
Orientation-aware plasma cell-free DNA fragmentation analysis in open chromatin regions informs tissue of origin
- Kun Sun1,2,
- Peiyong Jiang1,2,
- Suk Hang Cheng1,2,
- Timothy H.T. Cheng1,2,
- John Wong3,
- Vincent W.S. Wong4,
- Simon S.M. Ng3,
- Brigette B.Y. Ma5,
- Tak Y. Leung6,
- Stephen L. Chan5,
- Tony S.K. Mok5,
- Paul B.S. Lai3,
- Henry L.Y. Chan4,
- Hao Sun1,2,
- K.C. Allen Chan1,2,
- Rossa W.K. Chiu1,2 and
- Y.M. Dennis Lo1,2
- 1Li Ka Shing Institute of Health Sciences, The Chinese University of Hong Kong, Hong Kong SAR, 999077, China;
- 2Department of Chemical Pathology, The Chinese University of Hong Kong, Hong Kong SAR, 999077, China;
- 3Department of Surgery, The Chinese University of Hong Kong, Hong Kong SAR, 999077, China;
- 4Department of Medicine and Therapeutics, The Chinese University of Hong Kong, Hong Kong SAR, 999077, China;
- 5Department of Clinical Oncology, The Chinese University of Hong Kong, Hong Kong SAR, 999077, China;
- 6Department of Obstetrics and Gynaecology, The Chinese University of Hong Kong, Hong Kong SAR, 999077, China
Abstract
Cell-free DNA (cfDNA) in human plasma is a class of biomarkers with many current and potential future diagnostic applications. Recent studies have shown that cfDNA molecules are not randomly fragmented and possess information related to their tissues of origin. Pathologies causing death of cells from particular tissues result in perturbations in the relative distribution of DNA from the affected tissues. Such tissue-of-origin analysis is particularly useful in the development of liquid biopsies for cancer. It is therefore of value to accurately determine the relative contributions of the tissues to the plasma DNA pool in a simultaneous manner. In this work, we report that in open chromatin regions, cfDNA molecules show characteristic fragmentation patterns reflected by sequencing coverage imbalance and differentially phased fragment end signals. The latter refers to differences in the read densities of sequences corresponding to the orientation of the upstream and downstream ends of cfDNA molecules in relation to the reference genome. Such cfDNA fragmentation patterns preferentially occur in tissue-specific open chromatin regions where the corresponding tissues contributed DNA into the plasma. Quantitative analyses of such signals allow measurement of the relative contributions of various tissues toward the plasma DNA pool. These findings were validated by plasma DNA sequencing data obtained from pregnant women, organ transplantation recipients, and cancer patients. Orientation-aware plasma DNA fragmentation analysis therefore has potential diagnostic applications in noninvasive prenatal testing, organ transplantation monitoring, and cancer liquid biopsy.
Presence of circulating cell-free DNA (cfDNA) in human plasma was first reported by Mandel and Metais (1948). Later on, discoveries of fetal-derived DNA in the plasma of pregnant women (Lo et al. 1997), donor-derived DNA in transplantation patients (Lo et al. 1998), and tumor-derived DNA in cancer patients (Stroun et al. 1989) opened up the door of cfDNA-based noninvasive prenatal testing (van Opstal et al. 2017), transplantation monitoring (Schütz et al. 2017), and cancer liquid biopsies (Chan et al. 2017; Phallen et al. 2017; Cohen et al. 2018). cfDNA has thus become a biomarker class that is actively researched globally.
Recent studies had demonstrated the clinical feasibility of cfDNA analysis for sensitive cancer screening (Chan et al. 2013b, 2017; Cohen et al. 2018). For future development in this field, it is necessary to develop a robust approach for localizing the site of the tumor following a positive liquid biopsy test. Exploiting the differences in DNA methylation patterns between tissues, we have previously demonstrated that circulating fetal-derived DNA in maternal plasma originated predominantly from the placenta (Chim et al. 2005). This work was based on the detection of unmethylated SERPINB5 sequences as a placental marker in maternal plasma (Chim et al. 2005). More recently, a similar approach has been applied to the detection of cfDNA derived from the brain (Lehmann-Werman et al. 2016), cells of the erythroid lineage (Lam et al. 2017), the heart (Zemmour et al. 2018), and the liver (Gai et al. 2018; Lehmann-Werman et al. 2018).
We have further developed a general DNA methylation-based approach for determining the contributions of multiple tissue types into the cfDNA pool, a method that we have named “plasma DNA tissue mapping” (Sun et al. 2015). This principle has also been utilized to predict the tissue origin of tumors by other researchers (Kang et al. 2017; Li et al. 2018). These published approaches used whole-genome bisulfite sequencing (BS-seq) of plasma DNA (Lister et al. 2008; Lun et al. 2013; Chan et al. 2013a). However, BS-seq has the disadvantage that bisulfite conversion is associated with degradation of input DNA (Grunau et al. 2001) and also introduces GC content changes which may lead to biases in the sequencing data (Olova et al. 2018).
Besides DNA methylation, recent studies demonstrated that cfDNA molecules retained signatures of their nucleosomal origin, showing a size distribution with a dominant peak at 166 bp and a ∼10-bp periodicity (Lo et al. 2010). cfDNA has been shown to carry a nonrandom fragmentation pattern that provides a window into epigenetic regulation across the genome (Ivanov et al. 2015). Considering that nucleosome positioning across the genome is highly related to the cell identity (Radman-Livaja and Rando 2010), such fragmentation patterns thus hold the potential of tracing back the tissue origin of cfDNA molecules. Snyder et al. (2016) showed that plasma DNA molecules carried nucleosomal footprints. These authors further constructed a “nucleosome track” and found that the nucleosome spacing pattern could be used to infer the tissue origin of cfDNA. They also demonstrated the potential of this approach in predicting the tumor origin in cancer patients. Ulz et al. (2016) reported that plasma DNA coverage in the promoters could be used to predict the expression of genes. Our group had demonstrated the existence of tissue- and tumor-specific preferred end sites in cfDNA which showed clinical utility in predicting the fetal DNA fractions in maternal plasma (Chan et al. 2016; Sun et al. 2018) and tumor load in hepatocellular carcinoma (HCC) patients (Jiang et al. 2018). Taken together, these proof-of-principle studies demonstrated the potential clinical applications of fragmentation pattern analyses in inferring the tissue origin of cfDNA. However, this field is still in its infancy and further developments are necessary before clinical implementations. To this end, we further explored the clinical potential of cfDNA fragmentation patterns in this work. We hypothesized that cfDNA derived from tissue-specific open chromatin regions would carry information that allows one to infer the tissue origin of plasma DNA and predict the tumor location in cancer patients.
Results
Conceptual framework and nomenclature
Figure 1 shows the conceptual framework of our approach. Figure 1A shows an illustration of nucleosome positioning in the genome. In eukaryotic chromatin, the nucleosome is the basic unit for DNA packaging which consists of a DNA segment wrapped around histone proteins. Nucleosomes are generally connected to each other by a relatively short linker DNA, except in active regulatory elements (e.g., open chromatin regions), where nucleosomes are evicted and the nearby nucleosomes will be connected by a much longer stretch of DNA. It is believed that a significant proportion of cfDNA molecules are released following cell apoptosis (Jahr et al. 2001; Lo et al. 2010). During apoptotic DNA fragmentation, it is proposed that endonuclease enzymes prefer cutting internucleosomal DNA (Fig. 1B; Samejima and Earnshaw 2005; Sun et al. 2018). As a result, when cfDNA molecules are subjected to sequencing, the DNA wrapped on the histones are preserved. On the other hand, DNA originating from the linkers and open chromatin regions, as they are relatively unprotected, will be cleaved into small pieces and may not be efficiently sequenced (Fig. 1C; Jiang and Pugh 2009; Snyder et al. 2016; Ulz et al. 2016). Therefore, the genomic coverage of cfDNA would be high in the nucleosomes, and low in the linkers and open chromatin regions (Fig. 1D). In addition, we took advantage of the orientation information of the cfDNA fragment ends and defined those ends based on their alignment to the reference genome. Hence, an upstream (U) end represented one that had a lower value in the genome coordinate, whereas a downstream (D) end represented one that had a higher value in the genome coordinate. As a consequence, DNA wrapped on the nucleosomes will result in a pair of U and D ends at the upstream and downstream borders of the nucleosomes, respectively (Fig. 1E). For the linkers or open chromatin regions, however, there would be D ends flanking their upstream boundaries and U ends flanking their downstream boundaries (Fig. 1E). In this regard, the U and D end signals could be used to infer the positioning of the nucleosomes, linkers, as well as the open chromatin regions (Fig. 1F).
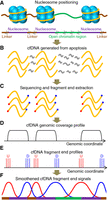
Conceptual framework of cell-free DNA (cfDNA) fragmentation analysis. (A) Illustration of nucleosomes with wrapped DNA (yellow line), linkers (brown line), and open chromatin regions (green line). An abstraction of nucleosome positioning and illustration of cutting events (scissors) during apoptosis were also provided. (B) Illustration of cfDNA generated from apoptotic DNA fragmentation. DNA wrapped around the nucleosomes is preserved, and that in the linkers and open chromatin regions will be cleaved into small pieces (gray line) that cannot be sequenced efficiently. (C) Illustration of the sequenced reads and extraction of the ends. Red and blue represent upstream (U) and downstream (D) fragment ends, respectively. (D) Genomic coverage; (E) U and D end profiles of cfDNA in relation to the reference genome. (F) Smoothened cfDNA end signals and deduced nucleosome positioning. Purple, brown, and green lines represent nucleosomes, linkers, and open chromatin regions, respectively.
Differentially phased cfDNA fragment ends in a nucleosomal array
To illustrate the above concept in a human genomic region, we first examined Chr 12p11.1, a region known to have well-positioned nucleosomes in almost all tissue types (Valouev et al. 2011; Gaffney et al. 2012; Snyder et al. 2016). To do this, we pooled cfDNA data from 32 healthy nonpregnant subjects generated in a previous study (Jiang et al. 2015) then profiled the coverage and fragment ends in this region. As shown in Figure 2A, plasma DNA coverage showed a strong periodicity pattern of ∼190 bp, and the regions with higher and lower coverages corresponded to the nucleosomes and linkers, respectively (Snyder et al. 2016). The U and D ends showed a similar ∼190-bp periodicity pattern and both were enriched in the linkers. The U and D end signals were then smoothened using the locally weighted scatterplot smoothing (LOWESS) algorithm (Cleveland 1979) for better data presentation. As shown in Figure 2B, the distance between any U end peak to its nearest downstream D end peak was ∼170 bp, which was roughly the size of a nucleosome (Struhl and Segal 2013). The distance between any U end peak to its nearest upstream D end peak was ∼20 bp, which was roughly the size of a linker (Struhl and Segal 2013). The data thus were highly concordant with our conceptual framework (Fig. 1) and showed that differentially phased cfDNA fragment ends indeed reflected the nucleosome positioning in this region (Fig. 2C). Notably, with the separation of U and D ends, we were able to resolve the positioning of both the nucleosomes and linkers, a development that represents an advance over previous studies that mostly focused on predicting the positions of nucleosome centers (i.e., loci with maximum nucleosome protection) (Fig. 2B; Gaffney et al. 2012; Pedersen et al. 2014; Snyder et al. 2016).
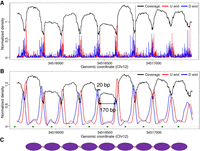
cfDNA fragmentation patterns in a nucleosome array region (Chr 12p11.1) in pooled healthy nonpregnant subjects. (A) Raw signal. (B) Smoothened signal. Green dots at the bottom represent the predicted nucleosome center loci by Snyder et al. (2016). (C) Deduced nucleosome positioning.
Differentially phased cfDNA fragment ends in tissue-specific open chromatin regions
Open chromatin regions are regulatory elements that are known to have a paucity of nucleosomes in the center and are flanked by well-phased nucleosome arrays (Gaffney et al. 2012; Schep et al. 2015). Therefore, we hypothesized that cfDNA derived from such regions might also exhibit differentially phased fragment end signals. We first investigated the common open chromatin regions shared by T cells and the liver, considering that these tissues are important contributors to the plasma DNA pool in various clinical scenarios. Hence, DNA derived from the T cells was one example of cfDNA released from the hematopoietic system, which is the major source of plasma DNA in healthy individuals (Lui et al. 2002). The liver is another major source of plasma DNA in healthy individuals, liver transplantation recipients, and liver cancer patients (Lo et al. 1998; Gai et al. 2018; Lehmann-Werman et al. 2018).
Open chromatin data for T cells was obtained from the Roadmap Epigenomics Project (Roadmap Epigenomics Consortium et al. 2015), and data for the liver was obtained from the ENCODE Project (The ENCODE Project Consortium 2012) and International Human Epigenome Consortium (IHEC) (Bujold et al. 2016; Methods). We identified the open chromatin regions that were shared by T cells and the liver as the common open chromatin regions (Supplemental Table S1). We then performed fragmentation analysis on these regions in the pooled cfDNA data. As shown in Figure 3A, characteristic fragmentation patterns, including coverage imbalance and differentially phased fragment ends, could be observed. We interpreted these results as the consequence of a nucleosome-depleted region in the center and the presence of neighboring well-phased nucleosomes. These results thus showed that differentially phased cfDNA fragment ends could inform the nucleosome positioning pattern in the open chromatin regions.
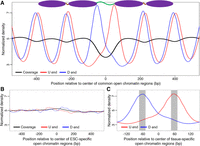
cfDNA fragmentation patterns in pooled healthy nonpregnant subjects in common open chromatin regions (shared by T cells and the liver; deduced nucleosome positioning was also plotted) (A) and embryonic stem cell (ESC)-specific open chromatin regions (B). (C) Illustration of the concept of orientation-aware cfDNA fragmentation (OCF) value. We focused on the center of tissue-specific open chromatin regions and measured the differences between U and D signals in the shaded regions as the OCF value for the corresponding tissue.
As a negative control, we used the same data set to analyze the cfDNA fragmentation patterns around open chromatin regions that were specific to embryonic stem cells (ESCs) (Supplemental Table S1; Methods). We reasoned that no plasma DNA would come from ESCs in healthy adults. Indeed, nucleosome positioning patterns (e.g., nucleosome-depletion in the center) could not be seen in the ESC-specific open chromatin regions (Fig. 3B).
We further hypothesized that cfDNA would only show the characteristic fragmentation patterns at open chromatin regions where the corresponding tissues contributed DNA into the plasma. To test this hypothesis, besides T cells and the liver, we mined tissue-specific open chromatin regions for five additional major human tissues (i.e., the placenta, lungs, ovary, breast, and small intestines) (Methods). The selection of these tissues was based on data availability and previous knowledge that they would contribute DNA into the plasma in selected clinical scenarios. Hence, in previous works, researchers had shown that the placenta-, lung-, ovary-, and breast-derived DNA could be found in the plasma of pregnant women, lung cancer, ovarian cancer, and breast cancer patients, respectively (Lo et al. 1997; Chim et al. 2005; Christie et al. 2017; Hulbert et al. 2017; O'Leary et al. 2018). In addition, colonic DNA could be found in the plasma of colorectal cancer patients (Strickler et al. 2018). We could not find any publicly accessible open chromatin data for colonic tissues with adequate quality in the present study. Hence, we used the data from the small intestines to represent the gastrointestinal system and considered small intestine-specific open chromatin regions as a surrogate for colonic ones. These open chromatin regions were mentioned as “intestine-specific” thereafter. We believed that our decision was justified because the epigenomic profiles of the small intestines and the colon shared much similarity (Roadmap Epigenomics Consortium et al. 2015).
In total, ∼25,000 tissue-specific open chromatin regions were obtained for each tissue type (range: 7540–55,537) (Supplemental Table S1). We then investigated the cfDNA fragmentation patterns in these tissue-specific open chromatin regions in the plasma of healthy individuals. The results from a typical case were shown in Supplemental Figure S1. As expected, plasma DNA showed nucleosome-depletion and well-phased nucleosome arrays in the T cell- and liver-specific open chromatin regions, but not in other tissue-specific open chromatin regions. These results were consistent with the fact that the hematopoietic system and the liver were the major contributors of plasma DNA in healthy individuals (Lui et al. 2002; Sun et al. 2015), suggesting that differentially phased cfDNA fragment ends may be informative in inferring the tissue origin of cfDNA.
Quantification of differentially phased cfDNA fragment ends
To explore the potential in inferring the relative contributions of various tissues in the plasma DNA pool, we developed a novel approach to measure the differential phasing of upstream (U) and downstream (D) fragment ends in tissue-specific open chromatin regions. We called this strategy orientation-aware cfDNA fragmentation (OCF) analysis. OCF values are based on the differences in U and D end signals in the center of the relevant open chromatin regions (Methods). As shown in Figure 3A, for tissues that contributed DNA into plasma, one would expect much cfDNA fragmentation to have occurred at the nucleosome-depleted region in the center of the corresponding tissue-specific open chromatin regions. In such a region, U and D ends exhibited the highest read densities (i.e., peaks) at ∼60 bp from the center, whereas the peaks for U and D ends were located on the right- and left-hand sides, respectively. Conversely, this pattern would not be expected for tissue-specific open chromatin regions where the corresponding tissue did not contribute DNA into the plasma (e.g., ESC in Fig. 3B). We thus measured the differences of U and D end signals in 20-bp windows around the peaks (shadowed regions in Fig. 3C) in the tissue-specific open chromatin regions as the OCF value for the corresponding tissue.
As a result, for tissues that contributed DNA into the plasma, positive OCF values would be expected. Otherwise, the OCF values should be zero or negative. OCF values for the seven tissue types in the 32 healthy individuals were shown in Figure 4 and Supplemental Table S2. All subjects showed positive OCF values for T cells and the liver; in addition, OCF values for T cells were higher than those for the liver in all cases (P < 0.001, Wilcoxon signed-rank test). OCF values for other tissue types were much lower and were close to or below zero. These results were consistent with previous data showing that in healthy individuals, the majority of plasma DNA originated from the hematopoietic system and the liver, with the former being the most dominant source (Lui et al. 2002; Sun et al. 2015). Our results thus showed utility of the OCF values in measuring the relative contributions of various tissues into the plasma DNA pool.
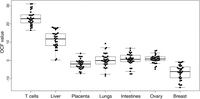
Quantification of cfDNA fragmentation patterns (OCF values) among various tissues in healthy nonpregnant subjects.
Application in noninvasive prenatal testing
To demonstrate the utility of our approach in noninvasive prenatal testing, we retrieved maternal plasma DNA data from a previous study (Chan et al. 2016). As discussed in “Introduction,” circulating fetal DNA in the plasma of pregnant women mostly originated from the placenta (Chim et al. 2005). cfDNA fragmentation patterns around the placenta-specific open chromatin regions in a third-trimester pregnant case were shown in Figure 5A. A strong nucleosome positioning pattern similar to that of common open chromatin regions in healthy nonpregnant individuals (Fig. 3A) could be observed, suggesting that cfDNA fragmentation pattern analysis could indeed detect the fetal/placental DNA in maternal plasma.
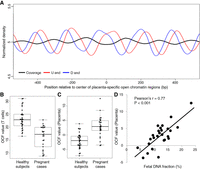
Application of cfDNA fragmentation pattern analysis in noninvasive prenatal testing. (A) cfDNA fragmentation patterns in the placenta-specific open chromatin regions in one pregnant case. (B,C) Comparison of OCF values between healthy nonpregnant subjects and pregnant women: (B) T cells; (C) placenta. (D) Correlation between OCF values for the placenta and fetal DNA fractions in a cohort of 26 pregnant women.
We further investigated the cfDNA fragmentation patterns using previously published data from a cohort of 26 first-trimester pregnant cases (Chan et al. 2016). Each case in this cohort was carrying a male fetus. Hence, the fetal DNA fraction in plasma DNA could be determined by analyzing the reads aligned to the Y Chromosome (Chan et al. 2016). When compared to the nonpregnant healthy subjects, OCF values for T cells were significantly decreased in the pregnant samples, and only OCF values for the placenta showed significant elevation (both P < 0.001, Mann–Whitney U test) (Fig. 5B,C; Supplemental Table S2). In addition, a strong positive correlation between OCF values for the placenta and fetal DNA fractions could be observed (R = 0.77, P < 0.001, Pearson correlation) (Fig. 5D). Notably, this R-value was higher than that obtained by our previous fetal-specific preferred end sites approach (which was 0.66) (Chan et al. 2016). The fetal DNA fraction is one of the most important parameters governing the performance of noninvasive prenatal testing. These results thus demonstrated the potential utility of differentially phased cfDNA fragment ends in noninvasive prenatal testing.
Application in liver transplantation and hepatocellular carcinoma patients
To investigate the performance of cfDNA fragmentation pattern analysis in predicting the contribution of the liver tissue, plasma DNA sequencing results from a previously reported cohort of 14 liver transplantation patients were retrieved (Gai et al. 2018). For each case, both the donor and recipient were genotyped such that donor-specific informative SNP sites could be identified to deduce the donor DNA fraction in recipient's plasma (Gai et al. 2018). When the cfDNA fragmentation pattern analysis was performed on this data set, a positive correlation between OCF values for the liver and donor DNA fractions could be observed (R = 0.74, P = 0.0022, Pearson correlation) (Fig. 6A; Supplemental Table S2).
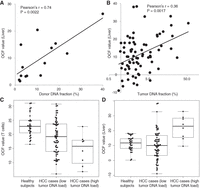
Application of cfDNA fragmentation pattern analysis in liver transplantation and hepatocellular carcinoma (HCC) patients. (A) Correlation between OCF values for the liver and donor DNA fractions in liver transplantation patients; (B) tumor DNA fraction in HCC cases. (C) Comparison of OCF values for T cells; (D) the liver among healthy nonpregnant subjects and HCC cases (separated into two groups based on the tumor DNA load in plasma).
In addition, we also retrieved plasma DNA data from a previously published cohort of 90 HCC patients (Jiang et al. 2015). For these HCC patients, the tumor DNA fractions in plasma were estimated by copy number aberration (CNA) analyses (Jiang et al. 2015). Through such analyses, 74 HCC samples showed evidence of presence of tumor DNA in plasma and were used in the following analyses. Notably, in these HCC patients, the tumor-derived cfDNA molecules were considered as of liver origin (Sun et al. 2015; Gai et al. 2018). A positive correlation between OCF values for the liver and the tumor DNA fractions could be observed (R = 0.36, P = 0.0017, Pearson correlation) (Fig. 6B; Supplemental Table S2). Moreover, a previous study had reported that in HCC patients, Chr 1q and Chr 8q frequently exhibited copy number gains, whereas Chr 1p and Chr 8p frequently exhibited copy number losses (Jiang et al. 2015). To explore the effect of copy number changes on our approach, we recalculated the OCF values for the regions with copy number gains and losses separately. As a result, similar correlations between the recalculated OCF values for the liver and the tumor DNA fractions were obtained (Pearson's r = 0.32 and 0.37 for copy number gain and loss regions, respectively), suggesting that CNA profile was not a significant confounder for this analysis.
Furthermore, we separated the HCC patients into two subgroups based on the tumor DNA fraction: “Low tumor DNA load” group contained those with tumor DNA load <10%, and “high tumor DNA load” group for the remaining cases. This separation was based on the knowledge that the liver contributes ∼10% plasma DNA in healthy subjects (Sun et al. 2015). The median tumor DNA loads for these two groups were 2.1% (range: 0.9%–9.9%) and 19.4% (range: 10.8%–53.2%), respectively (Supplemental Table S2). As shown in Figure 6C,D, when compared to the healthy subjects, OCF values for T cells were significantly decreased for both HCC patient groups (P = 0.0016 and P < 0.001 for low and high tumor DNA load groups, respectively, Mann–Whitney U test). OCF values for the liver showed no statistical difference in low tumor DNA load group patients but they were significantly elevated in high tumor DNA load group patients (P = 0.32 and P < 0.001 for low and high tumor DNA load groups, respectively, Mann–Whitney U test). We further explored the performance of the OCF values in differentiating HCC cases from healthy subjects (e.g., in cancer testing) and found that 50 of 74 HCC cases (sensitivity = 67.6%) and 30 of 32 healthy subjects (specificity = 93.8%) could be correctly classified. Such performance was comparable to our previous study based on CNA and DNA methylation analyses (Chan et al. 2013a). Taken together, these results suggested that our method might have applications in liver transplantation monitoring and cancer testing.
Application in colorectal cancer and lung cancer patients
A cohort of 11 colorectal cancer (CRC) patients was newly recruited in this study. For each case, plasma DNA was bisulfite sequenced (Methods) such that the colonic contribution could be determined using the plasma DNA tissue mapping approach (Sun et al. 2015). These results allowed us to explore the use of cfDNA fragmentation pattern analysis in BS-seq data. In the cfDNA of such individuals, we observed characteristic fragmentation patterns in the intestine-specific open chromatin regions. One typical case was shown in Supplemental Figure S2A. When compared to the healthy subjects, OCF values for T cells were significantly decreased, whereas OCF values for intestines were significantly elevated in the CRC patients (both P < 0.001, Mann–Whitney U test) (Fig. 7A,B; Supplemental Table S2). A positive correlation between OCF values for intestines and colonic contributions (measured by the plasma DNA tissue mapping approach) (Sun et al. 2015) could be observed (R = 0.89, P < 0.001, Pearson correlation) (Fig. 7C).
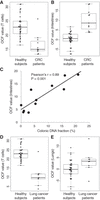
Application of cfDNA fragmentation pattern analysis in colorectal cancer (CRC) and lung cancer patients. (A,B) Comparison of OCF values between healthy nonpregnant subjects and CRC patients: (A) T cells; (B) intestines. (C) Correlation between OCF values for intestines and colonic DNA fractions (deduced by plasma DNA tissue mapping method) in CRC patients. (D,E) Comparison of OCF values between healthy nonpregnant subjects and lung cancer patients: (D) T cells; (E) lungs.
In addition, plasma DNA data for nine lung cancer patients were obtained from a data set generated by Snyder et al. (2016). We found that plasma DNA showed characteristic fragmentation patterns in the lung-specific open chromatin regions in these patients. One typical case was shown in Supplemental Figure S2B. OCF values for T cells were decreased, whereas OCF values for lungs were elevated when compared to the healthy subjects (P < 0.001 and P = 0.025 for T cells and lungs, respectively, Mann–Whitney U test) (Fig. 7D,E; Supplemental Table S2). Moreover, two lung cancer cases in this cohort had also been analyzed by Snyder et al. (2016); however, there were no significant correlations between our OCF values and the results from the most relevant tissue/cell types in Snyder et al. (2016) for these two cases (both P > 0.05, Spearman's correlation) (Supplemental Table S2).
Discussion
In this proof-of-principle study, we proposed a new method for nucleosome positioning profiling and quantitative determination of the relative contributions of various tissues in plasma DNA by fragmentation pattern analyses. We also demonstrated the diagnostic potential of this approach in noninvasive prenatal testing, organ transplantation monitoring, as well as cancer testing. We showed that cfDNA fragmentation patterns bore characteristic profiles in the nucleosome-depleted region and well-phased nucleosome arrays around the tissue-specific open chromatin regions. The results could be regarded as in vivo counterpart of previous studies using in vitro micrococcal nuclease or transposase digestion of genomic DNA (Schones et al. 2008; Gaffney et al. 2012; Schep et al. 2015). In the meantime, one could see that nucleosome positioning patterns in the tissue-specific open chromatin regions (e.g., Fig. 5A) were not as strong as those in the common open chromatin regions (Fig. 3A). The reason was that in the common open chromatin regions, most of the tissues that contribute plasma DNA possess well-positioned nucleosomes. However, for the tissue-specific ones, only the corresponding tissue possesses well-positioned nucleosomes. Such tissue-specific signals would be diluted by other tissues that did not have that pattern. Our data showed that a higher OCF value generally indicated a higher contribution of the corresponding tissue.
The ability to trace the tissue origin of cfDNA is of great interest in liquid biopsy, especially in predicting the tumor origin in cancer patients. We showed that by quantifying the cfDNA fragmentation patterns in cancer patients, OCF values for T cells would decrease and OCF values for the tissue origin of the tumor would increase. These observations were consistent with the fact that in these patients, the tumor (and peri-tumoral) tissues release DNA into the plasma which would (1) increase the contribution from the tissue origin of the tumor, and (2) dilute the contribution of the hematopoietic system. In addition, the results on the CRC cases (Fig. 7C) showed that this approach was highly concordant with the plasma DNA tissue mapping method (Sun et al. 2015). It is interesting to note that cfDNA fragmentation patterns are preserved among the bisulfite-converted DNA molecules. This is likely to be partly related to our library preparation protocol whereby sequencing adaptors were first ligated to plasma DNA molecules before bisulfite treatment (Lun et al. 2013). It would be interesting to explore if there would be additive value of using both OCF measurement and methylation-based tissue mapping in a synergistic manner to further enhance the performance of the tissue-of-origin analysis. Nonetheless, here we demonstrated that OCF analysis is an approach that provides tissue-of-origin information without reliance of methylation analysis. Compared to BS-seq, DNA-seq experiments were cheaper and involve simpler protocols.
Furthermore, there are important differences between our approach and previous methods. For instance, Snyder et al. (2016) had correlated the nucleosome spacing patterns in gene bodies with the transcriptome of various cell/tissue types and used the Pearson correlation coefficients to rank the relative contribution of each cell/tissue type. However, it has not yet been demonstrated that the Pearson correlation coefficients could be used for measuring the relative quantitative contributions for each cell/tissue type. In contrast, we used a different strategy based on fragment end profiles in the tissue-specific open chromatin regions and demonstrated that the OCF values were quantitatively correlated with relative contributions of the corresponding tissues, as validated in pregnancy, liver transplantation, and cancer models. Moreover, no significant correlations between our approach and Snyder et al. (2016) were found in the lung cancer cases that were analyzed by both methods. It is possible that the approach by Snyder et al. (2016) might be better for ranking analysis, whereas ours might have some advantages for quantification. As shown in Figure 3C, by taking the orientation information into account, we were able to differentiate the informative end signal from the uninformative one, which procedure might benefit the quantification performance. Ranking and quantification approaches are both valuable and may find preferential uses in different scenarios. However, considering the small sample size investigated here, comprehensive performance evaluations using larger sample cohorts may be needed. In addition, our method was based on open chromatin profiles, where data availability is not currently as accessible as the transcriptome data utilized by Snyder et al. (2016). Such relative paucity of data may limit the immediate applications of our approach. To this end, we think that newer approaches such as assay for transposase-accessible chromatin using sequencing (ATAC-seq) (Buenrostro et al. 2013) may provide open chromatin data that could be used by our approach. On the other hand, Ulz et al. (2017) demonstrated the potential of plasma DNA coverage pattern analysis in inferring the expression of genes thus revealing the tissue origin of tumors in cancer patients. However, the authors estimated that a 75% tumor DNA fraction in the plasma might be required for this purpose (Ulz et al. 2017), which was unlikely to be seen in most clinical cases. In contrast, our approach could work on cases with a much lower fraction of DNA from the tissue of interest. For instance, in CRC cases, elevated OCF values for intestines were already apparent when the colon contribution was only 5% (Fig. 7B). These results thus suggested that our approach might have the potential to work on relatively early cancer cases in which the tumor DNA load in the plasma might not be high.
The selection of a shortlist of tissues is important for the performance of our approach. In this study, we only considered tissues with known contributions to plasma DNA in certain clinical scenarios and reported tissue-level contributions. For instance, we used T cells as a representative of the hematopoietic system when considering the overall contribution from the hematopoietic system into the circulating DNA pool. We have adopted this approach because it is difficult to mine highly abundant and specific open chromatin regions if more than one blood cell type is included due to the high epigenomic similarity among these cells. However, for cells from different tissues where the epigenomes are generally distinct from each other (Roadmap Epigenomics Consortium et al. 2015), we could find many specific open chromatin regions that one could use. In this regard, we think that our approach has the potential to be applied to larger compendiums that contain additional tissue types (e.g., prostate and kidney).
In the future, our approach can be integrated with targeted massively parallel sequencing technology (Mertes et al. 2011) to analyze plasma DNA. Because the tissue-specific open chromatin regions only account for a very small proportion of the human genome, through designing hybridization probes to capture these regions, the cost of our approach can be substantially reduced. There is also a need to generate open chromatin data for many tissues of interest and to validate our approach on large sample sets covering different clinical scenarios.
Methods
Plasma DNA data collection and availability
This study was approved by the Joint Chinese University of Hong Kong and Hospital Authority New Territories East Cluster Clinical Research Ethics Committee. All participating subjects in this study gave written informed consent. All the plasma DNA data analyzed in this study were generated in paired-end sequencing mode. Plasma DNA data for healthy individuals, HCC patients, and pregnant cases were retrieved from the European Genome-phenome Archive (EGA; accession numbers EGAS00001001024 and EGAS00001001882) (Jiang et al. 2015; Chan et al. 2016); data for the liver transplantation patients (Gai et al. 2018) have been submitted to EGA (accession number EGAS00001003116); data for the lung cancer cases were obtained from Gene Expression Omnibus (GEO; accession number GSE71378) (Snyder et al. 2016).
CRC patients were newly recruited in this study. Peripheral blood samples were collected into EDTA-containing tubes. Blood samples were centrifuged at 1600g for 10 min at 4°C. The plasma portion was harvested and recentrifuged at 16,000g for 10 min at 4°C to remove the blood cells. Bisulfite conversion was performed as previously described (Lun et al. 2013). DNA libraries were prepared using the KAPA HTP Library Preparation Kit (Kapa Biosystems) according to the manufacturer's instructions (Chan et al. 2013b) and sequenced on a HiSeq 2000 system (Illumina) in 75 × 2 cycles mode with the TruSeq SBS Kit v3 (Illumina). Analysis of the BS-seq data, including quality control, sequence alignment, methylation status determination, and colon contribution inference were performed as previously described (Jiang et al. 2014; Sun et al. 2015). The median sequencing depth for these samples was 3.2-fold (range: 0.6-fold to 6.4-fold; Supplemental Table S2) haploid human genome coverage. The data have been submitted to EGA (accession number EGAS00001003117).
Tissue-specific open chromatin regions
We used the publicly available DNase-seq (DNase I hypersensitive sites sequencing) (Song and Crawford 2010) data to mine open chromatin regions. DNase-seq data for T cells, the placenta, lungs, the ovary, the breast, small intestines, and embryonic stem cells were obtained from the Roadmap Epigenomics Project (Roadmap Epigenomics Consortium et al. 2015); data for the liver was obtained from the ENCODE Project (The ENCODE Project Consortium 2012) and IHEC (Bujold et al. 2016). Accession numbers for the DNase-seq data can be found in Supplemental Table S1. For each tissue type, raw sequencing data were downloaded and aligned to the reference human genome (UCSC hg19) using Bowtie with default parameters (Langmead et al. 2009). Then, open chromatin regions were determined using model-based analysis for ChIP-seq (MACS) with default parameters (Zhang et al. 2008). For such analyses, chromatin immunoprecipitation followed by massively parallel DNA sequencing (ChIP-seq) input data from the corresponding tissue types were used as negative controls and a Q-value (i.e., adjusted P-value reflecting the false discovery rate) cutoff of 0.01 was used to call peaks. For the liver, DNase-seq data from ENCODE and IHEC were both analyzed and only the peaks that existed in both samples were kept; similarly, for lungs, DNase-seq data for IMR-90 (human fetal lung) and human lung fibroblast (HLF) cell lines were both analyzed and only the peaks that existed in both samples were kept. Then, for each tissue type, we compared its peaks with all the other tissues and only kept those unique to this tissue and within a size range of 50–200 bp as the final tissue-specific open chromatin regions. The identified open chromatin regions can be found in Supplemental Table S1. We think that reanalysis of the data using the GRCh38 (UCSC hg38) human reference genome would not affect the results significantly because the biggest difference between these two versions of human reference genome is the annotation of centromeric DNA, which is normally in a heterochromatin state and therefore should not contain open chromatin regions that have been used in our current analysis.
Quantification of cfDNA fragmentation patterns
To quantify the cfDNA fragmentation patterns around tissue-specific open chromatin regions, we focused on the nucleosome-depletion
signal at the center because it was one of the key characteristics (Jiang and Pugh 2009). In such signal, upstream (U) and downstream (D) ends exhibited peaks at ∼60 bp from the center of the open chromatin regions
but in different directions (Fig. 3C). Specifically, the D end peak was on the left-hand side, whereas the U end peak was on the right-hand side (Fig. 3C). We quantified the differences of read densities of the U and D ends in 20-bp windows around the peaks as follows: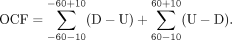 We named this parameter orientation-aware cfDNA fragmentation (OCF) value. Note that this calculation was performed using
the raw, unsmoothed U and D end signals. For each case, OCF values were calculated for the seven tissue types investigated
in this study using their tissue-specific open chromatin regions separately.
We named this parameter orientation-aware cfDNA fragmentation (OCF) value. Note that this calculation was performed using
the raw, unsmoothed U and D end signals. For each case, OCF values were calculated for the seven tissue types investigated
in this study using their tissue-specific open chromatin regions separately.
Data access
Plasma DNA sequencing data from 14 liver transplantation recipients and 11 CRC patients (all had consented to data archiving) have been submitted to the European Genome-phenome Archive (https://www.ebi.ac.uk/ega/) hosted by the European Bioinformatics Institute under accession numbers EGAS0000 1003116 and EGAS00001003117. Implementation codes for the method descripted in this study are provided in Supplemental Code.
Competing interest statement
K.C.A.C., R.W.K.C., and Y.M.D.L. hold equities in DRA, Take2 Health, and Grail, are consultants to Grail, and receive research funding from Grail/Cirina. P.J. is a consultant to Grail. Y.M.D.L. is a scientific cofounder and a member of the scientific advisory board for Grail. A patent application based on the method described in this manuscript has been filed.
Acknowledgments
We thank Ms. Coral Lee and Mr. Tao Liu for technical assistance, and Ms. Qi Wang for encouragement. This work was supported by the Research Grants Council of the Hong Kong SAR Government under the Theme-based research scheme (T12-403/15-N and T12-401/16-W), a collaborative research agreement from Grail and the Vice Chancellor's One-Off Discretionary Fund of The Chinese University of Hong Kong (VCF2014021). Y.M.D.L. is supported by an endowed chair from the Li Ka Shing Foundation.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.242719.118.
-
Freely available online through the Genome Research Open Access option.
- Received August 7, 2018.
- Accepted January 25, 2019.
This article, published in Genome Research, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








