates of the PAS sites used in this study are included in a BED file as Supplemental Material.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
The psiCHECK2 vector was kindly provided by Prof. Haijian Wang. We thank Prof. Hongyan Wang for luciferase assay instrument support. This work is financially supported by the National Key R&D Program of China (2018YFC1003500); the National Natural Science Foundation of China (31771408, 31771336, 31521003, 81730025, 81670864, and 81525006); and the Excellent Academic Leaders of Shanghai (18XD1401000).
Author contributions: T.N., Z.S., C.Z., M.X., and G.W. designed the study. R.W. performed the bioinformatics and statistical analyses. M.C. and T.N. performed the experiments. M.X. and Z.S. supervised the bioinformatics and statistical analyses. The manuscript was initially drafted by M.C. and R.W. and then was revised by T.N., M.X., and G.W. All authors read and approved the final manuscript.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.270256.120.
-
Freely available online through the Genome Research Open Access option.
- Received August 15, 2020.
- Accepted March 2, 2021.
This article, published in Genome Research, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








