
Glucocorticoid receptor recruits to enhancers and drives activation by motif-directed binding
- Ian C. McDowell1,2,
- Alejandro Barrera2,3,
- Anthony M. D'Ippolito2,4,
- Christopher M. Vockley2,3,14,
- Linda K. Hong2,5,15,
- Sarah M. Leichter2,3,16,
- Luke C. Bartelt2,3,
- William H. Majoros1,2,
- Lingyun Song2,5,
- Alexias Safi2,5,
- D. Dewran Koçak2,6,
- Charles A. Gersbach2,4,6,7,
- Alexander J. Hartemink1,2,8,9,
- Gregory E. Crawford1,2,5,10,
- Barbara E. Engelhardt11,12 and
- Timothy E. Reddy1,2,3,4,6,10
- 1Graduate Program in Computational Biology and Bioinformatics, Duke University, Durham, North Carolina 27708, USA;
- 2Center for Genomic and Computational Biology, Duke University, Durham, North Carolina 27708, USA;
- 3Department of Biostatistics and Bioinformatics, Duke University Medical Center, Durham, North Carolina 27708, USA;
- 4University Program in Genetics and Genomics, Duke University, Durham, North Carolina 27708, USA;
- 5Department of Pediatrics, Duke University Medical Center, Durham, North Carolina 27708, USA;
- 6Department of Biomedical Engineering, Duke University, Durham, North Carolina 27708, USA;
- 7Department of Orthopaedic Surgery, Duke University Medical Center, Durham, North Carolina 27708, USA;
- 8Department of Computer Science, Duke University, Durham, North Carolina 27708, USA;
- 9Department of Biology, Duke University, Durham, North Carolina 27708, USA;
- 10Department of Molecular Genetics and Microbiology, Duke University, Durham, North Carolina 27708, USA;
- 11Department of Computer Science, Princeton University, Princeton, New Jersey 08540, USA;
- 12Center for Statistics and Machine Learning, Princeton University, Princeton, New Jersey 08540, USA
Abstract
Glucocorticoids are potent steroid hormones that regulate immunity and metabolism by activating the transcription factor (TF) activity of glucocorticoid receptor (GR). Previous models have proposed that DNA binding motifs and sites of chromatin accessibility predetermine GR binding and activity. However, there are vast excesses of both features relative to the number of GR binding sites. Thus, these features alone are unlikely to account for the specificity of GR binding and activity. To identify genomic and epigenetic contributions to GR binding specificity and the downstream changes resultant from GR binding, we performed hundreds of genome-wide measurements of TF binding, epigenetic state, and gene expression across a 12-h time course of glucocorticoid exposure. We found that glucocorticoid treatment induces GR to bind to nearly all pre-established enhancers within minutes. However, GR binds to only a small fraction of the set of accessible sites that lack enhancer marks. Once GR is bound to enhancers, a combination of enhancer motif composition and interactions between enhancers then determines the strength and persistence of GR binding, which consequently correlates with dramatic shifts in enhancer activation. Over the course of several hours, highly coordinated changes in TF binding and histone modification occupancy occur specifically within enhancers, and these changes correlate with changes in the expression of nearby genes. Following GR binding, changes in the binding of other TFs precede changes in chromatin accessibility, suggesting that other TFs are also sensitive to genomic features beyond that of accessibility.
The human glucocorticoid (GC) hormone cortisol, once released from the adrenal glands, pervades every organ system in the body (Chrousos and Kino 2009). At the cellular level, cortisol permeates cell membranes and binds to GC receptor (GR). GR subsequently translocates into the nucleus, where it acts as a transcription factor (TF). GR binds to tens of thousands of locations across the human genome and regulates the expression of hundreds to thousands of target genes (Wang et al. 2004; Chen et al. 2008; Reddy et al. 2009; Biddie et al. 2011; John et al. 2011; Sacta et al. 2016). GC exposure leads to transcriptional activation and repression in roughly equivalent proportions (e.g., Reddy et al. 2009). While target genes are often involved in metabolism and immunity (Yamamoto 1985), the specific genomic binding sites and effects of GR vary greatly across cellular contexts (Chrousos and Kino 2009; John et al. 2011; Gertz et al. 2013).
Genomic responses to external stimuli like GCs are believed to vary across cell types largely due to preprogrammed differences in the chromatin landscape (Heinz et al. 2010, 2015; Zhang and Glass 2013). GR has been shown to bind predominantly to sites with high DNase accessibility (John et al. 2008, 2011). At these sites, GR also commonly cobinds with certain pioneer factors, such as AP-1 TFs (Biddie et al. 2011) and CEBPB (Grøntved et al. 2013), which are so designated for their ability to bind sequence motifs in condensed chromatin and to remodel chromatin to increase accessibility (Zaret and Carroll 2011). The model that has emerged from these findings posits that cell-specific pioneer factor binding creates a chromatin landscape that predetermines GR binding (John et al. 2011; Biddie and John 2014). It has also been reported that GR can act as a pioneer factor at a minority of binding sites, which have stronger GR motifs (John et al. 2008).
The prevailing model of GR binding—in which accessibility is the preeminent determinant—partially explains differences in GR binding across cell lines (John et al. 2011). However, there are typically more than 100,000 accessible chromatin sites in a cell type (Thurman et al. 2012) but only thousands to tens of thousands of GR binding sites. We hypothesized that accessibility is one of many genomic and epigenomic features—including TF binding, histone modifications, and 3D conformation—that influence GR binding. The relative influence of those features on GR binding has not been systematically investigated.
Because GR predominantly binds distal enhancers, we focused on investigating how the state of those enhancers prior to GC treatment directs GR binding and activity after GC treatment. Enhancers are regions of the genome where TFs bind and regulate expression of distal target genes (Kleinjan and van Heyningen 2005). During differentiation, combinations of pioneer factors bind and prime the chromatin landscape for enhancer formation and, ultimately, cellular identity (Heinz et al. 2010; Heinz and Glass 2011). In addition to opening chromatin, certain pioneer factors can increase methylation of the histone H3 lysine 4 (H3K4; SPI1 [Ghisletti et al. 2010; Heinz et al. 2010]; FOXA1 [Sérandour et al. 2011; Jozwik et al. 2016]). Once established, multiple genomic and epigenetic features mark the state of an active enhancer, including enhanced chromatin accessibility (Gross and Garrard 1988; Thurman et al. 2012), binding of the histone acetyltransferase EP300 (Heintzman et al. 2009; Visel et al. 2009), acetylation of H3 lysine 27 (H3K27ac) (Creyghton et al. 2010), and monomethylation of H3K4 (H3K4me1) (Heintzman et al. 2007; Koch et al. 2007). At steady state, TFs bind in dense clusters at enhancers (Moorman et al. 2006; Garber et al. 2012; Gerstein et al. 2012; Yan et al. 2013; Siersbæk et al. 2014) to a degree that cannot be explained by accessibility or motif strength alone (Morgunova and Taipale 2017).
To characterize the relationship between the enhancer landscape and GR binding, we tracked the dynamics of (1) TF and histone modification occupancy by ChIP-seq, (2) chromatin accessibility by DNase-seq, and (3) gene expression by RNA-seq across a 12-h time course of GC exposure in a well-studied human epithelial lung cell line, A549.
Results
To investigate the dynamics of the GC response in human cells, we measured changes in genomic and epigenomic state across a time course of GC exposure. To do so, we treated human A549 cells, a type II lung carcinoma cell line commonly used to study the GC response, with the synthetic GC dexamethasone (dex) for between 30 min and 12 h. Then, at 12 points along that time course, we measured chromatin accessibility with DNase-seq (Song and Crawford 2010), covalent histone modification occupancy with ChIP-seq (Johnson et al. 2007), and gene expression with RNA-seq (Mortazavi et al. 2008). We also measured the occupancy of several TFs with ChIP-seq. Those TFs include CEBPB, which colocalizes with GR (Grøntved et al. 2013) and is strongly dex-induced in A549 cells (false-discovery rate [FDR] < 0.01) (Supplemental Fig. S1A); the AP-1 subcomponents FOSL2, JUN, and JUNB, which also commonly colocalize with GR (Biddie et al. 2011); and additional TFs induced by dex in A549 cells, namely, BCL3 and HES2 (Reddy et al. 2009). In total, we completed more than 700 genome-wide assays and generated more than 21 × 109 sequencing reads (Fig. 1A; Supplemental Tables S1–S4). To limit potential artifacts due to differences in cell density, all cell cultures were grown to confluence before initiating treatments (Supplemental Fig. S1B).
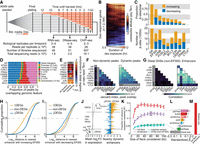
Dynamics of the genomic GC response are highly coordinated in enhancers and correlate with changes in gene expression. (A) Schematic shows experimental design for dex exposure time course and summary of sequencing effort. (B) Heatmap shows log2 fold change in gene expression for all dex-responsive genes over the time course as assayed by RNA-seq, quantified at the gene-level, and hierarchically clustered by complete linkage. (C) Bar plots show the proportion (above) and the total number (below) of differential sites across all ChIP-seq peak sets and DHSs, split into sets with increased and decreased signal with respect to the pre-dex time point. (D) Bar plot shows the proportion of ChIP-seq peaks and DHSs by genomic annotation with respect to protein-coding genes. (E) Heatmap shows the enrichment of differentially bound (or accessible) peaks in distal regions (distal intergenic or intron from D) versus nondynamic peaks. (N/A means not applicable.) (F) Heatmaps show Jaccard index of overlap for sets of nondynamic peaks (left) and for dynamic peaks (right). (G) Heatmap represents mean Pearson correlation coefficients in log2 fold change in binding of TFs within distal non-EP300-bound DHSs (left) and enhancers (right) as measured by ChIP-seq. (H) Cumulative distribution of distance of protein-coding genes by dynamics class to nearest neighboring enhancer with increased EP300 upon dex exposure. (I) Same as H, except distances are to enhancers with decreased EP300. (J) Plot shows mean log2 fold change of all differentially expressed genes and 1000 randomly selected nondifferentially expressed genes, all sorted in descending order (left). Data points for selected genes discussed in text are annotated and colored. Bar plot shows number of enhancers within increased EP300 and decreased EP300 within 20 kb (right) of the genes ranked at left. (K) Proportion of variance explained in mean log2 fold change in expression (R2; y), and standard error of R2, as a function of size of the window (x) within which the ChIP-seq and DNase-seq data were summed. Epigenomic data were summed either in the full flank or in subsets of the full flank as indicated. Mappings in the 3-kb TSS-proximal region were ignored in order to focus solely on distal regions. (L) Standardized estimated coefficients shown for the elastic net model in which change in expression was regressed on change in control-subtracted ChIP-seq and DNase-seq signal in enhancers within 60 kb of genes (see K). Standard deviations of coefficients are shown, which were computed by estimating coefficients from 1000 bootstrap replicates. (M) Bar plot shows relative size of the genomic region covered by the enhancer set within 100 kb of all tested gene TSSs compared with the full 100-kb windows.
We identified 2184 differentially expressed protein-coding genes at a FDR <0.01. Approximately equivalent numbers of genes were up-regulated and down-regulated (Fig. 1B). Up-regulated genes were enriched for lipid metabolic process gene ontology terms, including TFCP2L1 and ACSL1. Down-regulated genes were enriched for cell division–related terms and response to stress terms, including the genes PLK2, JUN, and IER5 (Supplemental Fig. S1C). These results are consistent with the known effects of GCs on lipid metabolism and stress response (Sapolsky et al. 2000; Macfarlane et al. 2008).
We also tested for changes in TF binding and chromatin accessibility in comparison to the pre-dex time point and found widely varying proportions and numbers of differential sites depending on the factor assayed (FDR < 0.1) (Fig. 1C). Dex stimulated a universal increase in GR occupancy over the time course, as anticipated. For nearly all other TFs assayed, dex both increased and decreased occupancy at a substantial fraction of binding sites (Fig. 1C). Three quarters of the approximately 20,000 binding sites for the transcriptional coactivator EP300 showed dynamic binding over the time course, and similar numbers of sites had increased EP300 occupancy as had decreased EP300 occupancy (differential and dynamic here and throughout refer to a significant change from the pre-dex time point). In sharp contrast, binding of the insulator protein CTCF remained largely unchanged throughout the time course (Fig. 1C).
TF binding dynamics are coordinated specifically within enhancers
To characterize the relationship between changes in TF binding and changes in gene expression, we first annotated TF binding sites by genomic location with respect to protein-coding gene features. GR is known to bind distal to promoters (Reddy et al. 2009), and here, 86% of GR binding sites occurred >3 kb from the nearest protein-coding gene transcription start site (TSS) (Fig. 1D). For comparison, in the same cell line, 26% of POLR2A binding sites occurred in such distal regions (The ENCODE Project Consortium 2012). While the relative ratio of promoter-distal to promoter-proximal sites varied widely across TFs (Fig. 1D), for all TFs differential binding were enriched in promoter-distal (except for GR and CTCF, which had too few nondynamic or dynamic sites, respectively, to perform a comparison; Fisher's exact test, P < 1.5 × 10−5) (Fig. 1E). Thus, GC-responsive TF binding sites were preferentially located in nonpromoter regions.
Because TF binding sites cluster in the genome (MacArthur et al. 2009; Yip et al. 2012), we hypothesized that differential TF binding sites should preferentially colocate. In all pairwise comparisons across all TFs and the set of DNase I hypersensitive sites (DHSs; again, with the exception of GR and CTCF for the same reasons as above), we found that sets of dynamic sites co-occurred more often than did sets of nondynamic sites (Fisher's exact test, P < 10−100) (Fig. 1F).
Given that dynamic TF binding sites were enriched for co-occurrence and were enriched in promoter-distal regions, we hypothesized that the coordinated changes in TF binding that we observed were concentrated specifically within enhancers. To test this, we first defined active enhancers as sites distal to protein-coding gene TSSs and with EP300 binding, open chromatin, and flanking enrichment of H3K4me1 and H3K27ac (Supplemental Fig. S1D). By these criteria, we identified approximately 16,000 enhancers active in at least one time point during the 12-h time course. Prior to dex exposure, the vast majority of enhancers were preprogrammed (H3K27ac+, 91%) and only a small minority of enhancers were poised (H3K27ac-, H3K4me1+, 3%) or latent (H3K27ac-, H3K4me1-, 5%), consistent with previous studies (Supplemental Fig. S1E,F; Biddie et al. 2011; John et al. 2011). However, we did not observe discrete clusters of enhancers based on initial premarking but instead observed a spectrum of initial activation in terms of flanking H3K4me1 and H3K27ac occupancy (Supplemental Fig. S1G).
To test whether TF binding sites were enriched in enhancers, we compared the numbers of overlapping TF binding sites in enhancers and in non-EP300-bound distal DHSs. To reduce confounding, we selected subsets of enhancers and non-EP300-bound distal DHSs that matched in chromatin accessibility (Supplemental Fig. S1H). A plurality of these enhancers bound seven of the eight TFs tested, with CTCF most commonly absent (95% of seven factor-bound sites), while a plurality of non-EP300-bound distal DHSs showed binding of a single factor, most commonly CTCF (81% of single factor-bound sites and 27% of distal DHSs in total) (Supplemental Fig. S1I). We hypothesized that the underlying characteristics that promote shared TF binding in enhancers would also promote similar TF binding dynamics across factors and within enhancers. We found that TFs had highly correlated binding dynamics specifically within enhancers (Fig. 1G; Supplemental Fig. S1J). Although change in GR binding poorly correlated with change in binding of other TFs, we later show that GR had an idiosyncratic binding profile poorly captured by linear associations and that its binding associated most strongly with a particular subclass of enhancer by activation dynamics. To ensure that our results of increased binding correlation in enhancers were not skewed by the presence of CTCF in distal DHSs, we repeated our analysis after excluding CTCF sites from the set of distal DHSs and rematching for accessibility obtained nearly identical results (Supplemental Fig. S1K). Our results suggest that the genomic GC response is largely promoter-distal, coordinated across a variety of TFs, and coordinated specifically within enhancers.
Changes in enhancer activity associate with changes in gene expression
Given that enhancers, by definition, increase the expression of target genes (Kleinjan and van Heyningen 2005), we hypothesized that changes in TF binding and histone modifications within enhancers would correlate with changes in gene expression of nearby genes. Up-regulated genes were nearer to enhancers with increased EP300 than were nondynamic and down-regulated genes (Mann-Whitney U test, P < 10−100, P = 2.2 × 10−55, respectively) (Fig. 1H), and down-regulated genes were nearer to enhancers with decreased EP300 than were nondynamic and up-regulated genes (Mann-Whitney U test, P = 1.2 × 10−7, P = 1.73 × 10−11, respectively) (Fig. 1I). Furthermore, the mean log2 fold change in expression associated with the number of enhancers with increasing and decreasing EP300 within 20 kb (t-test, P < 2.7 × 10−15), and this association was weaker when only considering the binary presence of a dynamic enhancer (χ2 test, P = 2 × 10−26) (Fig. 1J). Our results suggest that changes in enhancer activity associate with changes in gene expression and that multiple enhancers may contribute to dex-responsive expression dynamics of a target gene.
We next investigated the relationship, more generally, between promoter-distal (>3 kb) changes in TFs and histone modification occupancy and changes in gene expression. We modeled the log2 fold change in expression as a linear function of the log2 fold change of TF and histone modification occupancy summed in regions around the TSS. We shrunk coefficient estimates using elastic net (Zou and Hastie 2005). We found that the proportion of variance explained generally increased as wider TSS-flanking regions were considered yet plateaued after around 20–30 kb of flank was considered (Fig. 1K). After limiting the scope of TF and histone modification occupancy to enhancers, which comprised 1.6% of gene flank regions (Fig. 1M), the proportion of variance explained decreased by only 30% (Fig. 1K). Furthermore, the variance explained by enhancers was much greater than that explained by other size-matched sets of distal sites (e.g., at 60 kb, t-test, P < 10−100). These results suggest that the changes in TF and histone modification occupancy in promoter-distal regions most germane to changes in gene expression are largely concentrated within enhancers. The features most positively associated with gene expression within those enhancers were EP300, JUN, H3K27ac, and GR (Fig. 1L).
GR binds pervasively to enhancers
The increased coordination in GC-responsive TF binding dynamics in enhancers compared with non-EP300-bound distal DHSs led us to question the difference between the two sets in terms of GR binding. We first examined patterns in GR binding agnostic to other site-specific features. GR binding waned over the 12-h time course (Fig. 2A; Supplemental Fig. S2A), coincident with decreasing GR expression (FDR < 0.1) (Fig. 2A). GR binding was maximal at the earliest assayed time point (0.5 h of dex exposure) (Fig. 2A), suggesting that considerable binding dynamics occurred earlier than 30 min. We therefore assayed GR binding by ChIP-seq every 5 min for the first 25 min of dex exposure. That revealed approximately 44,000 additional GR binding sites and maximal GR binding at 15–20 min of dex exposure (Fig. 2B; Supplemental Fig. S2B). Nearly all of the GR binding sites present at later time points (0.5–12 h dex; 99.3%) were also present before 0.5 h of dex exposure (Fig. 2B).

Chromatin accessibility does not predetermine GR binding. (A) Line plot shows mean log2 fold change in GR binding in enhancers over time course (orange) as well as the expression of GR over the time course (black) as assayed by RNA-seq, quantified at the gene-level, and measured in transcripts per million (TPM). Error bars, SD across replicates. (B) Venn diagram shows overlap of GR ChIP-seq peaks called at early dex exposure time points (5–25 min) and at late time points (0.5–12 h). (C) Venn diagram shows the number of distal non-EP300-/non-CTCF-bound preaccessible DHSs and enhancers with initial EP300 binding within 500 bp of GR binding sites at 5 min of dex exposure. (D) Heatmaps show DNase-seq and input-subtracted ChIP-seq signal in reads per million (RPM) in chromatin accessibility-matched distal non-EP300-/non-CTCF-bound preprogrammed DHSs (left) and preprogrammed EP300 sites (right). Above, aggregate profile plots show mean RPM per base pair across sites in heatmaps. Regions shown range from −1 to +1 kb from site center. (E) Schematic of elastic net regression model (top) is shown along with estimated nonzero coefficients (below) across the time course. Error bars, SD of coefficient estimates across 1000 bootstrap samples. (F) Box-and-whisker plots show log2 fold change in standardized GR binding every 5 min for the first 25 min across the set of all enhancers split into quintiles either by initial enhancer activation (left) or by GR motif strength (right). Observations greater than 1.5× interquartile range beyond the first or third quartiles are shown as outliers. Ordinary least squares regression lines are shown in green and red.
We next compared distal DHSs and enhancers in GR binding. GR bound to nearly all enhancers with initial EP300 binding (98%) within 5 min of dex exposure, while at the same time point GR bound only 31% of distal non-EP300/non-CTCF DHSs with initial accessibility (Fisher's exact test, P < 10−100) (Fig. 2C). The observed difference in overlap with GR binding sites could not be attributed to the larger size of the distal DHS set (Supplemental Fig. S2C). Our results demonstrate that GR binds rapidly and pervasively to nearly all enhancers.
Based on prevailing models of GR binding site selection, we hypothesized that the preferential binding of GR to enhancers could be explained by differences in accessibility or motif strength. Enhancers were enriched for accessibility compared with non-EP300 DHSs (Supplemental Fig. S1H), yet after matching enhancers and distal DHSs in accessibility, GR binding remained overwhelming greater in enhancers (Fig. 2D, 1 h dex), suggesting that accessibility alone weakly explains GR binding preferences to enhancers. Furthermore, across the matched sets, enhancers did not have higher mean GR motif strength, demonstrating that the difference in GR binding could not be attributed to differences in motif strength (one-sided t-test, P > 0.05). To more directly test whether differences in GR binding could be attributed to differences in GR motif content, we searched for the GR motif in enhancers, in distal non-EP300 DHSs, and in random intergenic sequences. Nearly 800 sequences in the three sets had identical GR motifs (Supplemental Fig. S2D). Across these matched sets, GR binding was overwhelmingly greater in enhancers than in the other two sets at 1 h of dex exposure (paired t-test, P < 10−100) (Supplemental Fig. S2E). Together, our results demonstrate that factors beyond accessibility and motif content determine GR binding site preferences.
To identify those factors and to quantify their relative contributions to GR binding, we modeled GR binding to enhancers, to randomly chosen distal non-EP300-bound DHSs, and to randomly chosen size-matched intergenic intervals. We modeled GR binding per assayed time point as a binary response (peak or no peak) dependent on continuous measures of pre-dex TF occupancy, histone modification occupancy, accessibility, and GR motif strength. We used elastic net with cross-validation to limit the number of nonzero predictors and shrink coefficient estimates (for more details, see Methods; Zou and Hastie 2005). Elastic net is known to perform well in regression tasks with highly correlated predictors and tends to include or exclude groups of correlated predictors into the final fitted model (Friedman et al. 2010). The model predicted binary GR binding with high sensitivity and specificity (area under the receiver operating characteristic curve [AUC] across the time course >0.93) (Supplemental Fig. S2F). Early GR binding was predicted with greater accuracy than later GR binding (Supplemental Fig. S2F, inset), which may be explained by the fact that all predictors except GR motif strength were snapshots of pre-dex genomic features (Fig. 1C). However, early (<30 min) and late (≥30 min) GR binding was assayed in separate experimental batches, which may also explain the slight change in model performance between early and late time points. Initial EP300, JUNB, and CEBPB binding were most strongly positively associated with early GR binding (Fig. 2E). After 30 min of dex exposure and through the remainder of the 12-h time course, GR motif strength was most strongly positively associated with GR binding (Fig. 2E). It should be noted, however, that correlated features such as TF and histone occupancy share coefficient strength (jointly penalized) as a result of the penalization scheme (Friedman et al. 2010). H3K27ac was consistently positively associated with GR binding throughout the time course. When all sites that overlapped CTCF sites were excluded from the analysis, the trends held (Supplemental Fig. S2G). Although chromatin accessibility was greater in sequences bound by GR (Mann-Whitney U test, P < 10−100) (Supplemental Fig. S2H), after controlling for differences in other variables, accessibility remarkably did not associate with GR binding at most time points (Fig. 2E; Supplemental Fig. S2G). When TF occupancy data were excluded from the predictive model, initial DNase-seq signal was positively predictive of subsequent GR binding but was less predictive than enhancer features like H3K27ac (Supplemental Fig. S2I). Together, these results suggest that accessibility may serve as a proxy for TF binding but that accessibility is unlikely to be the primary means by which TF binding encourages GR binding. Although our correlative results cannot inform on whether chromatin accessibility is necessary for GR binding, they do suggest that chromatin accessibility is not sufficient for GR binding. More important determinants of GR binding may include EP300 binding, motif strength, pioneer factor binding, and histone modifications.
We observed two distinct modes of GR genomic recruitment in terms of the underlying determinants. These two modes of GR binding can be unified by a model in which initial enhancer marks strongly determine GR binding in the immediate term following dex exposure. GR increases activation at sites that also have strong motifs by, for example, recruiting EP300 (Kamei et al. 1996; Yao et al. 1996), which shifts the balance of GR recruitment toward those motif-driven sites. We assessed this model using ChIP-seq data. We first estimated initial activation for each enhancer. To do so robustly, we performed principal component analysis (PCA) on initial occupancy of EP300 and flanking H3K27ac and interpreted PC1-transformed occupancy data as representative of initial enhancer activation (Supplemental Fig. S2J,K). We found that GR initially bound most strongly to enhancers with the highest initial activation. However, the influence of initial activation gradually waned over the first half hour (Fig. 2F). Simultaneously, GR recruitment steadily increased in enhancers with stronger GR motifs (Fig. 2F), and these enhancers also gained EP300 binding (Supplemental Fig. S2L). We estimated the relative influence of initial enhancer activation and GR motif content by fitting a multivariate linear regression model to each time point and observed opposing dynamic trends in the influence of the two variables (Supplemental Fig. S2M). Our results suggest that enhancer activation and GR binding function together in a positive feedback loop, yet the initial enhancer landscape nevertheless has a substantial effect on the ultimate GR binding distribution.
Across cellular contexts, GR binds preferentially to enhancers
Our model of GR recruitment extends the received model in which chromatin accessibility is preeminent to include other important factors, including marks and factors associated with enhancers. We reanalyzed published data sets (Supplemental Table S5) to test the updated model, including GR binding data under varied dex concentrations and in different cellular contexts. We recapitulated the finding of Reddy et al. (2012) that enhancers with GR binding at low dex concentration had increased initial accessibility (dex hypersensitive vs. low-sensitive; Mann-Whitney U test, P = 1.1 × 10−14); however, the difference was starker in terms of initial EP300 (dex hypersensitive vs. low-sensitive; Mann-Whitney U test, P < 10−100) (Fig. 3B,C). In fact, after controlling for differences in initial EP300 occupancy in a multivariate linear regression, initial chromatin accessibility only marginally associated with dex dosage sensitivity of enhancers (Fig. 3D).
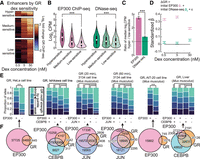
GR binds to enhancers across cellular contexts. (A) Change in GR binding shown for three classes of enhancers defined by GR binding responsiveness to varying dex concentrations. (B) Initial abundance of EP300 ChIP-seq (left) and DNase-seq (right) in enhancers by classes in A. Abundance in log2 counts per million (log2 CPM). (***) P < 0.001. (C) Ratio of log2 CPM EP300 ChIP-seq and DNase-seq in dex hypersensitive versus low-sensitive sites. (***) P < 0.001. (D) Estimated coefficients of a series of linear models regressing change in GR binding by dex concentration on initial EP300 ChIP-seq abundance and initial DNase-seq abundance after standardizing all variables (equation shown above). (E) Sets of GR binding sites across a variety of cellular contexts were split into quintiles by GR motif strength. Bar plots show the proportions of GR sites by intersection with EP300 sites and selected TF sites that fall within each quintile. (***) P < 0.001. (F) Venn diagrams show overlap of GR binding sites with EP300 sites and selected TF sites across a variety of cellular contexts corresponding to E.
A corollary of the updated model of GR recruitment is that GR binding sites that do not overlap preprogrammed enhancers will have stronger GR motif scores than binding sites that do overlap preprogrammed enhancers. We tested this hypothesis across diverse cellular contexts for which GR and EP300 binding data were available in stimulated and unstimulated treatments, respectively, and included pioneer factor (AP-1 factor, CEBPB) binding site intersections where available. As expected and across all cellular contexts, GR binding sites with initial EP300 binding had weaker median GR motif strength than did GR binding sites without initial EP300 binding (Mann-Whitney U test, P < 1.77 × 10−49) (Fig. 3E). While the proportion of EP300 binding sites to which GR recruited varied across cell line (Fig. 3F), our model of enhancer-driven GR recruitment applies across cellular contexts. For the one examined cell model in which GR binding was assayed at more than one time point, the Mus musculus 3134 mammary cell line, the proportion of GR binding sites that overlapped EP300 binding sites decreased significantly (74.5% to 67%) between 20 and 60 min of corticosterone exposure (Fisher's exact test, P = 1.88 × 10−66) (Fig. 3F), consistent with our model in which the influence of initial enhancer features on GR binding wanes over time.
GCs initiate a cascade of genomic events
Following GR recruitment, enhancers should undergo changes in TF binding and histone modifications reflective of differential activation. To study these changes, we tested for differential TF occupancy and accessibility within enhancers and differential histone occupancy in 1-kb enhancer flanks at each post-dex time point (compared with pre-dex and using negative binomial regression) (Heintzman et al. 2007; Nie et al. 2013). GCs caused differential occupancy of EP300 and H3K27ac in 73.4% and 72.8% of enhancers, respectively, across the time course (at 0.5–12 h dex) (Fig. 4A; Supplemental Fig. S3A,B). The majority of enhancers also had changes in AP-1 complex binding (JUN, 66.1%; JUNB, 65.3%; FOSL2, 40.3%) and 41.5% of enhancers had changes in CEBPB binding (Fig. 4A; Supplemental Fig. S3A,B). Our results demonstrate that saturating concentrations of dex can lead to global shifts in enhancer activation.

The extent and timing of GC-responsive changes in TF binding, histone modifications, and accessibility in enhancers. (A) Bar plot shows the proportion of enhancers with differential ChIP-seq or DNase-seq signal at some dex exposure time point compared with pre-dex levels. To be called differential, an enhancer also had to have enriched signal above background, i.e., a peak, at some point in the time course. (B) Line plot represents cumulative proportion of enhancers with increasing signal at or prior to each time point, scaled to unity, and smoothed by piecewise cubic Hermite interpolating polynomial splines. (C) Same as B, except for enhancers with decreasing signal.
To determine the order of changes within enhancers, we computed the proportion of enhancers that had differential signal prior to or at each dex exposure time point. We observed a temporal cascade of genomic events following GC stimulation in which rapid changes in GR binding was followed immediately by the redistribution of EP300, nearly simultaneous changes in pioneer factor binding and H3K27ac, gradual changes in chromatin accessibility, and, lastly, changes in H3K4 methylation (Fig. 4B,C; Supplemental Fig. S3A–E; Supplemental Table S6). Changes in TF binding generally preceded changes in accessibility (Supplemental Fig. S3F,G). These results imply that GR binding may enable recruitment of other TFs by modifying enhancer attributes, like EP300 binding and H3K27ac occupancy, prior to changes in accessibility, and again call into question the absolute preeminence of chromatin accessibility in the determination of TF binding.
Based on our observation that GC responses were highly coordinated across features, we reasoned that an integrated enhancer model that includes evidence from multiple assays and time points should enable the identification of dex-responsive enhancers. We trained a multivariate hidden Markov model (HMM) to classify dex-responsive enhancers as verified by a genome-scale high-throughput reporter assay (Arnold et al. 2013; Vockley et al. 2016). The trained model, which achieved high cross-validation accuracy (AUC = 0.80), benefitted from the integration of multiple epigenomic signals in a temporally and spatially aware manner (Supplemental Fig. S3H,I; Vockley et al. 2016).
Motif-directed binding drives changes in EP300 occupancy at enhancers
The observation that GR binding was common to nearly all enhancers, including those with divergent EP300 dynamics, led us to search for features that differentiate dynamic outcomes. We first tested whether the degree of change in GR binding associated with the degree of change in EP300 binding using simple linear regression. Change in GR signal in enhancers was strongly positively associated with change in EP300 signal across the entire time course (t-test, P < 10−100). This association generally increased over the time course. After adding an interaction term to regression model to allow for enhancers with different EP300 dynamics to vary in association with the change in GR binding, we found that the positive association was largely driven by changes in activated rather than deactivated enhancers (analysis of variance F-test, P < 10−100) (Fig. 5A,B). We further found that GR binding waned more rapidly in enhancers with decreased EP300 binding than in enhancers with increased EP300 (Mann-Whitney U test, ≥15 min dex exposure, P < 10−100) (Fig. 5B). Enhancers with increased EP300 were depleted for overlap with transient GR binding sites (<2 h, except 5 min; Fisher's exact test, P < 7.7 × 10−10) and were enriched for overlap with persistent GR binding sites (>2 h; Fisher's exact test, P < 1.2 × 10−6) in comparison to enhancers with unchanging EP300 (Fig. 5D,E). Differences in overlap with GR binding sites by duration were subtle between enhancers with decreased compared with unchanging EP300 (Fig. 5D,E). Together, our findings suggest that strong increases in GR binding lead to increases in EP300 binding. In our model of GR binding elaborated above, GR and enhancer marks such as EP300 are mutually reinforcing in the sense that increased GR binding leads to enhancer activation, which in turn leads to increased GR binding. At activated sites, this positive feedback should manifest as persistent GR binding, as observed here (Fig. 5C). Also consistent with this model is the observation that GR binding wanes at deactivated enhancers (Fig. 5C), which our model suggests may be due to the loss of enhancer marks.

Direct GR binding drives changes in EP300 binding in enhancers. (A) Lineplot displays estimated coefficients in regression of change in EP300 binding on change in GR binding in enhancers. Either all enhancers were used to fit model (black) or subsets of enhancers based on EP300 dynamics, which are displayed as indicated. Standard errors of coefficients shown as semitransparent ribbon. (B) Scatter plots compare log2 fold change in GR binding and in EP300 binding in enhancers at 0.5 and 12 h of dex exposure. Separate regression lines are shown with 95% confidence bands for each enhancer class indicated in A. (C) Heatmap shows the log2 fold change in GR binding in enhancers with increased, nondynamic, and decreased EP300 hierarchically clustered by complete linkage and sorted in descending order by change in binding (below). Discontinuity in x-axis label indicates where sampling frequency shifts from every 5 min to approximately every hour. Line plot shows mean across all enhancers by dynamics class (above). (D) Bar plot displays the proportion of enhancers, by EP300 dynamics class, that overlaps GR binding sites by the last time point at which GR was observed to bind above background. (E) Heatmap shows the log2 odds ratio of enrichment of overlap to the GR binding site sets in D for enhancers with dynamic EP300 versus enhancers with nondynamic EP300. (F) Heatmap shows elastic net logistic regression coefficients for RSAT-clustered JASPAR TF motifs with nonzero coefficients in the prediction of gains in EP300 by dynamic class. Each column represents results from an independent model where enhancer sets were split by time of increased EP300 binding as in Supplemental Figure S4B and the background set was all enhancers with decreased or nondynamic EP300.
Based on a recent finding that GR binding sites with GC-induced enhancer activity in a massively parallel reporter assay are strongly enriched for GR motifs (Vockley et al. 2016), we hypothesized that the direct binding of GR, and also of other TFs, may explain the observed differences in changes in EP300 occupancy across enhancers. We first wanted to establish which motifs contributed to initial EP300 binding, prior to dex exposure. Motifs for CEBP, forkhead box (FOX), hepatocyte nuclear factor/pou domain (HNF/POU), and AP-1 factors were the strongest predictors of the strength of initial EP300 binding (Supplemental Fig. S4A). The GR motif had no association with initial EP300 binding. To explore the mechanisms underlying dex-responsive EP300 binding dynamics, we first classified enhancers according to whether they were differentially bound by EP300 at early (E, 0.5–2 h), middle (M, 3–6 h), or late (L, 7–12 h) time periods or in combinations of these time periods (e.g., early, early-mid., mid.-late) (Supplemental Fig. S4B). Over half of EP300-dynamic enhancers had persistently increased or decreased EP300 binding (EML) (Supplemental Fig. S4C), and this class had much stronger changes in EP300 binding than transiently dynamic enhancers (EML vs. rest, Mann-Whitney U test, P < 10−100) (Supplemental Fig. S4D,E). We next used binary RSAT-clustered JASPAR motif calls (for motif naming scheme, see Supplemental Table S7; Sandelin et al. 2004; Castro-Mondragon et al. 2017) to predict membership in the EP300-dynamic classes. We used elastic net logistic regression with fivefold cross-validation (Zou and Hastie 2005) and controlled for initial EP300 binding by including it as a covariate in the model. Overwhelmingly, the most predictive DNA binding motif for changes in enhancer activation across the time course was the GR binding motif (Fig. 5F). Most of the other motifs positively predictive for enhancer activation were for factors that have been shown to participate with GR in genomic binding and/or transcriptional activation (e.g., HSF [Jones et al. 2004]; CEBP [Steger et al. 2010]; RAR/RXR [Tóth et al. 2011]; VDR [Zhang et al. 2013]; TEAD and FOX [Starick et al. 2015]). Thus, these factors may act in concert with GR, given the widespread direct binding of GR in activated enhancers. CEBP motif was positively predictive for enhancers with delayed activation (>2 h) and negatively predictive for enhancers transiently activated early in the time course (ML and L vs. E and EM in Fig. 5F). We confirmed these results with additional analyses (see Supplemental Results; Supplemental Fig. S4F). The AP-1 factor motif was most depleted in activated enhancers. We also analyzed combinations of motifs. Enhancer activation favored a combination of strong GR and CEBPB motifs as well as strong GR and weak AP-1 factor motifs (see Supplemental Results; Supplemental Fig. S4G,H).
As illustration of the effects of motif-directed binding on enhancer activation, several enhancers in the vicinity of the TSS of hippocalcin-like protein 1 (HPCAL1) with strong GR motifs had rapid and sustained increases in TF binding, chromatin accessibility, and activation-associated histone modifications following dex exposure (enhancers 1, 4, 6, 8, and 9 in Supplemental Fig. S5A). HPCAL1, accordingly, had sustained increases in expression (Supplemental Fig. S5D). Meanwhile, an enhancer 5 kb upstream of nuclear factor, interleukin 3 regulated (NFIL3) with strong CEBPB and GR motifs had delayed increases in activation-associated features following dex exposure (enhancer 2 in Supplemental Fig. S5B), while an intronic enhancer with a strong CEBPB motif but no GR motif instead had decreases in the same features (enhancer 1 in Supplemental Fig. S5B). NFIL3 had delayed increases in expression (Supplemental Fig. S5E). Two enhancers ∼20 kb upstream of the tissue factor pathway inhibitor 2 (TFPI2) gene with strong AP-1 factor and/or CEBPB motifs and no GR motifs had rapid decreases in JUN, CEBPB, and EP300 binding (Supplemental Fig. S5C), while TFPI2 concordantly decreased in expression (Supplemental Fig. S5F).
Enhancer dynamics are dependent across neighboring enhancers
Previous studies have shown that GR binding sites form local clusters in the genome, potentially via chromatin interactions between nearby enhancers (Kuznetsova et al. 2015; Vockley et al. 2016). GR binds to both pre-established chromatin loop anchors and induces novel chromatin interactions at sites with strong GR motifs (Kuznetsova et al. 2015). Based on these findings, we hypothesized that local interactions between enhancers should form clusters of nearby enhancers with similar activity dynamics. Enhancers with increased EP300 were nearest to other enhancers with increased EP300 (vs. nondynamic, Mann-Whitney U test, P = 9.7 × 10−13) (Supplemental Fig. S6A), and enhancers with decreased EP300 were nearest to other enhancers with decreased EP300 (vs. nondynamic, Mann-Whitney U test, P = 9.7 × 10−79) (Supplemental Fig. S6B). Change in EP300 binding between nearby enhancers was correlated more than would be expected by chance at distances of up to 50–100 kb (t-test, increased EP300, P = 8 × 10−4; decreased EP300, P = 2 × 10−3) (Supplemental Fig. S6C). Our results support the hypothesis that the GC-triggered dynamic activity of enhancers depends on the activity of nearby enhancers, which likely interact over tens of kilobases in genomic sequence space. We also found that persistent GR binding sites were significantly closer to (nonoverlapping) enhancers than were transient GR binding sites (Mann-Whitney U test, 12 h vs all other sets, P < 5.41 × 10−4) (Supplemental Fig. S6D), suggesting that enhancer interactions contribute to the stabilization of binding sites. We reasoned that GR binding sites present at low dex dosages may similarly depend on enhancer interactions, and we confirmed this with a comparable analysis (50 nM and 100 nM, 1 h dex; Mann-Whitney U test, P < 4.07 × 10–21) (Supplemental Fig. S6E; Reddy et al. 2012).
We hypothesized that spatial dependence of enhancer dynamics may explain, in part, why some enhancers bucked the general trends described above and, for example, had increased EP300 yet lacked a strong GR motif. We found that activated enhancers with the weakest GR motifs were closest to other activated enhancers (vs. those with the strongest GR motifs, Mann-Whitney U test, P = 8 × 10−9) (Fig. 6A), while deactivated enhancers with the strongest GR motifs were closest to other deactivated enhancers (vs. those with the weakest GR motifs, Mann-Whitney U test, P = 2.35 × 10−5) (Fig. 6B). Our results suggest that the dex-responsive activity dynamics of a given enhancer is jointly dependent on motif content and on the dynamics of neighboring enhancers. In particular, an enhancer lacking a strong GR motif, which will generally lose EP300 in saturating dex conditions, may instead gain EP300 in an activating local genomic environment. For example, two enhancers in the vicinity of HPCAL1 and in the vicinity of multiple enhancers with strong GR motifs gained EP300 despite the fact that they lacked strong GR motifs (enhancers 5 and 7 in Supplemental Fig. S5A).
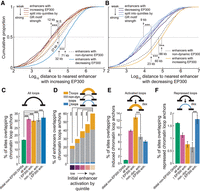
Enhancer dynamics are dependent across neighboring enhancers and coordinated by chromatin looping. (A) Cumulative distribution of distance to nearest neighboring enhancer with increased EP300 by EP300 dynamics class, where enhancers with increased EP300 were additionally split into quintiles by GR motif strength. (***) P < 0.001; N.S. = P > 0.01. (B) Cumulative distribution of distance to nearest neighboring enhancer with decreased EP300 by EP300 dynamics class, where enhancers with decreased EP300 were additionally split into quintiles by GR motif strength. (***) P < 0.001. (C) Barplot shows the percentage of sets of sites that overlap with chromatin loop anchors. Error bars, SE of percentage computed from the normal approximation of a binomial proportion. (***) P < 0.001. (D) Same as C, except with a specific focus on enhancers, which were split by initial enhancer activation as estimated by the principal component decomposition of flanking H3K4me1 and H3K27ac occupancy. (E) Same as C, except only anchors of induced loops considered. (F) Same as C, except only anchors of repressed loops considered.
Enhancer dynamics are coordinated by chromatin loops
If physical interactions explain the concordant activity of neighboring enhancers, we should expect clusters of enhancers to be demarcated by CTCF, which is known to anchor chromatin loops (Splinter et al. 2006; Rao et al. 2014). We found that the direction of EP300 occupancy changes were enriched for concordance across enhancers within domains demarcated by CTCF (Z-test, shuffled enhancers, P < 10−100; shuffled CTCF, P = 2.3 × 10−9) (Supplemental Fig. S6F,G). We found similar concordance in differential gene expression within such domains (80.2%; Z-test, shuffled genes, P = 9.3 × 10−11; shuffled CTCF, P = 1.1 × 10−10) (Supplemental Fig. S6H). These results demonstrate that enhancers or genes within regions demarcated by CTCF experience similar dynamics upon dex exposure.
To directly test for chromatin interactions between enhancers with coordinated dynamics, we mapped enhancers to chromatin loop anchors as identified in another study by in situ Hi-C performed in the same cells in pre-dex conditions and after 1, 4, 8, and 12 h of dex exposure conditions identical to this study (D'Ippolito et al. 2018). We found that enhancers were enriched in chromatin loops (see Supplemental Results; Fig. 6C,D). Furthermore, we found that enhancers with increased or decreased EP300 were enriched in loops with dex-responsive increases or decreases in interaction frequency, respectively, compared with nondynamic enhancers (Fisher's exact test, P = 5.8 × 10−11, P < 2.8 × 10−4) (Fig. 6E,F). For example, five enhancers with increased EP300 in the vicinity of the TSS of HPCAL1 overlapped three chromatin loop anchors that formed a clique of three loops—all of which had increased interaction frequency over the dex exposure time course (enhancers 1, 4, 7, 8, and 9 in Supplemental Fig. S6I).
Altogether, our results suggest that GR influences both the activity of enhancers at direct binding sites and the activity of neighboring enhancers connected through chromatin architecture. However, the direction and extent of the causal relationship between chromatin interactions, GR binding, and enhancer state remain unclear. Assaying chromatin interaction dynamics with increased spatial and temporal resolution may help to resolve this question.
Data visualization
Genome browsers present data statically, making it challenging to assess differences across time and data sets. For that reason, we developed a gene-centric website, GGR Visual, to animate the changes in multiple features of gene regulatory activity simultaneously (Supplemental Fig. S7; http://ggr.reddylab.org).
Discussion
We find that at saturating concentrations of GCs, GR binds pervasively to a largely preprogrammed landscape of enhancers and to a relatively small number of latent enhancers. In contrast to previous models of GR binding, we find that chromatin accessibility is not the most important factor influencing initial GR binding. Instead, a diversity of factors associated with active enhancers, such as EP300, H3K27ac, and pioneer factor binding—which exist in a spectrum of preprogramming—positively associate with GR binding. Motif-driven GR binding then activates enhancers, shifting the enhancer landscape to favor those sites with strong GR motifs.
The positive feedback loop between enhancer state and GR binding leads to long-lasting GR binding at sites with strong GR motifs and transient GR binding in the absence of strong GR motifs. This model has direct implications on experimental design and interpretations of past studies. Until a particular cell line and stimulus concentration is relatively densely sampled across time, it will be difficult to gauge a priori which regime of GR binding is being sampled at a particular time point. That is, an “early” time point—in terms of the GR-driven shift in enhancer landscape activation—may lead to the conclusion that GR passively binds to pre-established enhancers (or to accessible sites, if accessibility is the only auxiliary data available), while a “late” time point may lead to the conclusion that GR binding is largely motif-driven and leads to broad changes in enhancer state. Here, with dense sampling of GR binding and diverse genomic data sets, we resolve this apparent paradox and show that the data cohere with a model that unifies both enhancer-driven GR binding and GR-driven enhancer activation.
We observed that changes in GR binding and enhancer state are most strongly correlated at activated enhancers with a strong GR motif. It has been known that GR binding sites with effects on expression are largely activating and highly enriched for the GR binding motif (Vockley et al. 2016). Repressed enhancers, on the other hand, here had only subtle differences in GR binding from nondynamic enhancers. We also observed nearly equivalent numbers of activated and repressed enhancers. Together, these results are consistent with a model of cofactor squelching in which cofactors in limiting quantities are diverted from repressed to activated enhancers (Gill and Ptashne 1988; Kamei et al. 1996; De Bosscher et al. 2000). Our results suggest that transient GR binding sites lacking strong GR motifs may not be functional in themselves in terms of enhancer activation and repression but may instead be epiphenomenal to the nuclear search process or genomic recruitment process of GR. GR may follow a nuclear search mode described for other TFs in which the TF locally oversamples a nuclear space of reduced dimensionality, which may, for example, be defined by histone tails (Izeddin et al. 2014; Liu et al. 2014; Woringer et al. 2014). Such a search process would also help to explain the spatially correlated enhancer dynamics we observed here. More studies will be needed, however, to determine whether transient GR binding sites serve an independent function or whether they are simply a by-product of the nuclear search process of GR.
We show here that GC-responsive enhancer dynamics are refined by additional motif-directed binding, the effects of which differ in a TF-specific manner. In this way, the GR binding landscape specific to a particular cellular context—and the enhancer dynamics thereon—depend not only on steady-state preprogramming of the enhancer landscape but also on the specific relationships between GR and the cell-specific complement of expressed TFs. Based on our observations, these TFs likely vary in the degree and direction (antagonistic/facilitative) of collaborative interaction with GR, which further tunes the cell specificity of GR binding and activity.
We extend the model of GR-driven enhancer activation to include the effect of chromatin interactions, which encourage shared dynamics across interacting enhancers (D'Ippolito et al. 2018). As a consequence of shared dynamics, an enhancer with a weak GR motif may become activated in an activating context, such as several enhancers intronic to HPCAL1 (enhancers 5 and 7 in Supplemental Fig. S5A), and repressed in the absence of that context, such as upstream of TFPI2 (Supplemental Fig. S5C). This suggests that the flexible billboard model of enhancers (Arnosti and Kulkarni 2005) should be broadened to include the influence of TFs binding at linearly distant loci and brought into proximity through long-range interactions (Vockley et al. 2017). Targeted genome engineering could be used to directly test such dependencies between enhancers.
The pervasive binding of GR to the majority of enhancers raises the possibility of collaborative interactions between GR and its downstream TFs. Such interactions enable feed-forward loops (FFLs) in which GR and its downstream TFs physically interact at the same genomic loci to synergistically or antagonistically alter target gene expression. This model is in line with recent results on GR and multiple KLFs (Chen et al. 2013; Sasse et al. 2013; Chinenov et al. 2014) and has been proposed as a more general property of transcriptional regulation by nuclear receptors (Sasse and Gerber 2015). FFL architectures that require both GR and a downstream TF to be present for transcriptional regulation would reserve overarching regulatory control for GR, which is well attuned to ligand availability (Munck and Foley 1976). In this manner, transcriptional regulation by collaborative interactions with GR could be rapidly throttled as GC levels attenuate.
Methods
Cell culture, dex treatment, and sequencing
Our cell culture protocol was designed so that all cells were grown and passaged in the same manner up to the point of cell harvest (Fig. 1A). For more details on cell culture protocol, dex treatment, sequencing, and processing of sequencing data, see Supplemental Methods and Supplemental Figure S8.
Modeling change in gene expression data as a function of change in genomic occupancy
ChIP-seq and DNase-seq reads were quantified in flanks around the TSS of all GENCODE (v.22) (Harrow et al. 2012) protein-coding genes with mean TPM > 1 across the time course, after extending reads by half the median fragment length estimated by SPP (v.2.0) (Kharchenko et al. 2008) across all samples and within factor (e.g., GR). Flanks consisted in 1-kb bins from −100 to +100 kb from each TSS. For ChIP-seq, input control libraries were also mapped with fragment extension equivalent to the matched antibody condition. Read counts were converted to CPM. For ChIP-seq, input CPM was subtracted from antibody CPM, truncating bins with negative CPM at zero.
For each gene, normalized counts were then summed across flanks at varying widths around the TSS and log2-transformed after adding a small pseudocount of 0.01. We modeled standardized mean log2 fold change in gene expression as a linear function of the standardized mean log2 fold change in summed TSS-flanking occupancy. Elastic net regression (Zou and Hastie 2005) was performed using the Python package scikit-learn (Pedregosa et al. 2011). The alpha and l1_ratio parameters were determined by fitting the model across a grid of possible values (alpha = {10−8, 10−7,…, 100}, l1_ratio = {0.5, 0.75, 0.9, 0.95, 0.99, 1}) with stratified fivefold cross-validation to select the most sparse model with mean R2 within one standard error of the best-performing model (Breiman et al. 1984). Given these parameters, a model was fit to 1000 bootstrap replicates of the data to estimate standard errors on the model coefficients.
Elastic net is a regularized regression technique that combines L1 (least absolute shrinkage and selection operator or LASSO) and L2 (ridge) penalties to enable model selection and stable coefficient estimates in the case of highly correlated predictors (Zou and Hastie 2005). In such a case, LASSO regression will tend to include one of a set of highly correlated predictors and shrink all other predictors to zero. The resultant coefficient estimates may be unstable with small changes in the input data set. In ridge regression, on the other hand, all predictors—though shrunk—will likely be included in the final fitted model, precluding the possibility of model selection. In the case of highly correlated groups of predictors, elastic net will tend to include or exclude groups of correlated predictors. It should be noted, however, that correlated predictors tend to share coefficient strength. In the extreme case of p perfectly collinear predictors, for example, each predictor will have a coefficient estimate of 1/p if one is the coefficient strength in a model with only one of the p predictors (Friedman et al. 2010).
GR binding analysis
We modeled the binary binding of GR to all enhancers, 30,000 randomly selected distal non-EP300 bound DHSs, and 30,000 randomly selected non-EP300, non-DHS intergenic regions across the time course. We modeled binding as the output of a logistic function of standardized log2 CPM of all pre-dex DNase-seq and input-subtracted ChIP-seq data, as well as GR motif strength. Binary binding was defined as an overlap of at least 1 bp or book-ended with a GR peak in the union across the time course (0.5- to 12-h dex exposure). Sites were scored by GR motif strength by scanning for the GR motif (de novo motif discovered in the GR ChIP-seq peaks; see Supplemental Methods) using FIMO and a second-order Markov background model (see Supplemental Methods; Grant et al. 2011). FIMO P-values were negative log10 transformed and standardized. TF occupancy and accessibility data were quantified within site boundaries, while histone modification occupancy data were quantified in 1-kb upstream and downstream site flanks. Logistic elastic net regression (Zou and Hastie 2005) was performed using the Python package scikit-learn (Pedregosa et al. 2011). The alpha and l1_ratio parameters were determined as in “Modeling change in gene expression data as a function of change in genomic occupancy” above, except scoring function was log-loss instead of R2. The model was fit to 1000 bootstrap replicates of the data to estimate standard errors on the coefficients. The same series of models was run after either excluding all sites that overlapped with CTCF sites from the design matrix or excluding all TF ChIP-seq predictors.
GR binding and EP300 binding across cellular contexts
We downloaded a variety of GR, pioneer factor (CEBPB and JUN), and EP300 binding data from external studies (GEO accessions: GSE24518, GSE32465, GSE37235, GSE61911, GSE61236, GSE64516; SRA accessions: SRP007111) (see Supplemental Table S5). All GR sites were extended or trimmed to 1000 bp in width. We compared GR motif strength in GR binding sites by intersection (+/−) with EP300 binding sites and across cellular contexts. An extended GR site was considered overlapping an EP300 site if at least 1 bp overlapped between sites or sites were book-ended. Extended GR sites were scored by GR motif strength using FIMO and JASPAR motif MA0113.3 (Sandelin et al. 2004; Grant et al. 2011). FIMO motif P-values were then discretized into quintiles for display.
GR ChIP-seq data collected across varied dex dosages were downloaded from GSE92793 (Reddy et al. 2012; The ENCODE Project Consortium 2012). ChIP-seq reads were quantified in enhancers using featureCounts (v.1.4.6-p4) (Liao et al. 2014) as described in the Supplemental Methods (see “Sequencing data processing pipelines” section).
Hi-C data
Chromatin loop locations were taken directly from GSE92793. Differential loops were called using a method described previously (FDR < 0.05) (D'Ippolito et al. 2018).
Data access
All raw and processed sequencing data from this study have been submitted to the NCBI Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/) (Barrett et al. 2013) under accession numbers listed in Supplemental Table S3. These data are also available from the ENCODE DCC portal (http://www.encodeproject.org), which provides detailed summaries, quality metrics, and analysis pipeline schematics (Supplemental Table S3). Sequencing data processing pipelines are freely available in the Supplemental Material and at GitHub (https://github.com/Duke-GCB/GGR-cwl).
Acknowledgments
We thank the ENCODE DCC staff, especially I. Gabdank, C. Sloan, and A. Narayanan, for help in facilitating pipeline documentation and data release through the ENCODE project portal. We thank T. Konneker (NCSU), D.L. Aylor (NCSU), and C. Frank (formerly Duke) for contributing a custom script used to remove DNase-seq PCR artifacts. This work was mainly carried out at the Duke Center for Genomic and Computational Biology. This work was funded by the following grants: National Institutes of Health (NIH) U01 HG007900 (all authors), NIH F31 AI124563 (A.M.D.), NIH F31 HL129743 (C.M.V.), NIH R01 GM118551 (A.J.H.), NIH R01 DA036865 (D.D.K. and C.A.G.), NIH R00 HG006265 (B.E.E.), NIH R01 MH101822 (B.E.E.), and a Sloan Faculty Fellowship (B.E.E.).
Author contributions: Conceptualization was by T.E.R., G.E.C., B.E.E., A.J.H., C.A.G., and C.M.V.; supervision was by T.E.R., G.E.C., B.E.E., A.J.H., and C.A.G.; funding acquisition was by T.E.R., G.E.C., B.E.E., A.J.H., and C.A.G.; project administration was by L.K.H., T.E.R., G.E.C., and I.C.M.; investigation was by I.C.M. A.M.D., L.K.H., S.M.L., L.S., A.S., L.C.B., and C.M.V.; formal analysis was by I.C.M., T.E.R., W.H.M., A.B., and A.M.D.; data curation was by A.B. and I.C.M.; visualization was by I.C.M., A.B., and W.H.M.; writing of the original draft was by I.C.M., T.E.R., A.B., and W.H.M.; and review and editing was by all of the authors.
Footnotes
-
↵Present addresses: 14The Broad Institute of MIT and Harvard, Cambridge, MA 02142, USA; 15Sequencing and Genomic Technologies Shared Resource, Durham, NC 27701, USA; 16Laboratory of Genetics, University of Wisconsin-Madison, Madison, WI 53706, USA
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.233346.117.
-
Freely available online through the Genome Research Open Access option.
- Received December 4, 2017.
- Accepted July 5, 2018.
This article, published in Genome Research, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








