
Identifying clusters of cis-regulatory elements underpinning TAD structures and lineage-specific regulatory networks
- Seyed Ali Madani Tonekaboni1,2,
- Parisa Mazrooei1,2,
- Victor Kofia1,
- Benjamin Haibe-Kains1,2,3,4 and
- Mathieu Lupien1,2,4
- 1Princess Margaret Cancer Centre, Toronto, Ontario M5G 1L7, Canada;
- 2Department of Medical Biophysics, University of Toronto, Toronto, Ontario M5G 1L7, Canada;
- 3Department of Computer Science, University of Toronto, Toronto, Ontario M5T 3A1, Canada;
- 4Ontario Institute for Cancer Research, Toronto, Ontario M5G 1L7, Canada
Abstract
Cellular identity relies on cell-type–specific gene expression controlled at the transcriptional level by cis-regulatory elements (CREs). CREs are unevenly distributed across the genome, giving rise to individual CREs and clusters of CREs (COREs). Technical and biological features hinder CORE identification. We addressed these issues by developing an unsupervised machine learning approach termed clustering of genomic regions analysis method (CREAM). CREAM automates CORE detection from chromatin accessibility profiles that are enriched in CREs strongly bound by master transcription regulators, proximal to highly expressed and essential genes, and discriminating cell identity. Although COREs share similarities with super-enhancers, we highlight differences in terms of the genomic distribution and structure of these cis-regulatory units. We further show the enhanced value of COREs over super-enhancers to identify master transcription regulators, highly expressed and essential genes defining cell identity. COREs enrich at topologically associated domain (TAD) boundaries. They are also preferentially bound by the chromatin looping factors CTCF and cohesin, in contrast to super-enhancers, forming clusters of CTCF and cohesin binding regions and defining homotypic clusters of transcription regulator binding regions (HCTs). Finally, we show the clinical utility of CREAM to identify COREs across chromatin accessibility profiles to stratify more than 400 tumor samples according to their cancer type and to delineate cancer type–specific active biological pathways. Collectively, our results support the utility of CREAM to delineate COREs underlying, with greater accuracy than individual CREs or super-enhancers, the cell-type–specific biological underpinning across a wide range of normal and cancer cell types.
More than 98% of the human genome consists of sequences lying outside of gene coding regions that harbor functional features, including cis-regulatory elements (CREs) that are important in defining cellular identity by establishing lineage-specific gene expression profiles (Lupien et al. 2008; Heintzman et al. 2009; Ernst et al. 2011). CREs, such as enhancers, promoters, and anchors of chromatin interactions, are predicted to cover 20%–40% of noncoding sequences of the human genome (Kellis et al. 2014). Current methods to annotate CREs in biological samples include ChIP-seq for histone modifications (e.g., H3K4me1, H3K4me3, and H3K27ac) (Heintzman et al. 2007, 2009; Lupien et al. 2008; Ernst and Kellis 2010), chromatin binding protein (e.g., MED1, EP300, CTCF, and ZNF143) (Heintzman et al. 2007; Bailey et al. 2015), or chromatin accessibility assays (e.g., DNase-seq and ATAC-seq) (Thurman et al. 2012; Buenrostro et al. 2013). CREs are unevenly distributed across the genome, suggesting distinct biological underpinning to genomic coordinates based on CRE density, namely, between clusters of CREs (COREs) and individual CREs. Indeed, high CRE density, such as those reported as super-enhancers or stretch-enhancers, is associated to cell identity and is bound by transcription regulators with higher intensity than individual CREs (Hnisz et al. 2013; Whyte et al. 2013; Dowen et al. 2014; Boeva et al. 2017). In addition, such high-density CRE regions from cancer cells lie proximal to oncogenic driver genes (Lovén et al. 2013; Chipumuro et al. 2014; Northcott et al. 2014; Kron et al. 2017). Together, these features showcase the utility of classifying CREs into clusters versus individual CREs.
Here, we present a new methodology termed clustering of genomic regions analysis method (CREAM) relying on chromatin accessibility profiles, either from DNase-seq or ATAC-seq assays, as a unifying model to identify COREs in any cell type (Fig. 1). CREAM is a computational method relying on unsupervised machine learning that considers the distribution of distances between CREs in a given biological sample to systematically identify COREs consisting of at least two individual CREs. By conducting a comprehensive comparative study, we introduce CREAM as a new systematic way for the identification of COREs, outperforming other widely used CRE annotations, such as super-enhancers. We compared the enrichment of essential and highly expressed genes in the proximity of CREAM-identified COREs and individual CREs. We further compare the binding intensity of master transcription regulators between COREs and individual CREs. We also show the utility of COREs in studying the three-dimensional structure of the genome. Finally, we report on the clinical value of identifying COREs in tumor samples to discriminate the cancer type and the biological underpinning specific to each sample.
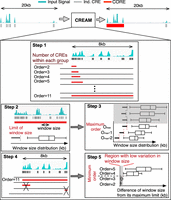
Schematic representation of the five main steps of the clustering of genomic regions analysis method (CREAM). For step 1, CREAM identifies all groups of two, three, four, and more neighboring CREs. The total number of CREs in a group defines its “Order.” Step 2 is identification of the maximum window size (MWS) between two neighboring CREs in group for each Order. The MWS corresponds to the greatest distance allowed between two neighboring CREs in a given cluster. Step 3 is identification of the maximum Order limit of COREs from a given data set. Step 4 is CORE reporting according to the criteria set in step 3 from the highest to the lowest Order. Step 5 is identification of the minimum Order limit of COREs based on the identified COREs in step 4.
Results
CREAM detects COREs from chromatin accessibility profiles
We developed CREAM as a new computational approach for the systematic identification of COREs. CREAM is designed to identify COREs from chromatin accessibility profiles through five iterative learning steps described in detail in the Methods section. Overall, these steps include the following: (1) grouping the individual CREs in clusters of varying number of individual CREs (referred to as Order); (2) identifying the threshold for the stitching distance between individual CREs within the clusters of the same Order; (3) identifying the maximum Order of COREs; (4) clustering individual CREs as COREs starting from the highest Order; and (5) filtering out low Order COREs with a stitching distance close to the corresponding stitching distance threshold of the same Order.
Applying CREAM across the DNase-seq data, aligned using human genome assembly GRCh37/hg19, from 102 cell lines available through the ENCODE Project Consortium (The ENCODE Project Consortium 2012) reveals between 1022 and 7597 COREs per cell line (Supplemental Fig. S1A), correlated with the total number of CREs identified in each cell line (Supplemental Fig. S1B). However, the fraction of CREs called within COREs is independent of the number of individual CREs (Supplemental Fig. S1C) and does not impact the median width of COREs across cell lines (Spearman's correlation ρ < 0.25) (Supplemental Fig. S1D), supporting the specificity of CORE widths with respect to each biological sample irrespective of the total number of CREs. We further show the ability of COREs to classify samples according to their tissue of origin using the ENCODE Project Consortium cell lines. Our results specifically show that COREs identify the tissue of origin for the 78 DNase I profiles of the ENCODE Project Consortium cell lines with high accuracy (Matthews correlation coefficient [MCC] of 0.85 for tissues with four or more cell lines) (Supplemental Fig. S1E). In agreement, close to 40% of the 32,997 COREs found across the ENCODE Project Consortium cell lines are unique to one cell line, and only a very small number are shared across all cell lines (Supplemental Fig. S2A). Furthermore, even COREs common to >50% of cell lines (12% of all COREs found in the ENCODE Project Consortium cell lines) (Supplemental Fig. S2) are not enriched at housekeeping genes (P-value >0.05) (Hsiao et al. 2001). Collectively, these results emphasize the cell line specificity of COREs.
COREs are unique cis-regulatory units of biological significance
We next used the DNase-seq data from the ENCODE Project Consortium tier I cell lines (GM12878, K562, and H1-hESC) to further characterize the biological underpinnings of COREs versus individual CREs. We focused on the ENCODE Project Consortium tier I cell lines because of their extensive characterization (The ENCODE Project Consortium 2012), inclusive of expression profiles and DNA–protein interactions assessed by ChIP-seq assays, allowing for a comprehensive biological assessment of COREs identified across different cell lines.
We first assessed the signal intensity for chromatin accessibility at COREs versus individual CREs. Our results show that COREs have a higher average chromatin accessibility signal per base pair compared with that of individual CREs across the three tested cell lines (GM12878: fold change [FC] = 1.9; K562: FC = 8.4; H1-hESC: FC = 1.1) (Fig. 2A). We next examined the difference in the expression level of genes proximal to COREs versus those proximal to individual CREs. We found that COREs are proximal to genes expressed at higher levels than those near individual CREs in the GM12878, K562, and H1-hESC cell lines (Wilcoxon signed-rank test FDR < 0.001; GM12878: FC = 4.6; K562: FC = 6.8; H1-hESC: FC = 1.3) (Fig. 2B). Up to 52%, 59%, and 39% of COREs overlap with active transcription start sites (TSSs) (TSSs harboring peaks of chromatin accessibility) in the GM12878, K562, and H1-hESC cell lines, respectively (Supplemental Fig. S2B). The association of COREs compared with individual CREs with highly expressed genes remains significant (FDR < 0.05) even when focusing on COREs distal to TSS (up to ±25 kb away from the TSSs; Supplemental Fig. S3), although differences in the expression of genes proximal to COREs and individual CREs decreases with increasing distance (Spearman's correlation ρ < −0.8; Fig. 2C). Hence, COREs are in proximity of genes with higher expression with respect to genes proximal to individual CREs irrespective of the distance and overlap between the CREs and gene TSSs.
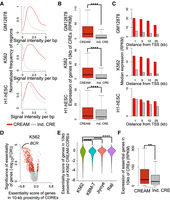
Comparison of genomic characteristics of the COREs identified by CREAM versus individual CREs in the GM12878, K562, and H1-hESC cell lines. (A) Distribution of DNase I signal intensity in individual CREs and COREs (signal per base pair). (B) Expression level of genes in 10-kb proximity of individual CREs or COREs. (****) P-value <0.0001. (C) Median expression of genes according to distance to the closest individual CRE (gray) or CORE (red). (D) Volcano plot of significance (FDR) and effect size (essentiality score) of genes in proximity of CREAM-identified COREs in the K562 cell line (red indicates significant fold change; gray, insignificant fold change). (E) Essentiality score from K562, KBM-7, Jiyoye, and Raji cell lines for genes proximal (±10 kb) to COREs identified by CREAM in the K562 cell line. (****) P-value <0.0001 using Wilcoxon signed-rank test. (F) Expression level of essential genes associated with individual CREs versus COREs. (**) P-value <0.01.
We next assessed the relevance of COREs versus individual CREs in bookmarking genes essential for growth. For this, we combined the CRISPR/Cas9 gene essentiality screen data reported in the K562 cell line (Wang et al. 2015) with CORE identification from the K562 cell line, revealing the enrichment of gene essential for growth proximal to COREs (FDR < 0.001 using permutation test) (Fig. 2D). This is exemplified at the BCR gene that is the most essential gene and proximal to a CORE in K562 chronic myelogenous leukemia (CML) cell line (Fig. 2D), positive for the BCR-ABL gene fusion reported in CML (Ren 2005), By extending our analysis to essentiality scores from other cell lines tested by Wang et al. (2015), we show that the essentiality score of genes proximal to K562 COREs is less in the KBM-7, Jiyoye, and Raji cell lines compared with the K562 cell line (FDR < 0.001) (Fig. 2E). We further show that the expression of genes essential for growth in K562 proximal to COREs is higher than the expression of essential genes associated with individual CREs (FDR < 0.001) (Fig. 2F). These results support the cell-type–specific nature of COREs and their association with essential genes and argue in favor of COREs accounting for a greater regulatory potential relevant to cell type essentiality than individual CREs.
CREAM identifies COREs bound by master transcription regulators
Transcription regulators (TRs) bind CREs to modulate the expression of cell-type–specific gene expression patterns. Quantifying the binding intensity of transcription regulators over COREs in the GM12878, K562, and H1-hESC cell lines reveals that >20% of ChIP-seq data of transcription regulators (GM12878: 92/237; K562: 256/325; H1-hESC: 24/119) show binding intensity higher over COREs compared with individual CREs when normalizing the ChIP-seq signal over COREs to the size of each CORE (FC > 2, FDR < 0.001) (Fig. 3A). The higher enrichment of TR binding intensity in COREs can be also seen using COREs excluding the CRE-free gaps (Supplemental Fig. S4A) regardless of whether COREs overlap active TSSs (TSSs harboring peaks of chromatin accessibility) or not (Supplemental Fig. S4B). This higher transcription regulator binding intensity at COREs is showcased in GM12878 by the master transcription regulators TCF3 and EBF1 (Somasundaram et al. 2015). Specifically, we observed a greater than threefold difference in binding intensity for TCF3 and EBF1 in the GM12878 cell line over COREs compared with individual CREs (Fig. 3B), exemplified at the CORE proximal to the ZFAT gene (Fig. 3C). Similarly, the master transcription regulators GABPA and CREB1 (Shankar et al. 2005; Yang et al. 2013) bind with a more than threefold greater intensity over COREs compared with individual CREs in the K562 cell line (Fig. 3B), exemplified at the CORE overlapping the LMBR1, NOM1, and MNX1 genes (Fig. 3C). Finally, in the H1-hESC cell line, the master transcription regulators NANOG and MYC (Pan and Thomson 2007) bind with higher intensity at COREs (FC > 1.2, FDR < 0.001) (Fig. 3B) in the H1-hESC cell line, exemplified at the HOXA locus CORE (Fig. 3C).
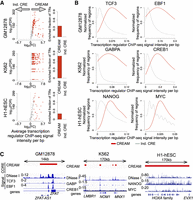
Transcription regulator (TR) binding intensity in individual CREs and COREs. (A) Enrichment of TR binding intensity from ChIP-seq data in COREs identified by CREAM versus individual CREs from DNase-seq in the GM12878, K562, or H1-hESC cell lines. Volcano plots represent −log10(FDR) versus log2(fold change [FC]) in ChIP-seq signal intensities. Each dot is one TR (colored indicates significant FC; gray, insignificant FC). The barplots show how many TRs have higher signal intensity in COREs or individual CREs (FDR < 0.001 and log2[FC] > 1). FC is defined as the ratio between the average signal per base pair in COREs versus individual CREs. (B) Distribution of ChIP-seq signal intensity at COREs and individual CREs for TCF3 and EBF1 as examples of master TRs in GM12878, for GABPA and CREB1 as examples of master TRs in the K562 cell line, and for NANOG and MYC as examples of master TRs in the H1-hESC cell line. (C) Examples of genomic regions with COREs (with different coverage) occupied by TRs presented in B.
CTCF- and cohesin-enriched COREs map to topologically associated domain boundaries
Beyond COREs, the human genome can be partitioned in various clusters including those based on contact frequencies between distal genomic coordinates that define topologically associated domains (TADs) (Ea et al. 2015). To assess the relation between COREs and TADs, we integrated the distribution of COREs with TADs reported from Hi-C data in the GM12878 and K562 cell lines (Rao et al. 2014). Our analysis reveals higher fraction of COREs compared with individual CREs at TAD boundaries (permutation test FDR < 0.001) (Fig. 4A,B; Supplemental Fig. S5A). Similar results are seen in the HeLa, HMEC, HUVEC, and NHEK cell lines (Supplemental Fig. S5B; Rao et al. 2014; Ea et al. 2015). Together, this suggests that COREs are preferentially found at TAD boundaries.
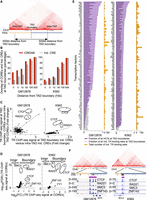
Arrangement of COREs and individual CREs with respect to TAD boundaries. (A) Schematic representation of TAD boundaries and intra-TAD regions (25-kb Hi-C resolution). (B) Comparison of fraction of COREs and individual CREs from DNase-seq that lie at TAD boundaries with increasing distance from TAD-boundary cutoffs in the GM12878 and K562 cell lines. (C) Enrichment of TR binding intensities within COREs over individual CREs that lie in proximity of TAD boundaries (±10 kb) versus COREs and CREs farther away from TAD boundaries (intra-TAD elements) in the GM12878 or K562 cell line. (D) Enrichment of TR binding intensity in COREs proximal to TAD boundaries (±10 kb) versus intra-TAD domains. (E) Fraction of HCTs (purple) and individual TR binding regions (gray) at TAD boundaries (±10 kb). The total number of individual binding regions for each TR in the GM12878 and K562 cell lines is also reported (orange). (F) Examples of HCTs for CTCF, RAD21, SMC3, and ZNF143 at the TAD boundary for the MYC and BCL6 genes (10-kb Hi-C resolution).
CTCF, cohesin (RAD21 and SMC3), YY1, and the ZNF143 transcription regulators preferentially bind chromatin at anchors of chromatin interactions, inclusive of TAD boundaries (Heidari et al. 2014; Rao et al. 2014; Bailey et al. 2015; Weintraub et al. 2017). We therefore assessed whether these transcription regulators were enriched within COREs at TAD boundaries based on their ChIP-seq signal intensity. CTCF and RAD21 were preferentially enriched within COREs compared with individual CREs restricted to TAD boundaries in both the GM12878 and K562 cell lines (FC > 1.5 for both COREs and individual CREs; FC at COREs more than 1.5 times the FC at individual CREs) (Fig. 4C). No enrichment over COREs at TAD boundaries was seen for ZNF143 and YY1 or for any of the 82 and 94 additional transcription regulators with ChIP-seq data in the GM12878 and K562 cell lines, respectively. Together, this argues that CTCF and cohesin behave differently from all other transcription regulators at TAD boundaries, mapping more to COREs as opposed to individual CREs. Furthermore, we show that CTCF and cohesin bind at TAD-boundary COREs with higher intensity than at intra-TAD COREs, defined as COREs within TADs found ≥10 kb away from boundaries, in both the GM12878 and K562 cell lines (FC > 2, FDR < 0.001 for CTCF and RAD21; FC > 1.7, FDR < 0.001 for SMC3 in GM12878 and K562 respectively) (Fig. 4D). ZNF143 also preferentially occupied TAD-boundary COREs as opposed to intra-TAD COREs but only in the K562 cell line (FC = 1.42, FDR < 0.001) (Fig. 4D). We observed lesser differences in the binding intensity of YY1 at TAD-boundary COREs versus intra-TAD COREs in the GM12878 and K562 cell lines (FC < 1.25 in both cell lines) (Fig. 4D). Extending this analysis to the remaining ChIP-seq data for transcription regulators in the GM12878 and K562 cell lines (The ENCODE Project Consortium 2012) revealed 69% and 35% of transcription regulators with increased binding intensity at TAD-boundary COREs versus intra-TAD COREs but with low effect size in the GM12878 and K562 cell lines, respectively (FC > 1, FDR < 0.001) (Fig. 4D).
The enrichment of CTCF and cohesin within COREs at TAD boundaries led us to assess if they were themselves forming homotypic clusters of transcription regulator binding regions (HCTs) (Gotea et al. 2010) at TAD boundaries. Using CREAM on the 86 and 98 ChIP-seq data from the GM12878 and K562 cell lines, respectively, identified 41 and 59 transcription regulators in each cell line forming at least 100 HCTs (Supplemental Table S1). Comparing the distribution of HCT at TAD boundaries versus intra-TADs revealed that >50% of CTCF, RAD21, SMC3, and ZNF143 HCTs lie at TAD boundaries (Fig. 4E), exemplified at the MYC and BCL6 gene loci (Fig. 4F). This contrasts with other transcription regulators, such as SP1 and GATA2 with <10% of HCTs mapping to TAD boundaries in the GM12878 and K562 cell lines, respectively (Fig. 4E). The differences in fraction of HCTs at TAD boundaries is not biased to the GC content of the individual binding regions within HCTs (Supplemental Fig. S5C). Taken together, these results suggest that clusters of CTCF and cohesin binding regions establishing HCTs are preferentially found at TAD boundaries.
COREs and super-enhancers are two distinct biological features of cells
Similar to COREs, super-enhancers were introduced as high-signal intensity regions identified from ChIP-seq data from features, such as H3K27ac or MED1, typical of a subset of CREs, including promoters and enhancers (Hnisz et al. 2013; Lovén et al. 2013; Vahedi et al. 2015). Although the concept of clusters of CREs was introduced before super-enhancers (de Laat and Grosveld 2003; Gaulton et al. 2010; Song et al. 2011), the computational method developed for super-enhancer calling, known as ROSE (Hnisz et al. 2013; Lovén et al. 2013; Whyte et al. 2013), accelerated the inclusion of super-enhancer identification across numerous studies. We therefore comprehensively compared CORE identification by CREAM with super-enhancer mapping from ROSE using the data from the GM12878, K562, and H1-hESC cell lines.
Our comparison of super-enhancers, identified either by ROSE or its latest version (ROSE2) (Stratton et al. 2016; https://github.com/linlabbcm/rose2), with COREs revealed limited overlap in all the three cell lines (Jaccard index <0.5) (Fig. 5A). Moreover, the pathway enrichment analysis based on genes within 10 kb of COREs or super-enhancers shows higher enrichment of phenotypic-specific pathways for COREs. For instance, enrichment for the B CELL RECEPTOR SIGNALING PATHWAY term is 2.6-fold more significant based on COREs as opposed to super-enhancers found in the lymphoblastoid GM12878 cell line (Fig. 5B). Similarly, the CHRONIC MYELOID LEUKEMIA term is 3.07-fold more enriched in genes proximal to COREs compared with super-enhancer in the chronic myeloid leukemia K562 cell line (Fig. 5B). Finally, the WNT SIGNALING PATHWAY term is 2.36-fold more enriched in genes proximal to COREs compared with super-enhancers in the H1 human embryonic stem cell line (Fig. 5B).
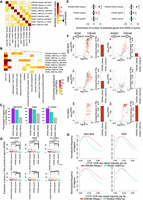
Comparison of CREAM-identified COREs and super-enhancers of the GM12878, K562, and H1-hESC cell lines. (A) Similarity of COREs and super-enhancers based on their genomic loci overlap. (B) Top five enriched biological pathways using genes in 10-kb proximity of the identified COREs and super-enhancers in each one of the GM12878, K562, and H1-hESC cell lines. (C) Percentage of COREs and super-enhancers containing two or more individual CREs. (D) Expression of genes in 10-kb proximity of both COREs and super-enhancers or exclusively in proximity of COREs or super-enhancers. (E) Enrichment of essential genes among genes in proximity of both COREs and super-enhancers or exclusively in proximity of COREs or super-enhancers. (F) Enrichment of TR binding intensity from ChIP-seq data in COREs identified by CREAM versus super-enhancers. Volcano plots represent −log10(FDR) versus log2(FC) in ChIP-seq signal intensities. Each dot is one TR (colored indicates significant FC; gray, insignificant FC). The barplots show how many TRs have higher signal intensity in COREs or super-enhancers (FDR < 0.001 and log2[FC] > 1). FC is defined as the ratio between the average signal per base pair in COREs versus super-enhancers. (G) Distribution of ChIP-seq signal intensity of CTCF at COREs and super-enhancers in 10-kb proximity of TAD boundaries.
We further compared the structure of COREs and super-enhancers according to their proportion reported to harbor two or more CREs. Although all COREs consisted of at least two CREs, between 75% and 90% of super-enhancers identified by ROSE were composed of at least two CREs in the GM12878, K562, and H1-hESC cell lines (Fig. 5C). This number plummets to <65% for super-enhancers called by ROSE2 in these same cell lines (Fig. 5C). This argues for a greater similarity between COREs and super-enhancers identified by ROSE than ROSE2. We next compared the relationship between gene expression versus COREs and super-enhancers. Our results reveal the higher expression of genes located in proximity to both COREs and super-enhancers, called either using ROSE or ROSE2, as opposed to genes exclusively proximal to COREs or super-enhancers in the GM12878 and K562 cell lines (Fig. 5D). Similarly, in the H1-hESC cell line, genes commonly assigned to COREs and super-enhancers called by ROSE show higher expression compared with genes assigned uniquely to either COREs or super-enhancers (FDR < 0.001) (Fig. 5D), but this does not apply to super-enhancers called by ROSE2, in which CREAM alone serves to identify COREs near genes with the highest level of expression (FDR < 0.001) (Fig. 5D).
Moreover, the expression of CORE-specific genes was higher in the GM12878 and K562 cell lines compared with genes exclusively in proximity of ROSE or ROSE2 super-enhancers (FDR < 0.001) (Fig. 5D). Regarding H1-hESC, the expression of CORE-specific genes was higher than ROSE2-specific genes (FDR < 0.001) (Fig. 5D) but lower than ROSE-specific genes (FDR < 0.001; Fig. 5D). Expanding our analysis to genes essential for growth in the K562 cell lines revealed that genes located in proximity of both COREs and super-enhancers have the highest enrichment for essential genes, followed by genes only proximal to COREs and, finally, genes only proximal to super-enhancers (Fig. 5E). Collectively, these results show a level of similarity between COREs and super-enhancers and also highlight differences in which COREs are more associated with biological functions than super-enhancers. This argues that COREs identified using CREAM are a more precise reflection of cellular identity and function.
As a final comparison, we assessed the enrichment of transcription regulators according to their ChIP-seq profiles within COREs versus super-enhancers. Our analysis reveals that >60% of transcription regulators are enriched in COREs compared with ROSE-super-enhancers in the GM12878 and H1-hESC cell lines (FC > 2 and FDR < 0.001) (Fig. 5F). In the K562 cell line, >30% of transcription regulators are more enriched in COREs compared with ROSE-super-enhancers (FC > 2 and FDR < 0.001) (Fig. 5F). In contrast, <2% of transcription regulators are more enriched in ROSE-super-enhancers compared with COREs in any of the three cell lines (Fig. 5F). Similar results are obtained with comparing COREs to ROSE2-super-enhancers in the H1-hESC cell line, with lower enrichment reported in GM12878 and K562 cell lines (FC > 2 and FDR < 0.001) (Fig. 5F). CTCF and the cohesin complex are among the transcription regulators preferentially enriched in COREs as opposed to super-enhancers in each cell line tested. This led us to assess the enrichment of CTCF at COREs versus super-enhancers located at TAD boundaries, inclusive of a 10-kb window around these boundaries. Our analysis revealed the strong binding intensity of CTCF within COREs at TAD boundaries, as well as weaker binding intensity within super-enhancers at TAD boundaries, in the GM12878 and K562 cell lines (FDR < 0.001) (Fig. 5G). Collectively, these results support the unique biological nature of COREs compared with super-enhancers toward chromatin looping factors and TAD boundaries, of relevance to the three-dimensional organization of the genome.
Clinical utility of CREAM to identify COREs discriminating tumor type and underpinning biological pathway
CRE identification on the human genome assembly GRCh38/hg38 was recently completed through ATAC-seq assays in 400 human tumor samples from 23 different cancer types part of The Cancer Genome Atlas (TCGA) (Corces et al. 2018). Using the k-nearest neighbor method (k = 3) on the COREs identified by CREAM classified these TCGA ATAC-seq profiles according to their tumor type (MCC = 0.86) (Fig. 6A). Out of 22 cancer types with more than four patient samples with available ATAC-seq profiles, 17 had balanced accuracy of >85% (Fig. 6A). We found that, in patient tumor ATAC-seq profiles, COREs were located in proximity of genes with higher expression than individual CREs (Fig. 6B) and were overrepresented in 49 out of 50 hallmarks of cancer gene sets (FDR < 0.05) (Fig. 6C; Liberzon et al. 2015). The TNF-α SIGNALING VIA NF-κB hallmark gene set was enriched for almost all of the TCGA samples, whereas other hallmark gene sets were tissue specifically enriched, such as the ANDROGEN RESPONSE hallmark gene set enriched in prostate adenocarcinoma (PRAD) tumor samples (Fig. 6C). Altogether our results show the potential utility of COREs in clinical setting to discriminate cancer types and identify hallmark gene sets within each tumor sample of biological relevance.
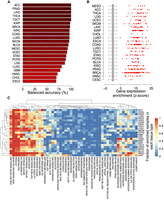
Biology of COREs in human tumor samples. (A) Balanced accuracy for classification of TCGA tumor samples based on their tissue of origin using CREAM-identified COREs. (B) Enrichment of highly expressed genes in proximity (±10 kb) of CREAM-identified COREs versus individual CREs for TCGA tumor samples. Boxplots show the null distribution corresponding to expression of randomly selected genes, and each dot corresponds to the expression of proximal genes to COREs for each tumor sample in TCGA. (C) Enrichment of hallmark gene sets relying on genes in proximity (±10 kb) of COREs versus genes in proximity (±10 kb) of individual CREs for TCGA tumor samples.
Discussion
Although the concept that CREs are not all equal is well established, their classification into clusters is recent and warrants the development of strategies for their classification according to the various approaches developed to map CREs. Here, we developed CREAM as the first unsupervised machine learning method providing a systematic approach to set the filters through an iterative learning process to identify COREs from chromatin accessibility profiles generated in any cell type. We show that CREAM identifies COREs that have higher transcription regulator binding intensity and that are enriched proximal to genes essential for growth compared with individual CREs. CREAM-identified COREs also classify cell types according to their tissue of origin, discriminating normal from cancer cells. These results support the utility of CREAM for reporting COREs from chromatin accessibility data of biological significance.
We also assessed the biological relevance of COREs with regards to the three-dimensional organization of the genome by comparing their distribution with regard to TADs. Our results show that COREs are enriched compared with individual CREs at TAD boundaries. These COREs are preferentially bound by a limited number of transcription regulators, namely, CTCF, the cohesin complex (RAD21, SMC3), and, to a lesser extent, ZNF143. These are transcription regulators previously shown to regulate contact frequencies between distal genomic coordinates defining the three-dimensional organization of the genome (Heidari et al. 2014; Rao et al. 2014; Bailey et al. 2015; Weintraub et al. 2017).
We further showed that COREs are distinct from super-enhancers defined by ROSE and ROSE2 when relying on DNase-seq data (of note, ROSE and ROSE2 were designed for identifying super-enhancers based on H3K27ac ChIP-seq data). Specifically, COREs show higher enrichment of biological pathways associated with phenotype of each cell type. Moreover, COREs compared with super-enhancers show higher enrichment in proximity of highly expressed and essential genes in binding of transcription regulators and association to CTCF-enriched regions at TAD boundaries.
Finally, we reveal the clinical value of CORE identification in 400 tumor samples to delineate their cancer type and enriched biological pathways based on genes proximal to COREs in each sample. In the process, we also provide the first pan-cancer CORE data set from 400 publicly released chromatin accessibility profiles (Corces et al. 2018) covering 23 distinct human cancer types. Overall, our results support the relevance of CREAM to classify CREs into COREs, and show the value of COREs, independently on genome assembly version, to delineate the biology unique to any sample profiled for its chromatin accessibility.
Methods
Statistical analysis of this paper has been conducted in R version 3.5.1 (R Core Team 2018).
CREAM
CREAM uses genome-wide maps of CREs in the tissue or cell type of interest generated from chromatin-based assays such DNase-seq and ATAC-seq. CREs can be identified from these data by peak calling tools such as MACS (Zhang et al. 2008). The called individual CREs then will be used as input of CREAM. Hence, CREAM does not need the signal intensity files (BAM, FASTQ) as input. CREAM considers proximity of the CREs within each sample to adjust parameters of inclusion of CREs into a CORE in the following steps (Fig. 1).
Step 1: grouping of individual CREs throughout the genome
CREAM initially groups neighboring individual CREs throughout the genome. Each group can have different number of individual CREs. Then it categorizes the groups based on their included CRE numbers. We defined Order (O) for each group as its included CRE number. In the next step, CREAM identifies the maximum allowed distance between individual CREs for calling a group as CORE of a given O.
Step 2: maximum window size identification
We defined maximum window size (MWS) as the maximum allowed distance between individual CREs included in a CORE. For each O, CREAM estimates a distribution of window sizes as the maximum distance between individual CREs in all groups of that O within the genome. Afterward, MWS will be identified based on the low stringent outlier threshold as follows:
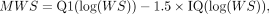 where MWS is the maximum allowed distance between neighboring individual CREs within a CORE. Q1(log[WS]) and IQ(log[WS]) are the first quartile and interquartile of distribution of window sizes (Fig. 1).
where MWS is the maximum allowed distance between neighboring individual CREs within a CORE. Q1(log[WS]) and IQ(log[WS]) are the first quartile and interquartile of distribution of window sizes (Fig. 1).
Step 3: maximum Order identification
After determining MWS for each Order of COREs, CREAM identifies the maximum O (Omax) for the given sample. Increasing the O of COREs results in the gain of information within the clusters, allowing the individual CREs to have further distance from each other. Hence, starting from COREs of O = 2, the O increases up to a plateau at which an increase of O does not result in an increase in MWS. This threshold is considered as the Omax for COREs within the given sample.
Step 4: CORE calling
CREAM starts to identify COREs from the Omax down to O = 2. For each O, it calls groups with window size less than MWS as COREs. As a result, many COREs with lower Os are clustered within COREs with higher Os. Therefore, remaining lower O COREs, for example, O = 2 or 3, have individual CREs with a distance close to MWS (Fig. 1). These clusters could have been identified as COREs because of the initial distribution of MWS derived mainly by COREs of the same O that are clustered in COREs of higher Os. Hence, CREAM eliminates these low O COREs as follows.
Step 5: minimum Order identification
COREs that contain individual CREs with a distance close to MWS can be identified as COREs owing to the high skewness in the initial distribution of MWS. To avoid reporting these COREs, CREAM filters out the clusters with (O< Omin) that do not follow monotonic increase of maximum distance between individual CREs versus O (Fig. 1). CREAM starts from the lowest order (O = 2) and checks changes of (MWS−median[WS])/median(WS), where WS is the distribution of maximum distance between individual CREs within COREs of that order. Then CREAM filters out called COREs with Order = 2 up to the point at which this parameter, (MWS−median[WS])/median(WS), is decreasing by increasing order.
Association with genes
A gene is considered associated with a CRE or a CORE if found within a ±10-kb window from each other. This distance was chosen to avoid a false-positive association of elements with gene TSSs based on previous reports (Sanyal et al. 2012). Expressions of genes with respect to distance of COREs and individual CREs with gene TSSs were conducted for different distances from ±1-kb up to ±25-kb windows as suggested by Sanyal et al. (2012).
Association with essential genes
The number of genes that are in ±10-kb proximity of COREs and are essential in the K562 cell line are identified (Wang et al. 2015). This number is then compared with the number of essential genes in 10,000 randomly selected (permuted) genes, among the genes included in the essentiality screen. This comparison is used to compute FDR, as the number of false discoveries in permutation test, and z-score regarding the significance of enrichment of essential genes among genes in ±10-kb proximity of COREs identified for the K562 cell line.
Gene expression comparison
RNA sequencing profiles of the GM12878, K562, and H1-hESC cell lines, available in the ENCODE Project Consortium database (The ENCODE Project Consortium 2012), are used to identify expression of genes in proximity of individual CREs and COREs. The expression of genes is compared using Wilcoxon signed-rank test.
Gene expression enrichment in TCGA
The expression of genes associated with COREs of each tumor sample in TCGA was compared with the expression of 100 randomly selected gene sets, with the same number of genes. The Z-score is calculated considering the null distribution generated relying on the average gene expression in the random gene sets. The P-values were calculated by comparing the expression of genes associated with COREs with genes associated with individual CREs using Wilcoxon signed-rank test.
Pathway enrichment analysis
A hypergeometric test was used to identify P-values for enrichment of hallmark gene sets using the dhyper function in the stats R package. CORE-associated genes for each sample and a catalog of genes associated with peaks were used as the query and background gene lists, respectively.
Housekeeping genes
The list of genes within the HSIAO_HOUSEKEEPING_GENES gene set (Hsiao et al. 2001) was used as the housekeeping genes.
Transcription regulator and input signal binding enrichment
bedGraph files of ChIP-seq data of transcription regulators are overlapped with the identified COREs and individual CREs in the GM12878, K562, and H1-hESC cell line using BEDTools (version 2.23.0) (Quinlan and Hall 2010). The resulting signals were summed over all the individual CREs or COREs and then normalized to the total genomic coverage of individual CREs or COREs, respectively. These normalized transcription regulator binding intensities are used for comparing TR binding intensity in individual CREs and COREs (Fig. 4). A Wilcoxon signed-rank test is used for this comparison.
Similar analysis is used, for enrichment of transcriptional regulators, to get overlap of DNase I signal data of the cell lines within individual CREs and COREs. The overlapped signal then normalized to the size of COREs and individual CREs. The distributions of these normalized signal per base within COREs and individual CREs were then compared for a given cell line.
We included the DNase I, ChIP-seq, and gene expression profiles available from all three tier I cell lines from the ENCODE Project Consortium (GM12878, K562, and H1-hESC) to provide a comprehensive analysis of COREs versus biochemical measurements across a diverse collection of cell types, acknowledging that differences in the significance in trends across cell lines could arise from cell-type–specific biology or variability in the quality of data between cell types.
Sample similarity
Similarity between two samples from the ENCODE Project Consortium or TCGA data sets was identified relying on the Jaccard index for the commonality of their identified COREs throughout the genome. Then this Jaccard index is used as the similarity statistics in a three-nearest-neighbor classification approach. We assess the performance of the classification using leave-one-out cross-validation. We used Matthews correlation coefficient for performance of the classification model (Smirnov et al. 2016). The phenotype of each tissue is considered as a class, and the obtained vector is used to calculate MCC using the implemented MCC function in PharmacoGx package in R (Smirnov et al. 2016). In this classification scheme, we considered the phenotype of the closest sample to an out of pool sample as its phenotype.
Multiple hypothesis correction
P-values were corrected for multiple hypothesis testing using the Benjamini-Hochberg procedure (McDonald 2009).
Software availability
CREAM is publicly available as an open source R package (https://cran.r-project.org/doc/FAQ/R-FAQ.html) on the Comprehensive R Archive Network (https://CRAN.R-project.org/package=CREAM) and as Supplemental Code.
Acknowledgments
DNase I sequencing profile of the HeLa cell line is used in this research. Henrietta Lacks, and the HeLa cell line that was established from her tumor cells without her knowledge or consent in 1951, have made numerous contributions to scientific progress and advances in human health. We are grateful to Henrietta Lacks, now deceased, and to her surviving family members for their contributions to biomedical research. We also acknowledge the ENCODE Project Consortium and the ENCODE Production Laboratories that generated the data sets provided by the ENCODE Data Coordination Center used in this manuscript. This study was conducted with the support of the Terry Fox Research Institute (New Frontiers Research Program PPG-1064, to M.L. and B.H.-K.), Canadian Cancer Research Society, and the Ontario Institute for Cancer Research through funding provided by the Government of Ontario. This work was also supported by the Canadian Institute for Health Research (CIHR: funding reference number 136963 to M.L. and 363288 to B.H.-K.) and Princess Margaret Cancer Foundation (M.L. and B.H.-K.). We acknowledge the Princess Margaret Bioinformatics group for providing the infrastructure assisting us with analysis presented here. M.L. holds an Investigator Award from the Ontario Institute for Cancer Research, a CIHR New Investigator Award, the Bernard and Francine Dorval Award of Excellence from CCSRI, and a Movember Rising Star Award from Prostate Cancer Canada and is proudly funded by the Movember Foundation (grant no. RS2014-04). S.A.M.T. was supported by Connaught International Scholarships for Doctoral Students. P.M. was supported by the Canadian Institutes of Health Research Scholarship for Doctoral Students. B.H.-K. is supported by the Gattuso-Slaight Personalized Cancer Medicine Fund at Princess Margaret Cancer Centre and the Canadian Institutes of Health Research.
Author contributions: S.A.M.T. developed CREAM. S.A.M.T. and V.K. prepared the CREAM R package. S.A.M.T. and P.M. performed the analysis. S.A.M.T., B.H.-K., and M.L. interpreted the results and conceived the design of the study. S.A.M.T., B.H.-K., and M.L. wrote the manuscript. B.H.-K. and M.L. supervised the study.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.248658.119.
-
Freely available online through the Genome Research Open Access option.
- Received January 19, 2019.
- Accepted August 14, 2019.
This article, published in Genome Research, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








