
Transcription factor activity and nucleosome organization in mitosis
- Nicola Festuccia1,2,5,
- Nick Owens1,2,5,
- Thaleia Papadopoulou1,2,
- Inma Gonzalez1,2,
- Alexandra Tachtsidi1,2,3,
- Sandrine Vandoermel-Pournin4,
- Elena Gallego1,2,
- Nancy Gutierrez1,2,
- Agnès Dubois1,2,
- Michel Cohen-Tannoudji4 and
- Pablo Navarro1,2
- 1Epigenetics of Stem Cells, Department of Developmental and Stem Cell Biology, Institut Pasteur, CNRS UMR3738, 75015 Paris, France;
- 2Equipe Labellisée LIGUE Contre le Cancer;
- 3Sorbonne Université, Collège Doctoral, F-75005 Paris, France;
- 4Mouse Functional Genetics, Department of Developmental and Stem Cell Biology, Institut Pasteur, CNRS UMR 3738, 75015 Paris, France
-
↵5 These authors contributed equally to this work.
Abstract
Mitotic bookmarking transcription factors (BFs) maintain the capacity to bind to their targets during mitosis, despite major rearrangements of the chromatin. While they were thought to propagate gene regulatory information through mitosis by statically occupying their DNA targets, it has recently become clear that BFs are highly dynamic in mitotic cells. This represents both a technical and a conceptual challenge to study and understand the function of BFs: First, formaldehyde has been suggested to be unable to efficiently capture these transient interactions, leading to profound contradictions in the literature; and second, if BFs are not permanently bound to their targets during mitosis, it becomes unclear how they convey regulatory information to daughter cells. Here, comparing formaldehyde to alternative fixatives we clarify the nature of the chromosomal association of previously proposed BFs in embryonic stem cells: While ESRRB can be considered as a canonical BF that binds at selected regulatory regions in mitosis, SOX2 and POU5F1 (also known as OCT4) establish DNA sequence-independent interactions with the mitotic chromosomes, either throughout the chromosomal arms (SOX2) or at pericentromeric regions (POU5F1). Moreover, we show that ordered nucleosomal arrays are retained during mitosis at ESRRB bookmarked sites, whereas regions losing transcription factor binding display a profound loss of order. By maintaining nucleosome positioning during mitosis, ESRRB might ensure the rapid post-mitotic re-establishment of functional regulatory complexes at selected enhancers and promoters. Our results provide a mechanistic framework that reconciles dynamic mitotic binding with the transmission of gene regulatory information across cell division.
During mitosis, the chromatin is drastically condensed and reconfigured to enable the equitable partition of the genetic material between the two daughter cells (Ma et al. 2015). This leads to a strong decrease in transcriptional activity and to the general reduction of transcription factor (TF) binding throughout the genome. Loss of TF binding is further accentuated by the stereotypical phosphorylation of many regulators during mitosis, leading to an intrinsic reduction of their ability to bind DNA. This is particularly well illustrated by the systematic phosphorylation of C2H2 zinc finger TFs such as YY1 (Rizkallah and Hurt 2009; Rizkallah et al. 2011) but has also been observed for other TFs such as POU5F1 (also known as OCT4) and SOX2 (Qi et al. 2016; Shin et al. 2016). Moreover, the breakdown of the nuclear envelope, and the consequent increase of the volume that TFs can freely explore, leads to a decrease of TF concentration. This process naturally inhibits the ability of TFs to scan DNA for their binding motifs. Therefore, many processes occur simultaneously to temporarily halt gene regulation and transcription during mitosis.
The mechanisms by which daughter cells accurately re-establish an environment permissive for efficient transcriptional activation early in interphase remain unknown (de Castro et al. 2016). One potential mechanism is known as mitotic bookmarking: Some TFs have the ability to interact with their DNA binding sites during cell division. These TFs, known as mitotic bookmarking factors (BFs), are believed to directly convey gene regulatory information from mother to daughter cells, as illustrated by GATA1 (Kadauke et al. 2012), FOXA1 (Caravaca et al. 2013), and ESRRB (Festuccia et al. 2016). Nonetheless, the molecular mechanisms underpinning this function remain to be elucidated (Festuccia et al. 2017). BFs are highly dynamic during mitosis and often exhibit reduced residence times on the chromatin. Therefore, the function of BFs is not simply mediated by their stable retention at enhancers and promoters. Instead, their transient binding activity may preserve specific chromatin features at bookmarked sites. These features would represent the inherited properties driving and accelerating the reassembly of functional regulatory complexes early in the following interphase. Although the chromatin is highly condensed during mitosis, gene regulatory elements remain globally accessible (Hsiung et al. 2015). This is particularly true at active promoters, perhaps reflecting their low but nevertheless significant mitotic activity, as recently reported (Palozola et al. 2017). Enhancers, in contrast, show more variable degrees of chromatin accessibility. Yet, mitotic chromatin accessibility does not seem to correlate with mitotic binding, at least in the case of bookmarking by GATA1 in erythroblasts (Kadauke et al. 2012). Moreover, the maintenance of chromatin accessibility does not preclude the possibility that nucleosome positioning in mitotic cells is highly modified, as previously suggested (Kelly et al. 2010; Javasky et al. 2018). Hence, further studies are required to clarify whether regulatory elements do indeed maintain a local chromatin architecture compatible with TF binding in mitotic cells and how mitotic bookmarking correlates with and ultimately drives nucleosome organization.
An essential condition to understand mitotic bookmarking processes is to accurately identify BFs and their mitotic binding sites. However, this has remained a difficult task because, as reported nearly 15 yr ago (Pallier et al. 2003), the most commonly used cross-linker, formaldehyde, leads to the artificial depletion of TFs from mitotic chromosomes (Pallier et al. 2003; Teves et al. 2016). To circumvent this problem, mitotic bookmarking activity has been explored using live imaging of tagged TFs. Even so, whether the global chromatin association of certain TFs detected by microscopy reflects the sum of site-specific interactions remains to be demonstrated. Diverse modes of binding, other than those involving base-specific interactions, may be responsible for the global decoration of the chromosomes by TFs, as we proposed earlier (Festuccia et al. 2017) and was clearly demonstrated for FOXA1 (Caravaca et al. 2013). These interactions with the chromatin, or with other constituents of mitotic chromosomes, might be extremely transient and not easily captured by formaldehyde. In support of this distinction between global and site-specific interactions, several TFs have been efficiently captured at their mitotic binding sites using formaldehyde (Festuccia et al. 2017), despite its seeming incapacity to cross-link TFs on mitotic chromosomes. Yet, it remains to be proven whether formaldehyde generally fails in capturing DNA sequence-specific interactions, leading to the loss of enrichment of BFs on the chromosomes, or whether the interactions sustaining the global retention of BFs are distinct from those involved in TF binding to DNA. This does not only represent an important technical question; rather, it directly interrogates the nature and, hence, the function, of the interactions established between TFs and mitotic chromosomes: While global, dynamic, and DNA sequence-independent interactions may increase the concentration of TFs in the vicinity of DNA, possibly facilitating the re-establishment of binding in the following interphase, authentic mitotic bookmarking of promoters and enhancers may confer specificity to these processes and provide robustness to the post-mitotic resuscitation of gene regulatory networks. In this work, we use mouse embryonic stem (ES) cells to study the capacity of different fixatives to cross-link pluripotency TFs either globally on the chromosomes or locally at gene regulatory regions during mitosis. We subsequently use this information to correlate TF binding with chromatin accessibility and nucleosome positioning and stability in interphase and mitosis.
Results
The global localization of ESRRB to mitotic chromosomes is preserved upon DSG fixation
Several TFs have been shown to seemingly coat the mitotic chromosomes when fusion proteins with fluorescent proteins or tags are used in live imaging approaches (Festuccia et al. 2017). This is the case of ESRRB (Festuccia et al. 2016), which we previously showed decorates mitotic chromatin using GFP (Fig. 1A), Tdtomato, and Snap-tag fusions in mouse ES cells. However, upon formaldehyde fixation, several TFs capable of coating the mitotic chromosomes seem to be globally de-localized and cross-linked outside of the chromosomes (Pallier et al. 2003; Teves et al. 2016). We first aimed to test whether this is also the case for ESRRB. As expected, we observed a clear depletion from the mitotic chromosomes (Fig. 1B), which were identified by DAPI (Supplemental Fig. S1) and MKI67 (also known as Ki-67) staining (Fig. 1B), a protein enriched on their periphery (Booth and Earnshaw 2017). We therefore aimed at identifying alternative cross-linking agents that would preserve the chromosomal enrichment of ESRRB. Among the different reagents and protocols that we tested (Supplemental Methods), we found two that clearly allow visualization of ESRRB coating of the mitotic chromosomes: first, the homobifunctional cross-linker disuccinimidyl glutarate (DSG), which, combined to formaldehyde (DSG+FA) (Fig. 1B; Supplemental Fig. S1), has been used to capture hyperdynamic protein–protein interactions due to its capacity to establish amide bonds via two NHS-ester groups (Tian et al. 2012); and second, glyoxal (Supplemental Fig. S1), a small bifunctional aldehyde that has been recently rediscovered for its use in fluorescent microscopy (Richter et al. 2018). With both fixatives, ESRRB is detected covering the entire area delimited by Ki-67, within the chromatin compartment (Fig. 1B; Supplemental Fig. S1). As a control, we stained ES cells for NANOG (Fig. 1A), a TF that is excluded from the mitotic chromatin (Festuccia et al. 2016). Upon DSG+FA or glyoxal fixation, we did not observe retention of NANOG on mitotic chromosomes (Fig. 1B; Supplemental Fig. S1), indicating that these two cross-linkers do not induce a specific aggregation on the chromosomes. We also tested whether DSG would allow us to visualize the global chromosomal retention of ESRRB in vivo. We have shown before that, upon microinjection of Esrrb-Tdtomato mRNA into mouse embryos, the produced fluorescent fusion proteins decorate the mitotic chromatin (Festuccia et al. 2016). Accordingly, when we fixed mouse blastocysts with DSG+FA, we could observe mitotic figures with a clear coating of the chromosomes by ESRRB but not by NANOG (Fig. 1C).
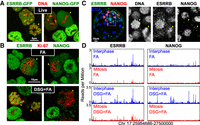
Capturing global ESRRB binding on mitotic chromosomes. (A) Localization of ESRRB-GFP (left) or NANOG-GFP (right) fusion proteins in live cells cultured with Hoechst 33342 (red). (B) ESRRB (left) and NANOG (right) immunofluorescence (green) after fixation with either FA (top) or DSG+FA (bottom). The chromosome periphery of mitotic chromosomes is identified by Ki-67 (red). (C) Immunostaining for NANOG (red) or ESRRB (green) performed on a mouse blastocyst fixed with DSG+FA. Counterstain with Hoechst 33342 is shown in blue. Close-up of two mitotic cells is shown in the right panels (dashed area delimits the selected region). Mitotic cells are indicated in A–C with yellow arrowheads. (D) Representative binding profiles of ESRRB and NANOG across 1.5 Mb in interphase (blue) or mitosis (red), obtained after fixation with either FA (top) or DSG+FA (bottom).
DSG fixation does not alter the profile of ESRRB binding in mitotic cells
Our finding that DSG and glyoxal maintain the global association of ESRRB with the mitotic chromosomes opens the possibility to test whether this binding results from the sum of site-specific interactions or from other mechanisms. Indeed, if the global staining reflected site-specific interactions exclusively, one should expect to identify a much larger number of ESRRB binding sites by chromatin immunoprecipitation (ChIP) after fixation with DSG or glyoxal than with FA. Yet, despite our efforts, we could not perform ChIP with these two reagents; in contrast, after a double cross-linking with DSG followed by FA (DSG+FA), which is frequently used in biochemical approaches (Tian et al. 2012), ChIP was particularly efficient. Therefore, we performed ChIP-seq in asynchronous (hereafter, interphase) and mitotic preparations of ES cells (>95% purity); after splitting the populations in two, we proceeded in parallel with either FA or DSG+FA cross-linking. We observed very similar profiles of ESRRB binding both in interphase and in mitosis, irrespective of whether the cells had been cross-linked with FA or with DSG+FA (Fig. 1D; Supplemental Fig. S2). Therefore, whereas ESRRB is globally cross-linked outside or within the mitotic chromosomes by FA and DSG, respectively, the mitotic ChIP signal does not vary dramatically. We note, however, that DSG+FA provides higher ChIP signal and a better signal-to-background ratio, both in interphase and in mitosis. In agreement with immunostaining and live imaging, NANOG binding is globally lost, both in FA and in DSG+FA (Fig. 1D; Supplemental Fig. S2). From this analysis, we conclude that the global coating and the interaction of ESRRB with specific sites are two distinct phenomena. While mitotic ESRRB bookmarking (i.e., binding to specific sites) can be revealed by FA and by DSG+FA, the global coating visible by microscopy is only preserved by DSG (or glyoxal).
DSG versus FA comparisons reveal different behaviors of other proposed BFs
In addition to ESRRB, other pluripotency TFs have been proposed to act as BFs in ES cells (Deluz et al. 2016; Teves et al. 2016; Liu et al. 2017), although evidence is contradictory. SOX2 has been consistently shown by microscopy to globally associate with mitotic chromosomes in three independent studies (Deluz et al. 2016; Teves et al. 2016; Liu et al. 2017). In contrast, while one study (Deluz et al. 2016) reported by ChIP-seq that SOX2 binds with poor efficiency to a few dozen regions in mitosis (compared to thousands of sites in interphase), another study claimed that SOX2 and POU5F1 remain bound to virtually all their interphase targets (Liu et al. 2017). In addition, SOX2 and POU5F1 were shown to be phosphorylated by Aurora kinases, which inhibit DNA binding in mitotic cells (Qi et al. 2016; Shin et al. 2016). These studies performed ChIP after FA fixation, which leads to an apparent depletion from mitotic chromosomes of both SOX2 and POU5F1 (Fig. 2A,C). In contrast, we found that SOX2 displays bright signal all over the chromosomal arms, within the Ki-67 delimited region, by immunostaining after DSG+FA and glyoxal fixation (Fig. 2A; Supplemental Fig. S1). We thus extended our ChIP-seq analysis based on DSG+FA fixation to SOX2. Whereas DSG+FA dramatically increases ChIP efficiency of SOX2 compared to FA, the profiles in mitosis are very similar for both fixatives, with little evidence for mitotic bookmarking activity (Fig. 2C; Supplemental Fig. S2). Therefore, while displaying a global behavior similar to ESRRB, mitotic SOX2 does not appear to be an efficient BF. Next, we analyzed POU5F1 binding. By immunofluorescence, we observed a nearly complete depletion from the chromosomal arms in mitosis, both after DSG+FA and glyoxal (Fig. 2C; Supplemental Fig. S1). In agreement, ChIP-seq analysis also showed almost complete loss of POU5F1 binding at its interphase targets (Fig. 2D; Supplemental Fig. S2). Our results are in agreement with a number of other studies (Deluz et al. 2016; Shin et al. 2016; Kim et al. 2018). However, even using DSG+FA, we could not reproduce recent results showing mitotic bookmarking by SOX2 and POU5F1 (Liu et al. 2017). The use of inhibitors of MEK/GSK3B in the conflicting publication, which leads to a reinforcement of the pluripotency network's activity, cannot account for these differences (Supplemental Fig. S3). Considering that only site-specific interactions mediate mitotic bookmarking activity (Festuccia et al. 2017) and that only residual signal can be detected at some POU5F1/SOX2 regions (Fig. 2B; Supplemental Fig. S2), we conclude from our data that neither SOX2 nor POU5F1 can be considered as potent BFs. Hence, while a global enrichment of TFs can be detected on the chromatin by microscopy, the establishment of site-specific interactions with regulatory elements is a property of selected bookmarking factors like ESRRB.
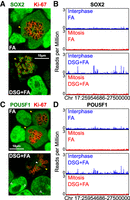
SOX2 and POU5F1 do not bind at regulatory regions in mitosis. (A) SOX2 immunofluorescence (green), after fixation with either FA (top) or DSG+FA (bottom). The mitotic chromosome periphery is identified by Ki-67 (red). (B) Representative binding profiles of SOX2 presented as in Figure 1D. (C,D) Results of the same analyses described in A and B are shown for POU5F1. Mitotic cells are indicated in A and C with yellow arrowheads. In C, the filled white arrowheads point to the centromeres, the empty white arrowheads to the PCH.
DSG enables capturing transient interactions at different chromatin compartments
The global retention of SOX2 on mitotic chromosomes was validated using live imaging of ectopically expressed SOX2-GFP (Fig. 3A, top). Similarly, the large exclusion of POU5F1 was also confirmed by live imaging (Fig. 3A, bottom). Careful examination of the POU5F1 stainings (after DSG+FA and glyoxal but not FA fixation) (Fig. 2C; Supplemental Fig. S1), as well as of unfixed POU5F1-GFP (Fig. 3A, bottom), did, however, unmask a previously unnoticed accumulation of this TF at DAPI-rich regions, the chromocenters (Saksouk et al. 2015), where several centromeres cluster together to form pericentric heterochromatin (PCH) (white arrowheads in Fig. 2C). Moreover, in mitotic cells we could also observe focal enrichment of POU5F1 at centromeric regions (filled white arrowheads in Fig. 2C and in Supplemental Fig. S1). This characteristic pattern of colocalization with the PCH was further validated by live imaging using endogenously expressed POU5F1-RFP fusion proteins (Supplemental Fig. S4A). The same results were obtained in cells cultured in regular conditions or with inhibitors of MEK/GSK3B (Supplemental Fig. S4A). In the latter conditions, PCH shifts from H3K9me3 to H3K27me3 (Tosolini et al. 2018), indicating that the PCH association of POU5F1 is independent of the presence of specific heterochromatic marks. Notably, Aurora kinase b, which has been shown to phosphorylate POU5F1 in mitotic cells to inhibit DNA binding (Qi et al. 2016; Shin et al. 2016), also stains PCH in interphase and the centromeres in mitosis (Supplemental Fig. S4B). Moreover, in the presence of the Aurora kinase inhibitor, Hesperadin, a slight increase of POU5F1 coating throughout the chromosomal arms could be observed (Supplemental Fig. S4C). Hence, using alternative fixatives to FA not only enables the visualization of the genuine mitotic localization of TFs but may also reveal additional activities in interphase. We then asked whether the interactions of SOX2 and POU5F1 unmasked by DSG and glyoxal are indeed dynamic, as generally reported (Teves et al. 2016). We observed highly dynamic interactions, both in interphase and in mitosis, for all three factors fused to GFP and analyzed in parallel experiments (Fig. 3B,C; Supplemental Fig. S5). ESRRB and POU5F1 displayed faster fluorescent recovery after photobleaching (FRAP) in mitosis than in interphase (Fig. 3B,C). This is particularly true for the interaction of POU5F1 with PCH, which is already very dynamic in interphase (Fig. 3C). In reciprocal experiments, we assessed fluorescence loss in photobleaching (FLIP) (Supplemental Fig. S5). We could not identify any significant remnant signal on mitotic chromatids after one minute of continuous bleaching of the freely diffusing TF molecules. Hence, DSG (and glyoxal) are capable of capturing the highly dynamic interactions established by ESRRB/SOX2 on the chromosomal arms and by POU5F1 in PCH.
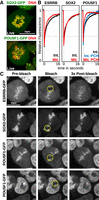
The interactions captured by DSG are dynamic. (A) Localization of SOX2-GFP (top) or POU5F1-GFP (bottom) fusion proteins (green) in live cells cultured with Hoechst 33342 (red). Arrowheads indicate pericentric heterochromatin foci (PCH) in interphase (yellow) and centromeres in mitosis (white). (B) Quantifications of FRAP experiments in interphase (black) and mitosis (red) performed in cells expressing ESRRB-GFP or SOX2-GFP. For cells expressing POU5F1-GFP, recovery of fluorescence at (blue) or outside of (black) pericentric heterochromatin foci is displayed for interphase. Recovery at PCH is displayed for mitosis (red). The y-axis shows the mean percentage of fluorescence relative to prebleach levels detected in multiple independent experiments; the x-axis shows the time after bleaching. (C) Representative examples of ESRRB-GFP, SOX2-GFP, and POU5F1-GFP signal on mitotic chromosomes before and after bleaching, at the indicated time. For POU5F1-GFP, the recovery of signal at PCH is also shown for cells in interphase (bottom).
ESRRB is the only prominent BF among ESRRB, SOX2, POU5F1, and NANOG
Using the collection of data sets generated for ESRRB, SOX2, POU5F1, and NANOG in interphase and in mitosis (Supplemental Table S1 provides an overview of the samples and their relevant statistics), we sought to comprehensively identify regions subject to mitotic bookmarking. To this end, we first identified the binding regions of individual TFs (listed in Supplemental Table S2; Supplemental Fig. S6) and confirmed that only ESRRB displays clear and frequent binding in mitosis (Fig. 4A); for POU5F1, SOX2, and NANOG, only the regions displaying very high levels of binding in interphase show residual ChIP signal in mitosis, especially in DSG+FA, where the number of detected peaks is increased in both conditions. Peaks that were called only in DSG+FA, and neither in our FA samples nor in other publicly available data sets (Chen et al. 2008; Marson et al. 2008; Aksoy et al. 2013; Whyte et al. 2013), tend to be smaller (Supplemental Fig. S7). Nevertheless, their signal is clearly above background in all the analyzed data sets of interphase cells fixed with FA (Supplemental Fig. S7). Hence, DSG helps capture regions displaying low levels of binding and increases the overall efficiency of the ChIP. Nonetheless, it does not specifically unmask new binding in mitosis. We then used a statistical differential occupancy approach to define regions as bookmarked or lost (see Supplemental Methods for details and Supplemental Table S2). We found 10,144 regions bookmarked by ESRRB, representing 29.9% of its interphase sites. All other factors displayed a drastic contraction in binding in mitosis: 574 regions for SOX2 (2% of interphase targets); 102 regions for POU5F1 (0.6%); and 18 regions for NANOG (0.07%). Strong ESRRB binding motifs were identified at the vast majority of ESRRB bookmarked regions (73.4%, score > 12) (Fig. 4B, left) but only at a smaller subset of the regions losing binding in mitosis (34.9%, score > 12). In contrast, regions losing ESRRB binding displayed an increased occurrence of POU5F1/SOX2 composite motifs (Fisher P < 7 × 10−45, score > 12). We observed a scaling relationship: Regions containing high quality POU5F1/SOX2 motifs exhibit a higher tendency to lose ESRRB binding in mitosis (Fig. 4B, right). Previously, we used titration experiments to investigate whether the binding levels seen for ESRRB in mitosis could be explained by contamination from interphase (Festuccia et al. 2016); all our mitotic preparations have <5% of remnant interphase cells and typically between 2 and 4%. We repeated this analysis for SOX2, given the relatively high number of low mitotic peaks that we detected in comparison to POU5F1 and NANOG. To produce SOX2-depleted chromatin, we generated an ES cell line with (1) both endogenous Sox2 alleles tagged with an auxin-inducible degradation domain (producing SOX2-AID fusion), and (2) a constitutive transgene expressing the TIR1 protein inserted at the Tigre locus (Madisen et al. 2015). Upon treatment with the auxin analog IAA for 2 h, a significant reduction of SOX2-AID levels was observed (Fig. 4C). To further deplete SOX2, cells were differentiated in the presence of retinoic acid (RA) and IAA for 4 d. Gradually increasing amounts of WT chromatin were then spiked into chromatin prepared from IAA-RA treated cells, and ChIP-seq analysis performed. We found that as little as 5% of WT chromatin was sufficient to detect clear SOX2 peaks of reduced enrichment (Fig. 4D). The amount of signal observed by adding 5% of contaminant chromatin was higher, on average, than that seen in mitosis at the regions potentially bookmarked by SOX2 (Fig. 4D). Therefore, it is possible that a significant fraction of the regions seemingly bound by SOX2 in mitosis, as well as the absolute levels of enrichment in mitosis, results from the small percentage of contaminant interphase cells in our preparations. To further corroborate that SOX2 is not an efficient bookmarking factor, we turned to a functional assay. Confirming our previous results, the set of ESRRB bookmarked regions identified here tend to be enriched in the vicinity of genes that are controlled by this TF in early G1 (Fig. 4E; Festuccia et al. 2016). We then introduced a GFP-CCNA cell-cycle reporter (Festuccia et al. 2016) into SOX2-AID cells, treated them with IAA for 2 h, and sorted early G1 cells to perform RNA-seq analyses. In comparison with ESRRB, we found a rather minor statistical association between the genes controlled by SOX2 in early G1 (Supplemental Table S3) and the regions potentially bookmarked by SOX2 (Fig. 4E). We conclude that, while we cannot fully rule out that SOX2 may display minimal bookmarking activity, only ESRRB represents a potent and functionally relevant BF among the tested pluripotency factors. This conclusion is particularly well illustrated when the ChIP signal measured at each region is plotted in interphase versus mitosis (Fig. 4A, bottom panels), or when the proportion of reads on peaks are calculated for each TF (Fig. 4F). Why SOX2 and POU5F1 have been previously found mitotically bound at most of their interphase targets (Liu et al. 2017) remains, therefore, unclear. This is particularly striking, taking into consideration that our DSG+FA data sets clearly display improved ChIP efficiency compared to several other published profiles (Fig. 4F). Despite our efforts and the addition of three and two additional independent replicates for SOX2 and POU5F1, respectively (red dots in Fig. 4F), we did not find strong evidence for SOX2 and POU5F1 bookmarking.
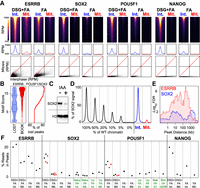
Comparative analysis of ESRRB, SOX2, POU5F1, and NANOG binding in interphase and in mitosis. (A) (Top) Heat maps of ChIP-seq signal at the indicated binding regions (±500-bp peak summit) identified in interphase (Int.) and mitosis (Mit.) for DSG+FA and FA alone. (Middle) Average binding profile of the regions shown in the heat maps. Heat maps and average binding profiles display reads per million (RPM); the y-axis differs for each factor and is scaled by median DSG+FA interphase binding: ESRRB: 0–1.5 RPM, SOX2: 0–3.2 RPM, POU5F1: 0–2.6 RPM, NANOG: 0–2.7 RPM. (Bottom) Scatter plots of ChIP-seq signal in reads per million at the above regions for interphase and mitosis (DSG+FA scale 0–40 RPM; FA scale 0–20 RPM). (B) Violin plots (left) depicting the FIMO-called best motif score per ESRRB peak in sites losing binding in mitosis (LOST) or retaining binding (BOOK). (Right) Percentage of lost peaks with a composite POU5F1/SOX2 motif of at least the given quality score. (C) Levels of SOX2-AID fusion protein in cells cultured in the absence (−) or presence (+) of the Auxin analog IAA for 2 h; H3 is shown as a control. (D) Percent of the SOX2 ChIP signal detected at binding regions after spiking increasing amounts of WT chromatin into chromatin prepared from SOX2-depleted cells shown alongside the average SOX2 binding profile at potentially bookmarked regions in WT cells in interphase and mitosis. (E) Enrichment of genes responsive to ESRRB (red) and SOX2 (blue) in early G1 as a function of the distance to ESRRB or SOX2 bookmarked regions, respectively, displayed as −log10 Fisher FDR. (F) Percentage of ChIP-seq reads in identified binding sites for ESRRB, SOX2, POU5F1, and NANOG, in both interphase (Int.) and mitosis (Mit.) and DSG+FA or FA fixation in our data (black labels) and public data sets (green labels). The red dots correspond to the samples that were added to our study to further corroborate our results.
Drastic changes in nucleosome organization characterize regulatory elements in mitosis
Recently, mitotic chromatin has been shown to maintain high levels of chromatin accessibility at virtually all regulatory elements that are active in interphase, in particular, at promoters (Hsiung et al. 2015; Teves et al. 2016). Accordingly, we observed that promoter accessibility in mitotic ES cells even surpasses the level observed in interphase, as evaluated by ATAC-seq (Fig. 5A; Supplemental Fig. S8A). However, distinct nucleosome organizations might characterize accessible chromatin in these two phases of the cell cycle (Kelly et al. 2010; Teif et al. 2014; Rhee et al. 2014; Mieczkowski et al. 2016; Voong et al. 2016; Mueller et al. 2017; Javasky et al. 2018). To address this, we inferred nucleosome positioning and stability in interphase and in mitosis from a series of experiments based on MNase-seq and H3 ChIP-seq using chromatin digested with titrated MNase activity. We first analyzed promoter regions and observed preserved nucleosome depleted regions (NDRs) around the transcription start sites in mitotic cells (TSSs) (Fig. 5B). Yet, the phasing of nucleosomes at both sides of the NDRs was drastically attenuated in mitotic cells (Fig. 5B), probably reflecting reduced transcriptional activity. Moreover, when we compared average H3 ChIP-seq signal between mitosis and interphase at different levels of MNase digestion (Fig. 5C; Supplemental Fig. S8B), a clear asymmetry was revealed: Upstream of the TSS, the sensitivity of the nucleosomes to MNase increased in mitotic cells (as shown by reduced signal with strong digestion); downstream, the +1 nucleosome displayed a similar stability as in interphase, while the following nucleosomes acquired increasing levels of fragility in mitosis. At the minimal promoter region (TSS and 150 bp upstream), we did not find evidence of a nucleosome displaying high occupancy either in interphase or in mitosis (Fig. 5B; Supplemental Fig. S8B). Nonetheless, the H3 signal detected over the minimal promoter tends to increase in mitosis, irrespective of the MNase conditions (Fig. 5C; Supplemental Fig. S8B). These results indicate that, globally, the nucleosomes at promoters are more fragile (Teif et al. 2014; Mieczkowski et al. 2016; Voong et al. 2016) in mitosis, except at the minimal promoter region, where they display unaltered stability and similar occupancy. Moreover, the differential behavior within and outside the transcription unit may potentially reflect the reduced transcriptional activity that has been recently detected in mitotic cells (Palozola et al. 2017). Therefore, promoters are subject to drastic nucleosome reorganization in mitotic cells. We then analyzed enhancers (identified here as EP300-bound elements, excluding TSSs and gene bodies). As previously shown (Hsiung et al. 2015), we found enhancers to partially lose accessibility in mitosis (Fig. 5D; Supplemental Fig. S8A) and display a profound reconfiguration in nucleosomal architecture (Fig. 5E) that is particularly well revealed with increasing MNase activity (Supplemental Fig. S8B): Nucleosomes resistant to our most aggressive digestion conditions can be detected at the site of EP300 recruitment exclusively in mitosis, and the phasing of the surrounding nucleosomes is altered (Fig. 5E; Supplemental Fig. S8B). Moreover, both upstream of and downstream from the stabilized nucleosome, increased fragility can be measured in mitotic cells (Fig. 5F; Supplemental Fig. S8B). Therefore, even though promoters and enhancers maintain significant levels of accessibility in mitotic cells, the arrangement of their nucleosomes changes substantially.
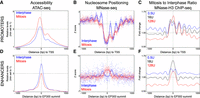
The nucleosome landscape of promoters and enhancers in interphase and in mitosis. (A) Accessibility profiles measured by ATAC-seq in the region surrounding the TSS of active genes in interphase (blue) or mitosis (red). Signal is the number of Tn5 cut sites for 0–100 bp fragments, normalized to minimum accessibility in ±1000-bp windows. Depth normalized data can be found in Supplemental Figure S8B. See Supplemental Methods for details. (B) Nucleosome positioning at the same set of promoters, established by MNase-seq. In this panel, the z-score of the number of midpoints of nucleosome-sized fragments (140–200 bp) per base, after digestion with 16 U of enzyme, are plotted. The lines represent a Gaussian process modeling nucleosome positioning (see Supplemental Methods) in interphase (blue) and in mitosis (red). (C) Mitosis over interphase ratio of MNase H3 ChIP-seq signal for nucleosomal fragments (as assessed by Gaussian process regression) (see Supplemental Methods). Ratios shown for MNase digestions with 0.5 U (blue), 16 U (black), and 128 U (red) of enzyme. Since this analysis aims at identifying quantitative changes in nucleosome occupancy, we used H3 data, as not all MNase-generated nucleosome-sized fragments are guaranteed to contain a nucleosome. (D–F) As in A–C but for regions centered on summits of interphase EP300 ChIP-seq peaks excluding promoters. Note that in E, MNase-seq signal is from 128 U digestions. Full data sets can be found in Supplemental Figure S8B.
Chromatin accessibility and nucleosome organization as a function of ESRRB bookmarking
We then focused on the analysis of the regions bound by ESRRB. While ESRRB-bookmarked regions partially lose accessibility (Fig. 6A), this reduction is significantly more pronounced at the regions where ESRRB binding is lost in mitosis (Fig. 6C). Hence, there is a clear correlation between the ability of ESRRB to bind to certain targets in mitotic cells and the partial maintenance of accessibility. Moreover, at bookmarked regions, we observed highly positioned nucleosomes both in interphase and mitosis: The ESRRB motif lies within a major NDR and phased nucleosomes spread both upstream of and downstream from the binding site (Fig. 6B; Supplemental Fig. S9A). This pattern contrasts markedly with that seen at EP300 enhancers (Fig. 5E), clearly establishing a strong correlation between ESRRB mitotic binding and the retention of well-structured nucleosome arrays. Moreover, in mitosis we observed a slight shrinking of the nucleosomal array converging toward the central ESRRB motif, leading to a modest change of position of the nucleosomes. When we calculated a frequency map of additional ESRRB motifs within these regions (gray histogram in Fig. 6B), we observed a small but clear enrichment precisely at the mitosis-specific inter-nucleosomal space between the −2/−1 and +1/+2 nucleosomes. This strongly indicates that in mitosis, the DNA binding activity of ESRRB becomes dominant in establishing nucleosome positioning. In contrast, at regions losing ESRRB binding in mitosis, the nucleosomes appeared barely organized compared to their bookmarked counterparts: A nucleosome occupies the ESRRB motif, particularly in mitosis, and clear phasing is lacking at both sides (Fig. 6D). Since high-quality ESRRB motifs are not particularly prevalent at these regions (Fig. 4B), we reanalyzed the data by recentering on ESRRB summits. We noted that POU5F1/SOX2 motifs are enriched in the vicinity of ESRRB summits (gray histogram in Fig. 6E) and therefore also recentered these regions on these motifs (Fig. 6F; Supplemental Fig. S9A). Both analyses unveiled a clear nucleosomal organization in interphase that is highly modified in mitotic cells (Fig. 6E,F). This indicates that ESRRB may be recruited indirectly and play a minor role in establishing nucleosome positioning over these regions. In accord, the nucleosome pattern at regions centered on ESRRB summits was also highly similar to that seen at the bulk of POU5F1/SOX2 binding sites (Fig. 6H; Supplemental Fig. S10A). These regions show a consistent reduction in accessibility in mitosis (Fig. 6G) and major nucleosome repositioning, with signs of shifting in the nucleosomal array and invasion at both flanks of the POU5F1/SOX2 motifs (Fig. 6H). At all these regions, a concomitant increase in occupancy by fragile nucleosomes could also be observed (Supplemental Figs. S9, S10). Of note, the presence of a more stable nucleosome at specific positions, like the ones we observed at TSSs and EP300 summits, could not be detected at ESRRB or POU5F1/SOX2 binding sites in mitotic cells (Fig. 5E,F; Supplemental Fig. S9B, S10B). Finally, at regions exhibiting low mitotic SOX2 ChIP-seq signal, we also observed major reorganizations of nucleosomes in mitosis. Nonetheless, the presence of a very narrow NDR in mitosis could not be ruled out (Supplemental Fig. S11), possibly reflecting minimal bookmarking activity. From these analyses, we conclude that TF binding is likely required to maintain nucleosome positioning at regulatory elements during cell division. ESRRB acts as a major organizer of the chromatin in both phases of the cell cycle (Fig. 6B).
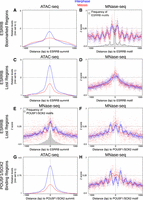
Binding of ESRRB at its cognate motif correlates with nucleosome organization in interphase and in mitosis. Accessibility (A,C,G) determined by ATAC-seq as in Figure 5A, and nucleosome positioning (B,D,E,F,H), established as in Figure 5B, at the regions indicated on the left and centered as shown on their corresponding x-axis, in interphase (blue) and mitosis (red). Histograms embedded in B and E depict rate of occurrence of the indicated binding motifs: (B) additional ESRRB motifs with FIMO score >8; (E) top scoring POU5F1/SOX2 composite motifs.
Discussion
Proposed around 20 yr ago (Michelotti et al. 1997), the idea that certain TFs mitotically propagate gene regulatory information had been, until recently, only sporadically explored. Instead, over the last few years, several publications have revealed a continuously growing number of candidate mitotic bookmarking TFs (Festuccia et al. 2017). Considering that FA, arguably the most used cross-linker, leads to an artificial depletion of TFs from the mitotic chromosomes, as visualized by microscopy (Pallier et al. 2003; Teves et al. 2016), many more TFs than those currently described are probably able to associate with the chromatin during division, as recently suggested (Cai et al. 2018; Ginno et al. 2018; Raccaud et al. 2018). However, whether all these TFs are engaged in site-specific interactions and therefore act as mitotic bookmarking factors remains unclear (Festuccia et al. 2017). Here, we identify cross-linkers that preserve the global mitotic localization detected by microscopy for several TFs, providing a simple experimental method to study the behavior of new transcriptional regulators during division and, more generally, visualize spatial organizations deriving from transient and fast binding events. Conversely, our results impose caution: We show that localization of a TF to the chromatin does not necessarily imply DNA sequence-specific binding in mitosis (Fig. 7A). This is exemplified by SOX2 and, as shown by others, by CTCF (Oomen et al. 2019): While these TFs are both chromosomally retained, they are largely evicted from the sites occupied in interphase. The functional consequences of this distinction are major: We failed to identify a strong relationship between the weak and sparse binding of SOX2 in mitotic ES cells and its transcriptional effects in early G1. Conversely, the functional relevance of site-specific mitotic binding (Fig. 7) has been documented for several canonical bookmarking factors, including GATA1 (Kadauke et al. 2012), FOXA1 (Caravaca et al. 2013), and ESRRB (Festuccia et al. 2016). Therefore, the emerging idea of a widespread mitotic bookmarking activity needs to be carefully considered and evaluated. At the same time, the potential function of a global chromosomal retention cannot be ignored and requires dedicated experimental setups. In this regard, our comparative analysis of fixatives reveals that distinct molecular mechanisms likely contribute to the overall mitotic localization of TFs (Fig. 7B). ESRRB displays highly correlated binding profiles by ChIP when the chromatin is fixed with FA or with DSG+FA. In contrast, only DSG captures global ESRRB enrichment on the chromatin. Given the ability of DSG to efficiently fix transient interactions, and in light of the results of FRAP and single molecule tracking studies (Caravaca et al. 2013; Deluz et al. 2016; Teves et al. 2016; Raccaud et al. 2018), this reveals that most likely the bulk of the molecules for a given TF bound to the chromatids during mitosis are not engaged in sequence-specific interactions with DNA. However, we showed previously that mutating 3 amino acids (aa) of the ESRRB DNA binding domain that are engaged in base-specific contacts with the binding motif dramatically decreases the global decoration of the mitotic chromosomes (Festuccia et al. 2016). It is possible that these amino acids of the ESRRB zinc-finger domain are also required for ESRRB to scan the DNA in search of its binding sites. Alternatively, these mutations may more generally alter the structure of ESRRB, preventing interactions with other proteins enriched on mitotic chromosomes. Notably, the bifunctional cross-linkers that we have used, DSG and glyoxal, are expected to increase the efficiency of fixation within large protein complexes, opening the possibility that the interactions driving the global enrichment of TFs on the chromatids are based on protein–protein rather than protein–DNA contacts. Thus, we suggest that the model previously proposed for FOXA1 regarding the existence of at least two distinct phenomena underlying the behavior of TFs in mitotic cells could be extended and applied generally to BFs: On the one hand, both DNA scanning and the ability to interact with other proteins of the chromatin sustains the bulk localization of TFs to the chromatids; on the other, bona fide bookmarking, understood here as the capacity to mediate site-specific binding, drives functionally relevant accumulation of TFs at regulatory elements (Festuccia et al. 2017). While FOXA1 is capable of binding nucleosomes directly (Cirillo et al. 1998), by virtue of its inherent structural properties (Clark et al. 1993; Ramakrishnan et al. 1993), the mitotic partners involved in protein–protein interactions with other TFs decorating mitotic chromosomes may be more diverse (Fig. 7B). These proteins could be part of the chromatin or restricted to the chromosomal periphery (Booth and Earnshaw 2017). While our costaining with Ki-67 excludes the possibility of such restricted localization for ESRRB, SOX2, and POU5F1, this may apply to other TFs. Indeed, a multitude of determinants of TF localization seem to exist. This is the case of POU5F1, that we report here as focally enriched within (peri)centric regions, both in interphase and in mitosis. Extending beyond mitosis, given the complexity revealed by the use of multiple cross-linking agents, this study directly calls for a general reassessment of TF localization and function as inferred from fixed samples.
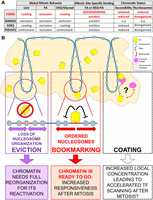
Model summarizing distinct behaviors of pluripotency TFs in mitotic cells and their relationships to nucleosome organization and post-mitotic gene regulation. (A) Summary table of the behavior of ESRRB, NANOG, SOX2, and POU5F1 in mitotic mouse ES cells. (B) Many TFs show global localization on the chromosomes in mitosis, such as ESRRB and SOX2. This localization is likely driven by sequence-independent interactions with DNA or other components of the chromatin or of the mitotic chromosomes and might serve a function in increasing the local concentration of TFs in proximity of their targets, in turn facilitating binding in G1. In contrast, during division, only few TFs remain dynamically bound to a subset of the sites they occupy in interphase, as exemplified by ESRRB. At bookmarked sites, the continued activity of these TFs maintains an ordered chromatin configuration, possibly limiting the extent of chromatin remodeling required to re-establish functional regulatory architectures in the following cell cycle. At sites losing TF binding, nucleosome positioning is disorganized, and increased occupancy by nucleosomes is detected at binding motifs. Although these sites do not become fully inaccessible, profound chromatin rearrangements are expected to be needed in early G1 to reinstate proper function.
Distinguishing TFs as enriched or depleted from mitotic chromosomes, and as binding or not at specific regulatory regions, will eventually allow us to establish a hierarchy of their contributions to the re-establishment of transcription after mitosis (Fig. 7). This will be particularly important in highly proliferative cells undergoing progressive implementation of new cell identities during development (Festuccia et al. 2017). To gain a full understanding of the importance of mitotic bookmarking, it is also crucial to elucidate the molecular mechanisms mediating its function. Different lines of evidence point to the lack of permanent TF binding during mitosis; even in the extreme case of the general TF TBP, the residence time on the mitotic chromatin is below 2 min (Teves et al. 2018). Therefore, static occupancy by single molecules of mitotic bookmarking factors does not physically transfer regulatory information from mother to daughter cells; to be functional, BFs may instead induce specific modifications around their mitotic target sites. However, regardless of their mitotic bookmarking status, most, if not all, active regulatory regions remain at least partially accessible in mitotic cells (Hsiung et al. 2015; Teves et al. 2016). This has now been shown, analyzing the binding sites of several TFs, including GATA1 (Kadauke et al. 2012), and, here, ESRRB, SOX2, and POU5F1 (Fig. 7A). Therefore, even if many other BFs remain to be identified, the general loss of TF binding characterizing mitosis is unlikely to completely abolish chromatin accessibility. In general, the presence of de-stabilized nucleosomes at regulatory elements could suffice to maintain these regions less refractory to the binding of transcriptional regulators. Nevertheless, TF binding might still contribute toward maintaining comparatively high accessibility at selected loci. This was originally proposed for the bookmarking factor FOXL1 (Yan et al. 2006) and is further supported by our observation that the regions bookmarked by ESRRB display a milder reduction of ATAC signal compared to those where ESRRB is evicted. More significantly, our nucleosome mapping studies indicate that ESRRB bookmarking plays a major role in preserving the fine patterns of nucleosome organization, rather than mere accessibility, at regulatory elements (Fig. 7). Indeed, at regions bookmarked by ESRRB, binding motifs are strongly associated with a nucleosome depleted region and are flanked by well-organized and phased nucleosomes. This configuration is detected in interphase but is significantly clearer in mitosis where even neighboring inter-nucleosomal spaces correlate with the presence of additional ESRRB motifs. We believe this reflects the loss of counteracting effects from binding of other TFs in mitosis and the consequent dominance of ESRRB over the organization of the nucleosomes at these sites. In this light, mitosis might represent a context of simplified interactions of TFs with the chromatin, where few fundamental activities are maintained. In contrast, in the complete absence of mitotic TF binding, nucleosomal arrays are largely reconfigured. This is true at enhancers marked by EP300, at regions losing ESRRB and/or POU5F1/SOX2 binding, as well as at CTCF binding sites (Oomen et al. 2019). At regions losing ESRRB in mitosis, a clear nucleosomal organization is only appreciated when regions are aligned relative to the ESRRB peak summit or the binding motifs for POU5F1/SOX2. Hence, at these regions, ESRRB might be recruited indirectly by other TFs that are not capable of binding in mitosis, such as POU5F1/SOX2: The nucleosomal organization of these regions, therefore, is not likely to be imposed by ESRRB. Together, these observations clearly indicate that mitotic bookmarking by ESRRB is essentially driven by sequence-specific DNA interactions through which this factor imposes specific constraints on nucleosomal organization. In this light, the nucleosomal landscape around TF binding sites in mitosis may be used as a proxy for mitotic bookmarking activity, further indicating that neither SOX2 nor POU5F1 are efficient bookmarking factors.
The recent observation of widespread chromatin accessibility in mitotic cells suggested that many TFs would act as bookmarking factors, a notion that is further supported by recent results derived from large imaging and proteomic screens (Cai et al. 2018; Ginno et al. 2018; Raccaud et al. 2018). In contrast, our analysis of TF binding, chromatin accessibility, nucleosome positioning, and stability in mitotic cells rather indicates that mitotic bookmarking can only be mediated by selected TFs, such as ESRRB in ES cells. Indeed, the stereotypical behavior of enhancers that we observe here indicates that a robust nucleosome is positioned at EP300 recruitment sites, with more fragile nucleosomes occupying the vicinities. These de-stabilized nucleosomes may explain the apparent accessibility of these regions. At promoters, we also observe a loss of phasing and a relative stabilization of the nucleosomes lying just upstream of the TSS as compared to those more distally located, which appear to be more fragile. While the identification of the molecular players de-stabilizing these nucleosomes will require further investigation, our data indicate that ESRRB, and potentially other bookmarking factors, may generally act by locally preserving specific nucleosome architectures. These configurations might, in turn, favor the re-establishment of functional regulatory complexes early after mitosis. We propose this mechanism to represent the molecular basis of the transmission of regulatory information by sequence-specific mitotic bookmarking factors (Fig. 7B).
Methods
Cell culture, embryos, and fixation
Mouse ES cells (E14TG2a) were grown on FCS and LIF, unless indicated, on gelatin-coated flasks. For imaging, they were plated on ibidi plates coated with poly-l-ornithine and laminin. Blastocysts were collected at E3.25 and cultured for 5 h in KSOM. GFP fusion proteins were ectopically expressed using CAG-driven vectors. Sox2-AID ES cells were generated in ES cells expressing a Ccna-GFP reporter to perform cell-cycle sorting as previously described (Festuccia et al. 2016). Fixation of ES cells was performed with either formaldehyde (FA; 1% for ChIP and 4% for stainings for 10 min), disuccinimidyl glutarate (DSG; 2 mM for 50 min) followed by FA, or glyoxal (3.1% for 30 min); embryos were fixed for 24 h in DSG followed by 20 min in FA. The detailed protocols are available in the Supplemental Methods.
Chromatin preparation from mitotic ES cells
E14TG2a cells were grown to 70%–80% confluency, incubated with nocodazole (50 ng/mL−1 for 4−5 h), and mitotic cells shake-off was performed as detailed in Supplemental Methods, reaching a purity above 95%. After fixation, chromatin was prepared as previously described (Festuccia et al. 2016) and either sonicated with a Bioruptor Pico (Diagenode) for TF binding analysis by ChIP-seq, digested with different amounts of MNase (0.5/16/128 U) for MNase-seq and H3 ChIP-seq, or processed for ATAC-seq analysis as previously described (Buenrostro et al. 2013). Detailed protocols and experimental procedures can be found in the Supplemental Methods.
Sequencing and bioinformatics analyses
TF ChIP-seq libraries were sequenced (SR50–75) at the BioMics facility of the Institut Pasteur; all other libraries were sequenced (PE150) by Novogene Co., Ltd. At least two biological replicates were sequenced per experiment. Reads were aligned with Bowtie 2 (Langmead and Salzberg 2012) to the mm9 genome to enable direct comparison with our previous study on ESRRB bookmarking (Festuccia et al. 2016) as well as with other TF localization studies in mouse ES cells (Supplemental Methods). Realigning to mm10 would not affect the conclusions as the regions with the most significant changes from mm9 to mm10 were discarded from our analyses (i.e., repetitive regions). ChIP-seq peaks were identified by MACS2 (Feng et al. 2012) and analyzed by a Generalized Linear Model with DESeq2 (Love et al. 2014) to identify genomic sites of mitotic bookmarking; TF binding motifs were identified with FIMO (Grant et al. 2011). Chromatin accessibility was analyzed counting the cut-sites of 0–100-bp paired-end fragments obtained by ATAC-seq across regions of interest, and smoothed with a Gaussian process regression. Nucleosome positioning was inferred from MNase-seq and MNase-H3 ChIP-seq, taking into consideration the midpoints of 140–200-bp paired-end fragments. Midpoints counts were corrected to the natural bias of MNase, assessed in each library by a k-mer approach. Subsequently, we used Gaussian process regression to evaluate nucleosome positioning signals. All the procedures and normalizations are described in detail in the Supplemental Methods.
Data access
All genome-wide data sets from this study have been submitted to the NCBI Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/) under accession number GSE122589.
Acknowledgments
We thank the Imagopole France–BioImaging infrastructure, supported by the French National Research Agency (ANR 10-INSB-04-01, Investments for the Future), for advice and access to the UltraVIEW VOX system. We also thank the Transcriptome and EpiGenome, BioMics, Center for Innovation and Technological Research of the Institut Pasteur for NGS. We thank Elphège Nora and Benoit Bruneau for sharing reagents and advice to establish the auxin-dependent degradation system. This work was supported by recurrent funding from the Institut Pasteur, the CNRS, and Revive (Investissement d'Avenir; ANR-10-LABX-73). P.N. acknowledges financial support from the Fondation Schlumberger (FRM FSER 2017), the Agence Nationale de la Recherche (ANR 16 CE12 0004 01 MITMAT), and the Ligue contre le Cancer (LNCC EL2018 NAVARRO). N.F. was supported by a Marie Curie IEF fellowship and a Pasteur-Cantarini Fellowship program. N.O. is supported by Revive.
Author contributions: N.F. performed or supervised most of the experimental work. N.O. analyzed sequencing data. T.P. performed ChIP-seq, ATAC-seq, confocal microscopy, and western blots. I.G. set up the ATAC protocol and performed ChIP-seq in 2i ES cells. A.T. performed live imaging, confocal microscopy, and all experiments relating to POU5F1 localization, with help from N.G. Embryo work was performed by S.V.-P. and M.C.-T. The fixation protocols were initially tested by E.G. and A.D. Data analysis and interpretation, as well as writing the manuscript, was performed by N.F., N.O., and P.N.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.243048.118.
- Received August 14, 2018.
- Accepted December 5, 2018.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see http://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








