
Transcriptional regulatory dynamics drive coordinated metabolic and neural response to social challenge in mice
- Michael C. Saul1,12,
- Christopher H. Seward1,2,12,
- Joseph M. Troy1,3,
- Huimin Zhang1,2,
- Laura G. Sloofman1,4,
- Xiaochen Lu1,2,
- Patricia A. Weisner1,5,
- Derek Caetano-Anolles1,2,
- Hao Sun1,
- Sihai Dave Zhao1,6,
- Sriram Chandrasekaran7,8,9,
- Saurabh Sinha1,4,10,11 and
- Lisa Stubbs1,2,5
- 1Carl R. Woese Institute for Genomic Biology, University of Illinois at Urbana–Champaign, Urbana, Illinois 61801, USA;
- 2Department of Cell and Developmental Biology, University of Illinois at Urbana–Champaign, Urbana, Illinois 61801, USA;
- 3Illinois Informatics Institute, Urbana, Illinois 61801, USA;
- 4Center for Biophysics and Quantitative Biology, University of Illinois at Urbana–Champaign, Urbana, Illinois 61801, USA;
- 5Neuroscience Program, University of Illinois at Urbana–Champaign, Urbana, Illinois 61801, USA;
- 6Department of Statistics, University of Illinois at Urbana–Champaign, Urbana, Illinois 61801, USA;
- 7Harvard Society of Fellows, Harvard University, Cambridge, Massachusetts 02138, USA;
- 8Faculty of Arts and Sciences, Harvard University, Cambridge, Massachusetts 02138, USA;
- 9Broad Institute of MIT and Harvard, Cambridge, Massachusetts 02142, USA;
- 10Department of Computer Science,
- 11Department of Entomology, University of Illinois at Urbana–Champaign, Urbana, Illinois 61801, USA
- Corresponding author: ljstubbs{at}illinois.edu
-
↵12 These authors contributed equally to this work.
Abstract
Agonistic encounters are powerful effectors of future behavior, and the ability to learn from this type of social challenge is an essential adaptive trait. We recently identified a conserved transcriptional program defining the response to social challenge across animal species, highly enriched in transcription factor (TF), energy metabolism, and developmental signaling genes. To understand the trajectory of this program and to uncover the most important regulatory influences controlling this response, we integrated gene expression data with the chromatin landscape in the hypothalamus, frontal cortex, and amygdala of socially challenged mice over time. The expression data revealed a complex spatiotemporal patterning of events starting with neural signaling molecules in the frontal cortex and ending in the modulation of developmental factors in the amygdala and hypothalamus, underpinned by a systems-wide shift in expression of energy metabolism-related genes. The transcriptional signals were correlated with significant shifts in chromatin accessibility and a network of challenge-associated TFs. Among these, the conserved metabolic and developmental regulator ESRRA was highlighted for an especially early and important regulatory role. Cell-type deconvolution analysis attributed the differential metabolic and developmental signals in this social context primarily to oligodendrocytes and neurons, respectively, and we show that ESRRA is expressed in both cell types. Localizing ESRRA binding sites in cortical chromatin, we show that this nuclear receptor binds both differentially expressed energy-related and neurodevelopmental TF genes. These data link metabolic and neurodevelopmental signaling to social challenge, and identify key regulatory drivers of this process with unprecedented tissue and temporal resolution.
Agonistic encounters represent challenges to wellbeing and require a swift, measured response. Animals must learn quickly from socially threatening situations and adjust their behavior during future encounters accordingly; failure to adapt can have serious consequences. Counterbalancing the necessity for normal fear reactivity are the physiological costs of a reaction: Repeated exposure to social challenge adversely impacts neurochemistry, behavior, and health in both recipients and perpetrators of threats (Hawker and Boulton 2000; Bjorkqvist 2001).
Though social challenges have stronger and more durable consequences when reinforced, neurochemical and behavioral differences are observed after even a single challenge. For instance, one round of the classic “resident-intruder” territorial challenge test is sufficient to rapidly raise blood glucose and serum corticosterone in both naïve residents and intruder mice while inducing alterations in neurochemistry associated with depressive and anxious behaviors (Meehan et al. 1987; Audet et al. 2010). Such a response triggers the first steps in fear learning, preparing an animal for future encounters by tempering subsequent behaviors (Duvarci and Pare 2014). Normal learning from social challenge is critical to establishing social rank, a ubiquitous element of social hierarchies across metazoans (Fernald and Maruska 2012), while abnormalities in social threat-based learning lead to maladaptive behaviors, including those associated with human neuropsychiatric disease (for reviews, see Pittenger 2013; Gilmartin et al. 2014).
Fear learning in response to social challenge is ubiquitous across metazoans, and our recent study demonstrated deeply conserved molecular mechanisms underlying the early steps in this response (Rittschof et al. 2014). These previous results documented rapid activation of closely related genes and strikingly similar pathways in brains of three classic behavioral models—honey bees, stickleback fish, and laboratory mice—very shortly after challenge exposure. These data revealed a rapid down-regulation of genes involved in oxidative phosphorylation, correlated with the activation of a conserved cohort of coexpressed neurodevelopmental transcription factors (TFs) and signaling pathways. The results suggested that energy metabolism and neurodevelopmental systems cooperate as central parts of a deeply conserved mechanism involved in coordinating behavioral plasticity and, ultimately, emotional learning after social threat.
While our initial study suggested a link between metabolism and neural signaling through development, it did not include the tissue and temporal resolution needed to resolve this link, describe the downstream transcriptional response, or reveal the factors involved in its regulation. To understand the molecular response within the animal's brain as it responded to a salient social challenge and to identify the factors regulating these neuronal events, we generated and then computationally integrated data sets reflecting gene expression and chromatin states as they changed across the brain and over time.
Results
Gene expression analysis reveals a complex interplay of metabolic signaling and developmental regulators
A generalized linear model identifies the systems-wide response
Our previous study (Rittschof et al. 2014) focused on gene expression in resident and control animals in the ventral hypothalamus (HYP), sampling gene expression quickly (15 min) after removal of the intruder mouse. To further explicate this response and its downstream consequences, we examined gene expression at 30, 60, and 120 min after intruder removal in three interconnected brain regions with key roles in fear learning and response to aggression: frontal cortex (FCX), whole HYP, and amygdala (AMY) (for review, see Takahashi and Miczek 2014). A table summarizing the gene expression and other genomics data sets presented in this article is available as Supplemental Table S1.
To maximize the utility of this rich expression data set, we modeled the system-wide interplay of the transcriptional response across these three tissues and over time. We used a generalized linear model (GLM)—essentially a three-way factorial ANOVA, but with different distributional assumptions—to detect differential expression related to four factors (predictors) of interest: a factor indicating whether the sample came from a challenge or control animal (challenge), factors representing the interaction between challenge and tissue assayed (tissue:challenge) or interaction between challenge and time point (challenge:time), and also the three-way interaction among all of the previous factors (tissue:challenge:time). Each factor provides different information about tissue and temporal patterning of response to social challenge, and each yielded many differentially expressed genes (DEGs) of interest at a false-discovery rate (FDR) < 0.10 (Supplemental Table S2).
The gene with the strongest P-value among challenge factor DEGs was Ide, encoding insulin degrading enzyme (edgeR FDR = 6.70 × 10−5) (Fig. 1A); mitochondrial tRNA gene mt-Tq was highest ranked among tissue:challenge DEGs (edgeR FDR = 1.3 × 10−4). However, functional analysis revealed no significant enrichments among DEGs in either of these factors (Supplemental Table S3). In contrast, all DEGs from factors with a temporal component were very highly enriched for functional terms related to mitochondria and oxidative phosphorylation, with the tissue:challenge:time DEGs being most highly enriched in this functional category, with the gene Ndufa7, encoding a mitochondrial NAD dehydrogenase, having the smallest P-value (edgeR FDR = 1.2 × 10−5) (Fig. 1B; Supplemental Table S2). As exemplified by the expression patterns of Ide and Ndufa7 (Fig. 1) and in agreement with our earlier study (Rittschof et al. 2014), the GLM analysis thus implies that a hallmark of the response to social challenge is the rapid down-regulation of oxidative phosphorylation, accompanied by up-regulation of insulin signaling, and, more generally, the modulation of energy metabolism across brain regions and time after challenge.
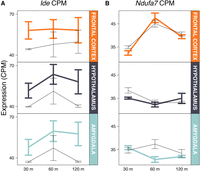
Expression patterns for Ide (A), the top DEG in the challenge factor of the GLM, and Ndufa7 (B), the top DEG in the tissue:challenge:time interaction factor of the GLM. Thin black lines are control samples, while thick colored lines are experimental samples. Error bars, SEM.
Pairwise comparisons reveal the spatiotemporal sequence of events
Pairwise post-hoc comparisons of gene expression between challenged and control animals provide a different perspective, revealing the specifics of response within each tissue and time point. In these comparisons, we found striking variation in the number of DEGs in different tissues over time (Fig. 2A; Supplemental Table S4). The transcriptomic response was most dramatic in FCX 30 min after challenge, while the largest responses in AMY and HYP occurred at 120 min. Though these DEG sets were mostly distinct, there were some important overlaps between tissues and time points (Fig. 2B,C). For example, Ide, the top gene associated with challenge in the factorial analysis (as described above), was consistently up-regulated in the HYP at all time points and up-regulated in all brain regions 120 min after challenge (Supplemental Table S4). Furthermore, the pairwise post-hoc comparisons generally overlapped with factors that included challenge in the model analysis (Supplemental Table S5).
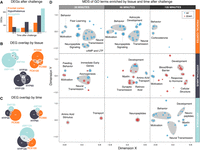
DEGs at FDR < 0.10 in the intruder versus control comparison for each combination of brain region and time point after intruder. (A) Plot of DEGs by time point after intruder. (B) Plots of overlap between time points after intruder within each brain region. (C) Plots of overlap between brain regions within each time point after intruder. (D) Semantic MDS plot projecting the spatial and temporal pattern of enriched GO biological processes (BPs). Distances represent GO semantic dissimilarity as measured by simRel. Point size represents log-fold-change of a GO BP term. Point color represents direction of differential expression of a GO BP term.
To describe systems responses over time, we tested these pairwise post-hoc DEG sets for enrichment of Gene Ontology (GO) Biological Processes (BPs) and visualized the results using nonmetric multidimensional scaling (MDS) on distances derived from GO term semantic dissimilarity (Supek et al. 2011). This approach revealed a complex tissue-specific and temporal patterning of functional response (Fig. 2D; Supplemental Table S6). In FCX, an early up-regulation of GO BPs related to neurotransmission (a cluster including a term related to insulin signaling), neuropeptide signaling, and learning was observed, which attenuated over the length of the experiment. In HYP, we detected a highly dynamic GO BP response over time, beginning with neuropeptide-related functions such as feeding behavior, followed by neural signaling and developmental functions. In AMY, few GO BPs were enriched in early time points, but strong, diverse enrichments were seen late after challenge. Similar to HYP, these functionally enriched categories ranged from neural signaling and transmission to development.
As a confirmation of the conclusions from the RNA-seq experiments, we performed RT-qPCR on a selection of key DEGs focusing on samples taken 120 min after social challenge. For each of these genes, we confirmed differential expression in the same direction and of the same approximate magnitude as in the RNA-seq study (Supplemental Fig. S1).
Taken together, the pairwise and system-wide expression analyses led to the hypothesis that early neuromodulatory signaling in FCX and HYP leads to a rapid shift in energy metabolism, which prefigures later neuronal plasticity in the amygdalar-hypothalamic axis. We explore this hypothesis further in following sections, but first we sought to associate these different transcriptomic signals to specific cell types across the brain.
Computational deconvolution analysis identifies cell-type–specific functions
Brain tissue is a complex mixture of cell types, complicating the interpretation of expression results. When we examined the overlap between our data set and genes linked to specific cell types in a previous RNA-seq experiment involving sorted mouse brain cells (Zhang et al. 2014), we found an enrichment of neuron-specific genes for most DEG sets, along with other cell types in specific brain regions and time points (Supplemental Table S7A). However, being limited to direct overlaps, this analysis included a relatively small subset of genes. To permit a more comprehensive analysis, we used “population-specific expression analysis” (PSEA) to deconvolve the signals from individual cell types in our data set (Kuhn et al. 2011). By using the same data set cited above (Zhang et al. 2014), we constructed reference signals for five different cell types: astrocytes, neurons, oligodendrocytes, microglia, and endothelial cells. We then classified DEGs from the GLM and from the pairwise post-hoc results based on these PSEA-defined cell categories. Because the reference data set utilized cortical tissue, we expect cell-type deconvolution to perform best on the FCX samples.
PSEA revealed strong overlaps between the pairwise post-hoc DEGs and genes associated with neurons from the reference data set (Supplemental Tables S7C, S8). In contrast, we identified significant overlaps between genes differentially expressed in the challenge-related GLM factors and genes associated with glia, most consistently oligodendrocytes, in all brain regions (Supplemental Tables S7D, S8). From these analyses, we infer that the system-wide energy metabolism shift detected by the GLM is correlated most highly with gene expression in oligodendrocytes, while the signals related to neuronal transmission and developmental pathways described in the post-hoc analysis derive primarily from neurons.
Coexpression reveals the organization of oxidative phosphorylation and neuronal signaling genes in opposing network clusters
To evaluate the interrelationships between genes and functions underlying challenge response, we used weighted gene correlation network analysis (WGCNA) to generate a coexpression network across tissues and time points (Supplemental Table S9). To visualize the most stable coexpression relationships, we extracted network edges with correlation coefficients ≥0.85 in absolute value and with the same sign in every tissue. The resulting high-correlation network displayed a topology wherein two distinct gene clusters were bridged by a strong predominance of anticorrelated edges (Fig. 3A). The opposing “left” (L) and “right” (R) gene clusters were very highly enriched in functionally related genes; the L cluster predominates in functions related to oxidative phosphorylation, while the R cluster is especially enriched in genes related to transcriptional regulation and neuronal signaling (Supplemental Table S9).
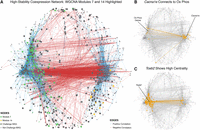
(A) Coexpression network of plotting the largest connected component with all relationships with correlations of an absolute value of 0.85 or better. The network topology includes two sides connected by a profusion of negative correlations. Module 7 nodes (green), which are associated with oxidative phosphorylation, inversely connect to module 14 nodes (yellow), which are associated with ion channels. (B) The gene with the highest betweenness centrality in module 14, Cacna1e, connects to many oxidative phosphorylation genes. Edges connecting out from Cacna1e and the nodes they connect to are highlighted in orange. (C) The gene with the highest betweenness centrality in module 7, Tceb2, implies a role for transcriptional regulatory dynamics. Edges connecting out from Tceb2 and the nodes they connect to are highlighted in orange.
Within this network structure, we identified two WGCNA modules with particularly significant relationships to social challenge, modules 7 and 14, whose eigenegenes are strongly anticorrelated (r = −0.90). High-stability module 7 genes were highly enriched for oxidative phosphorylation, while high-stability module 14 genes were enriched for ion channel activity. Indeed, the gene with the highest measure of betweenness centrality (BC; a metric of network connectivity) from module 14, Cacna1e, is a DEG in the tissue:challenge:time factor and in FCX pairwise analysis at 30 min and displays very strong and direct negative relationships with oxidative phosphorylation-related nodes (Fig. 3B). Cacna1e is particularly interesting in this context, connecting calcium signaling, insulin signaling, and synaptic plasticity (Pereverzev et al. 2002; Breustedt et al. 2003), and has been implicated in fear learning (Lee et al. 2002). The WGCNA network structure thus identified concrete bridges between the metabolism-related DEGs described in the GLM results and the neural signaling DEGs described in the pairwise post-hoc results, providing an explicit architecture of an oppositional relationship between brain oxidative phosphorylation and the neuronal response to social challenge.
We further identified the gene with the highest BC in module 7 and in the entire network structure as Tceb2, a subunit of the TF B complex that activates RNA Pol II activity (Fig. 3C). Together, the central role of Tceb2 in core transcription and the enrichment of TFs and chromatin modifiers in the L cluster imply that transcriptional regulation is a core component of the network, a prediction examined further below.
Transcriptional regulatory dynamics underlie the transcriptomic response to intruder
Transcriptional regulatory network reconstruction identifies a cohort of TFs central to intruder response
To identify TFs orchestrating this challenge response, we reconstructed a transcriptional regulatory network (TRN) across the expression data set using the ASTRIX approach. ASTRIX is also based on coexpression but centers on expressed TFs and highly correlated putative “target genes” (Chandrasekaran et al. 2011). The analysis yielded a TRN consisting of 4251 interactions between 253 TFs and 1211 target genes (Supplemental Table S10). We validated this TRN by comparing TF–gene interactions in our TRN with those constructed using DNase footprinting from the ENCODE Project (Stergachis et al. 2014), and found highly significant overlap between networks constructed using ENCODE data from mouse adult and fetal brains (hypergeometric overlap test P-values <10−11) compared with a random brain TRN with the same TFs and targets (for further details, see Supplemental Methods).
To identify the TFs most likely to drive the observed differential gene expression, we extracted those TFs with predicted target sets enriched in pairwise and model DEGs (Supplemental Table S11; pairwise results presented in Fig. 4). The TRN noted above includes TFs with related functions and outlines a regulatory architecture linking energy metabolism, developmental signaling, neuron plasticity, and social behavior (Table 1). Especially noteworthy is the presence of TFs associated with combined metabolic and behavioral phenotypes including ESRRA, PLAGL1, NR4A1, and NFATC2.
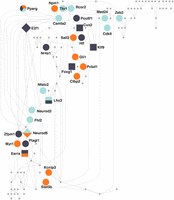
Network of TFs with TF–TF interactions in the TRN highlighting tissue-specific and time-specific regulators. TFs whose targets were enriched for DEGs in FCX are orange, TFs whose targets were enriched for DEGs in HYP are dark gray, and TFs whose targets were enriched for DEGs in AMY are blue. The times after social challenge where enrichment was found are depicted as shapes: Square nodes are enriched at 30 min, circular nodes are enriched at 120 min, and diamond-shaped nodes are enriched at both 30 min and 120 min after challenge. Multicolored TF modules such as Pparg and Lhx2 were enriched for DEG in multiple regions or time points. TFs that were not enriched for any tissue-specific or time-specific DEG—shown as small nodes—may represent homeostatic processes active across all conditions.
Metabolic and neurological functions for TFs in the social challenge TRN
Significant global chromatin remodeling accompanies intruder response
Gene expression patterns integrate TF activity with chromatin accessibility, and the magnitude and rapidity of the challenge-associated transcriptional response suggested that shifts in chromatin accessibility might underlie this response. Since accessible chromatin is generally marked by histone H3 acetylated at lysine 27 (H3K27ac) (Hon et al. 2009), we carried out chromatin immunoprecipitation followed by deep sequencing (ChIP-seq) with a validated H3K27ac antibody to map accessible chromatin in each brain region at different time points after challenge (Supplemental Table S12). We will refer to H3K27ac peaks shared by challenged and control animals as “baseline peaks” and peaks significantly altered in accessibility after challenge as “differential accessibility peaks” (DAPs).
We identified 23,763 DAPs with at least a twofold difference in H3K27ac enrichment (at HOMER P < 1 × 10−4) between challenged and control samples among all comparisons (Supplemental Table S12). Chromatin near DEGs generally transitioned from an open to a relatively more closed state after challenge in both FCX and HYP, while chromatin transitioned to a more open state in AMY samples over time, a pattern that mirrors the changes seen in expression (Fig. 5A; Supplemental Table S12). Despite the apparent match between accessibility and expression profiles, DAPs did not track pairwise post-hoc differential expression in matched regions and times after social challenge (Supplemental Table S13A).
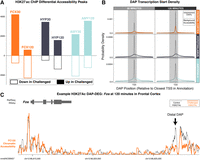
(A) H3K27ac DAPs in pairwise comparisons at a threshold of twofold. Solid bars above the x-axis represent peaks with increased accessibility in experimental samples relative to control. Outlined bars below the x-axis represent peaks with decreased accessibility in experimental samples. (B) Probability density of locations of differential accessibility in a 30-kb window around TSS sites. While the highest density of DAPs still lies within 5 kb upstream of or 2 kb downstream from (gray boxes) the TSS, the majority of DAPs are distal. Relative to background accessibility (thin black lines), DAPs have lower density near the TSS and higher density at distal sites. (C) Example distal differential accessibility peak in a known distal enhancer element for the immediate early gene Fos in FCX at 120 m after challenge.
We attribute this mismatch to two general processes. First, we believe there may be a mismatch in the speed of changes in accessibility and in changes in expression. For example, the only brain region in which DAPs significantly predict pairwise DEGs is the AMY, where genes near DAPs at 30 min after social challenge predict DEGs at 120 min after challenge (hypergeometric overlap test, FDR = 1.61 × 10−8). Therefore, DAPs detected at 120 min after challenge—by far the most numerous—may predict gene expression at later time points. Second, while baseline H3K27ac peaks generally clustered near promoters (41.2% of baseline H3K27ac peaks lie within 5 kb upstream of and 2 kb downstream from transcription start sites), DAPs in every brain region and time point displayed a highly significant preference for more distal locations relative to baseline peaks (Fisher's exact test P-values <10−76) (Fig. 5B; Supplemental Table S13B). Therefore, although it is more difficult to assign distal enhancers to target genes conclusively, correlations limited to promoter-proximal DAPs are likely to miss many significant regulatory relationships.
To identify cases of potentially causative links between differential accessibility and expression, we examined DAPs within longer distances (100 kb) of the TSS of the closest pairwise DEGs. We found 788 DAPs linked to 323 DEGs across brain regions and time points after social challenge (Supplemental Table S14). Among these DAPs were known distal enhancers, including an element located approximately 10 kb downstream from the immediate early gene Fos (Fig. 5C). This distal element, called “e5,” interacts with the Fos promoter through 3D chromatin interactions in response to neuronal stimulation of various types (Joo et al. 2016). Together these data suggest that the modulation of distal enhancers and the three-dimensional chromatin landscape are hallmarks of the social challenge response.
Motif enrichment within open chromatin regions reveals a role for pioneer factors and highlights ESRRA activity in intruder response
To explicate the interaction between active TFs and chromatin components, we asked whether known TF binding motifs (TFBMs) were enriched in baseline chromatin near DEGs or in DAP-DEGs. Although we did not find many DAPs near TSS positions, the highest density of baseline H3K27ac peaks was found near TSS positions. We examined motifs within −5-kb to +2-kb intervals, filtering data to include only motifs present in baseline-accessible regions within these intervals. By using this filter, we found significant enrichment of TFBMs, including motifs for TFs in the TRN (Supplemental Table S15A). For example, DEGs for HYP and FCX showed enrichment for ESRRA-related motifs, particularly in DEGs down-regulated at 60 min. Importantly, we did not detect significant TFBM enrichments without first filtering for accessibility; limiting motif searches to accessible windows thus greatly improved the accuracy of TFBM identification.
Focusing next on DAP-DEGs, we identified significant motif matches (binomial test P-value <0.001) primarily to “pioneer factors” (for review, see Iwafuchi-Doi and Zaret 2014), including TFAP2A, CEBPA, POU3F1, and MEIS1. We also identified enrichments for TFBMs associated with chromatin remodeling factors (HINFP1 [Ng et al. 1999]; ELK1 [Besnard et al. 2011]; EGR1 [Spaapen et al. 2013]) (Supplemental Table S15B). We regard these statistical associations as evidence that the regulatory response to challenge includes the action of pioneer and remodeling factors to reshape the accessibility landscape near target genes.
The combined analysis of gene expression, chromatin accessibility, network reconstruction, and DNA binding motif enrichment revealed several apparent mechanisms through which TFs and other key proteins participate in the challenge response (Table 2). For example, while the abundance of some TFs, like Fos and Neurod2, appears to be regulated transcriptionally, the differential expression may or may not be driven through differential accessibility. On the other hand, for TFs such as ESRRA whose nuclear location and DNA binding activities are regulated post-translationally (Rossi et al. 2011; Huss et al. 2015), their central action in the TRN appears without differential expression of the genes themselves.
Summary of key genomics results for selected genes reveals different modes of activation and participation in the challenge response
ESRRA protein binds to promoters of metabolically active genes in FCX
A central hypothesis to emerge from these studies is that the adaptive response to social challenge couples the down-regulation of brain oxidative phosphorylation to the activation of pathways associated with neuronal plasticity. Our data further indicate that this transcriptional response is correlated with alterations in the chromatin landscape coordinated with the activities of specific TFs. At the nexus of these expression and regulatory dynamics stands the TF ESRRA, a nuclear receptor known as a key regulator of metabolism and differentiation (for review, see Villena and Kralli 2008). Also highlighted in our cross-species study (Rittschof et al. 2014), ESRRA was implicated in both the TRN and baseline motif predictions presented here. Of particular interest, ChIP-chip experiments involving human cell lines (Deblois et al. 2009) and mouse heart and liver (Dufour et al. 2007; Charest-Marcotte et al. 2010) indicate that ESRRA is a direct regulator of energy metabolism genes. Consistently, ESRRA mutations are associated with both metabolic defects and social behavior deficits in humans and mice (Cui et al. 2015), suggesting a central role for ESRRA in coordinating metabolic and behavioral response.
We tested a key provision of this hypothesis by performing ESRRA ChIP-seq in FCX chromatin collected 30 min after challenge, identifying 636 ChIP-seq peaks (Supplemental Table S16). ChIP peaks were strongly enriched for the known ERRE motif shared by ESRRA and paralogs (HOMER ESRRB motif enrichment P-value: 1 × 10−356) and included a number of previously identified ESRRA binding targets. Notably, one of the strongest peaks, mapped near the Esrra promoter, marks a conserved multihormone response element that binds ESRRA and other nuclear receptors (Laganiere et al. 2004; Liu et al. 2005), indicating auto-regulation.
Analysis of genes nearest ESRRA ChIP-seq peaks showed especially high enrichment for mitochondrial and oxidative phosphorylation-related functions (DAVID annotation cluster scores = 26.95 and 14.85), as well as terms related to glycolysis and gluconeogenesis. Genes near ESRRA peaks were also highly enriched in DEGs from the tissue:challenge:time factor of the GLM results (hypergeometric overlap test P-value = 0.0053). Aside from energy metabolism genes, ESRRA FCX binding sites were enriched near TF loci; these include DEGs such as the genes encoding known ESRRA interactor and energy metabolism regulator GABPA and the TRN component and neurodevelopmental TF NEUROD2 (Supplemental Table S16; Sakkou et al. 2007). These data suggest that Esrra is central to the challenge-related response and potentially involved in coordinating the regulation of energy metabolism genes with expression of neurodevelopmental TFs.
ESRRA is highly expressed in oligodendrocytes and neurons in FCX
Determining where and when a gene or protein is expressed is central to deciphering its biological function, and ESRRA is a case in point. A recent study ruled out expression of mouse ESRRA in cortical astrocytes and concluded that the protein was neuronal, but did not examine markers to test this assumption (Cui et al. 2015). Furthermore, the cell-type–specific expression data upon which the PSEA model was based showed Esrra gene expression in multiple cortical cell types (Zhang et al. 2014). Since PSEA analysis tied the energy metabolism signal primarily to oligodendrocytes in the FCX, we hypothesized that ESRRA would also be expressed in that cell type.
To test this hypothesis, we used an antibody for ESRRA together with those for two cell-type–specific markers: CNPase (a marker of oligodendrocyte cytoplasm) and NeuN (a marker of neuronal nuclei) in immunohistochemistry on thick (200-µm) CLARITY-cleared mouse brain slices. ESRRA protein was expressed at high levels in the nuclei of cells across the cortex (Fig. 6A). Like many TFs and most nuclear receptors (Rossi et al. 2011), ESRRA was also present in the cytoplasm, including cellular processes. High-magnification images revealed ESRRA protein expression in oligodendrocyte nuclei, identified by being surrounded by thin layers of CNPase-positive cytoplasm (arrow in Fig. 6B); ESRRA also costained CNPase+ cytoplasm, including processes extending from these cells. However, a number of large ESRRA+, CNPase− nuclei were also found in the same regions (blue arrows in Fig. 6B,C). These large nuclei correspond to neurons, as evidenced by costaining with neuronal marker NeuN (green arrows in Fig. 6D,E). Together these data place ESRRA protein in oligodendrocytes as well as neurons in the cortex, suggesting that the protein serves regulatory functions in both types of cells.
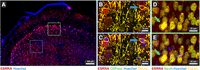
Localization of ESRRA. (A) Sagittal overview showing FCX distribution of ESRRA protein (red) with nuclei counterstained by Hoechst 33342 (blue). Blue and green boxes show approximate positions of panels B,C and D,E, respectively. (B,C) Fine detail in the FCX showing colocalization of ESRRA (red) and CNPase (green) in individual oligodendrocytes (blue arrow) without (B) and with (C) nuclear counterstain (blue). (D,E) Fine detail in the FCX showing costaining of ESRRA (red) and NeuN (green) in individual neurons (green arrow) without (D) and with (E) nuclear counterstain (blue).
Discussion
We describe a rich data set documenting gene expression and chromatin modulation in the mouse brain as animals respond to a salient social challenge in the form of a territorial threat. It is well known that threatened animals adjust their behavior after exposure to such a challenge (Fernald and Maruska 2012), and our goal was to develop a novel view of sequence of molecular events that direct this behavioral modulation. To do this, we measured the changing patterns of gene expression and chromatin profiles across the brain and over time and integrated these data to develop new insights to the regulatory mechanisms underlying a very complex biological response.
The completed analysis supports and significantly elaborates a model first suggested in our earlier study (Rittschof et al. 2014), in which social challenge induces a rapid shift in energy metabolism that is linked to activation of developmental signals driving neural plasticity in the brain. Here, we demonstrate that the energy shift is correlated primarily with glia, particularly oligodendrocytes, in this behavioral context and that the metabolic signal in these cells prefigures TRN activation, primarily in AMY neurons. Significant shifts in chromatin accessibility over time and across the brain, particularly affecting regulatory elements that are far distal from the TSS of nearest genes, are correlated with these events. TRN reconstruction and motif enrichment highlight the underlying network of interacting TFs, including many with known functions in energy homeostasis and neurodevelopment, and estrogen-related receptor alpha is highlighted for an early, central role. We show that ESRRA, which is expressed in both cortical oligodendrocytes and neurons, binds to energy homeostasis-related and TF genes in the FCX of challenged resident mice. Since ESRRA was also implicated in our cross-species analysis of the response to territorial threat (Rittschof et al. 2014), we propose ESRRA as a central node in a gene network coupling metabolic signaling with neuronal and behavioral plasticity in the context of social threat across a diverse array of animal species.
More specifically, these data provide new information about the traverse of these events across the brain over time. The deeply conserved metabolic signal observed rapidly after challenge (Rittschof et al. 2014) is known in honey bees to represent a shift in the balance of glucose metabolism away from oxidative phosphorylation and toward aerobic glycolysis, better known as the Warburg effect (Chandrasekaran et al. 2015). Warburg metabolism has been linked to learning in other contexts, including adaptive motor learning in mice (Shannon et al. 2016). An interesting corollary to this linkage is that glycolytic “hotspots” in the adult human brain—which are also hotspots of plasticity—display “transcriptional neoteny,” or gene expression patterns characteristic of the developing brain (Goyal et al. 2014). Thus, Warburg metabolism correlates with reawakening of genes associated with neuron differentiation and plasticity both spatially and temporally. Although astrocytes are a primary source of glycolytic activity in mammalian brains, other cell types, including neurons and oligodendrocytes, are sensitive to blood glucose levels and respond to other signals of metabolic state (Gundersen et al. 2015). Our study points to oligodendrocytes as the site of a Warburg-like metabolic shift and suggests that this shift is translated through metabolically-sensitive signaling pathways and TFs to neurons, activating the neotenous, plastic state.
Outlining the mechanism in further detail, our data reveal an early burst of neuropeptide signaling, very likely responsible for the metabolic shift, which is accompanied by calcium channel and neurotransmitter-related activity in the HYP and FCX. This initial phase is followed temporally by significant up-regulation of “neotenous” neurodevelopmental signaling genes, particularly in the AMY. Underlying this entire sequence of events, the modeling results revealed the persistent modulation of oxidative phosphorylation as the most robust system-wide transcriptional response. Therefore, we conclude that the down-regulation of oxidative phosphorylation-related genes we detected at an early time point (Rittschof et al. 2014), driven rapidly after challenge by neural signaling and neuropeptide release, has an enduring effect in the form of plasticity that follows social challenge response across the brain.
Second, we present new information regarding the cis- and trans-acting components that regulate these processes. The response to challenge is marked by a substantial level of global chromatin reorganization, with chromatin dynamics correlating strongly with overall patterns of gene expression across the brain. Unlike an earlier study focused on response to foot shock (Halder et al. 2016), we detected a global reorganization of chromatin in response to social challenge, even at the earliest time point. Although chromatin structure is generally thought to be stable in adult cells (Whitney et al. 2014), recent work shows that activity-dependent chromatin remodeling happens in neurons within an hour (Yang et al. 2016). Our data suggest that the chromatin landscape is altered very quickly in the adult brain after a salient emotional experience. Based upon data from AMY, where early differential accessibility appears to predict late differential expression, we conclude that differential chromatin accessibility may prefigure differential expression. The differentially accessible peaks we identified were enriched in motifs associated with pioneer factors and chromatin modifiers, suggesting an important role for these factors in determining social response; the fact that most DAPs were located far from TSS suggests the importance of three-dimensional chromatin architecture in this epigenetic process. In addition to known enhancers, these DAPs comprise a rich resource of novel candidate regulatory elements that are linked to social behavior by this study for the first time.
By using network-based and motif-enrichment tools, we predicted a cohort of coordinated TFs to interact with these novel regulatory elements to drive the transcriptional response. The TRN comprises TFs known to integrate metabolic and neurobiological functions, including the estrogen-related receptor ESRRA. ESRRA itself was not differentially expressed in this study, and the ESRRA motif was enriched near baseline but not differential accessibility peaks. Yet, Esrra expression, ESRRA motif enrichment, and ESRRA ChIP-seq peaks strongly predict the differential expression of other genes. These facts pose a puzzle on first consideration, but they are reconciled by the nature of this TF protein and its known modes of action. In particular, ESRRA is a nuclear receptor, the activity of which is regulated through ligand binding and post-translational modifications that impact nuclear localization and DNA binding properties (Ozcan et al. 2010). Indeed, many TFs, including some of those in our network, are known to be regulated by post-translational events that affect their localization, DNA binding, protein–protein interactions, and more (e.g., Reich and Liu 2006; Ivanova et al. 2007; Ozcan et al. 2010; Boldingh Debernard et al. 2012; Dayalan Naidu and Dinkova-Kostova 2017); TFs of this type thus need not to be significantly differentially expressed, or permitted de novo access to differentially accessible chromatin sites, to carry out differential regulatory roles.
Consistent with ChIP results in other tissues (Dufour et al. 2007; Charest-Marcotte et al. 2010), ESRRA binds primarily near genes involved in energy metabolism in cortical cells and is enriched near challenge-related DEGs. However, ESRRA binding sites are also enriched near TF genes, including key neurodevelopmental regulators, suggesting one possible mechanism through which metabolic and neurodevelopmental components of social challenge response might be transcriptionally coordinated. Known functions of ESRRA are consistent with this proposed role. Specifically, mouse Esrra mutations are associated with defects in insulin and glucose homeostasis as well as behavioral rigidity and social deficits (Luo et al. 2003; Dufour et al. 2011; Cui et al. 2015), and in human patients, ESRRA is linked to the combined metabolic and behavioral symptoms of eating disorders (Cui et al. 2013). On the cellular level, ESRRA regulates Warburg-like metabolic shifts in cancer cells and during cellular differentiation in mammals (Nie and Wong 2009; Charest-Marcotte et al. 2010) and coordinates a Warburg shift that is essential to Drosophila development (Tennessen et al. 2011). Together these data argue that ESRRA is an essential component in coordinating Warburg-like metabolism, developmental functions, and behavioral plasticity across the evolutionary spectrum.
It is not known how Warburg metabolism interfaces with neural plasticity, but two general hypotheses have been advanced. First, the redeployment of glycolysis characteristic of the Warburg response increases synthesis of raw materials like lipids, amino acids, and nucleotides. In cancer and development, these materials are essential to the rapid generation of new cells (for review, see Vander Heiden et al. 2009; Tech et al. 2015). In the brain, these materials would be directed toward neurite outgrowth, dendritic branching, synaptogenesis, synaptic remodeling, and myelination (Goyal et al. 2014). Alternatively or additionally, byproducts of aerobic glycolysis are shuttled between glia and neurons, interfacing with a range of signaling pathways (Yang et al. 2014; Gundersen et al. 2015; DiNuzzo 2016), which possibly include signaling cascades required to organize the raw materials into new neural structures (Agathocleous and Harris 2013).
These signaling pathways may, in turn, activate neuronal gene expression. For instance, the activities of some TFs, including ESRRA regulator and coactivator PPARGC1A, are regulated directly by metabolites and metabolic signaling (Scarpulla 2006). Beyond its role in energy homeostasis, ESRRA is known as an upstream regulator of Wnt (Auld et al. 2012), and Wnt intersects with ESRRA on both metabolic and behavioral axes. Notably, mutations in the Wnt effector, Tcf7l2, are associated with abnormal insulin and glucose homeostasis as well as deficits in fear learning (for review, see Nobrega 2013). Tcf7l2 is one of a small number of genes significantly up-regulated across the brain 120 min after challenge, suggesting an important late role in challenge response. Together these data suggest a model in which Wnt and neural plasticity downstream from Tcf7l2 is metabolically regulated through ESRRA in socially responsive regions of the brain.
We close by pointing to a clear limitation of this study: Our experiments were conducted in tissue homogenates, limiting our ability to assign expression or chromatin signals to any specific cell type. Our data, together with results of previous studies (Cui et al., 2015), assign ESRRA regulatory function primarily to neurons and oligodendrocytes in the cortex, suggesting these cells as the locus of the metabolic shift in this specific context. However, cell-type–specific methods of discovery will be required to address this hypothesis and add further depth to the discussion. Nevertheless, we provide a novel view of the brain's response to social challenge, including a large collection of novel challenge-associated enhancers and networked TFs with predicted metabolic and neurobehavioral roles. The data build support for a deeply conserved, fundamental, and mechanistic link between shifts in brain energy metabolism and subsequent redeployment of developmental factors for behavioral plasticity in the context of emotional learning.
Methods
Animals
All protocols were approved by the UIUC IACUC (protocol no. 15245) and undertaken in compliance with the NIH Guide for the Care and Use of Laboratory Animals. All reasonable efforts were undertaken to minimize animal suffering. Briefly, after a period of habituation to our animal room and cohousing with females to establish a territory, animals were confronted with an unfamiliar intruder for 5 min and then kept in a quiet, dark place for 30, 60, or 120 min until they were euthanized by cervical dislocation. Immediately after euthanasia we extracted FCX, HYP, and AMY. A schematic of the dissection is included as Supplemental Figure S1, and detailed methods for animal husbandry, behavior, and dissections are included in Supplemental Methods.
RNA extraction and library preparation and RNA sequencing
Dissected tissue was immediately flash-frozen in liquid nitrogen and held at −80°C. Total RNA was prepared and analyzed for quality as previously described (Bolt et al. 2016). RNA-seq libraries were prepared robotically from total RNA and then sequenced on an Illumina HiSeq 2500 sequencer. Detailed methods for RNA extraction, library preparation, and RNA-seq are included in Supplemental Methods.
RNA-seq bioinformatics, functional enrichment, and WGCNA analysis
FASTQ files were aligned to the Ensembl (Flicek et al. 2012) annotation of the NCBIM37 version of the mouse genome using TopHat2 (Kim et al. 2013) and counted using HTSeq (Anders et al. 2015), and differential expression analyses were done using the Bioconductor package edgeR (Robinson et al. 2010).
To enrich for biological systems and visualize the results of the pairwise comparisons, we used enrichment and visualization techniques similar to the online tool REVIGO (Supek et al. 2011). Other systems enrichments utilized DAVID (Huang da et al. 2009a,b).
We used WGCNA (Langfelder and Horvath 2007) in signed mode to find modules of coexpressed genes across the entire data set. To visualize only the coexpression edges representing the most stable relationships, we extracted only the edges where the absolute value of correlation coefficients was >0.85 and was the same sign in every tissue. We visualized these relationships, calculated betweenness centrality, and plotted modules using Cytoscape (Shannon et al. 2003).
Detailed methods for RNA-seq bioinformatics, functional enrichment, and WGCNA analyses are included in Supplemental Methods.
RT-qPCR confirmation
RT-qPCR confirmation was performed for seven genes: Dnajb1I, Ide, Igf2, Oxt, Pmch, Tcf7l2, and Zic1. Primer sequences, run conditions, and results are presented in Supplemental Table S17. A comparison of results to RNA-seq is presented in Supplemental Figure S1, and complete RT-qPCR methods are included in Supplemental Methods.
Cell-type deconvolution analysis
Because brain tissue is a heterogeneous mixture of multiple distinct cell types, we used a modification of PSEA to deconvolve our samples and identify genes associated with some of the many cellular components of brain tissue (Kuhn et al. 2011). We used reference signals from astrocytes, neurons, oligodendrocytes, microglia, and endothelial cells generated in a previous RNA-seq experiment on sorted cells from adult mouse FCX (Zhang et al. 2014). Briefly, after regressing expression values on these reference signals, we used high-throughput model selection to find the best model or models, a good indicator of cell-type specificity. Details of the best models are reported in Supplemental Table S8, and detailed methods for cell-type deconvolution are included in Supplemental Methods.
TRN reconstruction
ASTRIX (Analyzing Subsets of Transcriptional Regulators Influencing eXpression) was applied to infer a TRN as previously described (Chandrasekaran et al. 2011). The predicted targets of TFs were defined as those genes that share very high mutual information with a TF and can be predicted quantitatively with high accuracy. We identified the TFs with predicted target genes most significantly enriched in DEGs at specific time points, tissues, or a combination of those parameters to identify a cohort of the TFs most predictive of challenge-associated DEG sets (Fig. 3; Supplemental Table S10). Further, we validated these TFs and targets against references TRNs from ENCODE (Stergachis et al. 2014). Details of the methods used to reconstruct the TRN are provided in Supplemental Methods.
ChIP tissue preparation, chromatin immunoprecipitation, and library preparation
After husbandry, challenge, and dissection as in the RNA-seq experiment for 30- and 120-min samples, tissue samples dissected from five animals were pooled for ChIP-seq. Briefly, nuclei were isolated, chromatin was fragmented, and a rabbit polyclonal antibody raised against acetylated histone H3 lysine 27 (Abcam ab4729) or ESRRA (Santa Cruz sc-66882) was used to precipitate chromatin. After ChIP, immunoprecipitated DNA was checked for quality and then used to prepare libraries that were sequenced with an Illumina HiSeq 2500 sequencer. Detailed methods used for ChIP-seq tissue preparation, library preparation, and sequencing are included in Supplemental Methods.
ChIP-seq bioinformatics
Sequence data were mapped with Bowtie 2 (Langmead and Salzberg 2012) to the UCSC Mus musculus mm9 genome and then analyzed for peaks using HOMER (Hypergeometric Optimization of Motif EnRichment, Heinz et al. 2010). Differential chromatin peaks were identified as any peaks that changed at least twofold between conditions with a significance cutoff of 1 × 10−4.
To identify differential accessibility peaks associated with differential expression (DAP-DEGs), a chromatin domain was defined for each gene in the mouse mm9 genome. First, for each Ensembl-annotated splice variant arising from the gene, a window was defined that began 100 kb upstream of the transcription start site and ended 100 kb downstream from the transcription end site. Next, this window was truncated so that it did not intersect with any transcript of any other gene. A DAP that overlaps with any window of any transcript of a gene, along with that corresponding gene, constitutes a DAP-DEG.
Detailed methods used for ChIP-seq bioinformatics and statistics used for DAP calling are reported in Supplemental Methods.
Cis motif analysis
We used the Stubb algorithm (Sinha et al. 2003) to identify sequence segments with significant presence of a TF binding motif (position weight matrix), scanning the genome with 500-bp windows with a 250-bp shift size. To incorporate accessibility information (captured by H3K27ac ChIP-seq data), we required that the motif score of a 500-bp window be considered only if the window is deemed “accessible”; i.e., the windows either overlapped with identified H3K27ac ChIP peaks or were proximal to such peaks and had read count above input control. All such windows were assigned scores equaling their average read count, and the resulting profile of genome-wide scores was smoothed as previously described (Kazemian et al. 2013). Finally, windows scoring in the top three percentile were considered accessible. Detailed methods used for cis motif analysis are reported in Supplemental Methods.
Identification of motifs in DAP-DEGs
To test for cis motif enrichment in DAP-DEGs, we adapted the method above as follows: For each motif, the Stubb score P-value corresponding to the best-scoring 500-bp window that overlapped each DAP-DEG was assigned as the score of that DAP-DEG. The number of DAP-DEGs with scores <0.05 were counted and subjected to a binomial test where the success probability parameter was learned from the frequency of such motif scores in size-matched background sequences that were sampled from gene deserts and did not contain the H3K27ac mark.
Thick-slice CLARITY immunohistochemistry
To assess the cellular and subcellular localization of specific TFs, we used a CLARITY protocol modified to work on 200-µM-thick slices of brain (Chung et al. 2013). Briefly, male animals were transcardially perfused, and then brains were extracted and fixed in the perfusion solution before being sectioned in 200-µm slices on a vibrating microtome. Sections were embedded in hydrogel and then cleared overnight using electrophoretic tissue clearing. After washing, tissue was incubated with primary antibodies (ERR-alpha: Santa Cruz sc-66882; CNPase: Millipore no. MAB326) for 3 d. After three more washes, cleared tissues were incubated with fluorescent secondary for 3 d before being washed. The final wash included the nuclear counterstain Hoechst 33342. The tissue slices were then cleared in RIMS made from 70% Histodenz (Sigma no. D2158) in PBS with Triton-X100 and mounted on lifter slides before being imaged on a Zeiss LSM 710 confocal microscope. Detailed methods used for thick slice CLARITY are reported in Supplemental Methods.
Data access
The sequencing data from this study have been submitted to the NCBI Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE80345.
Acknowledgments
We thank Alvaro Hernandez for expert oversight in sequencing in this project. We thank Mayandi Sivagaru for his expert advice on imaging. We thank Alison Bell and Gene Robinson for helpful discussions and Gene Robinson and Elbert Branscomb for critical comments on the manuscript. This work was primarily funded by a grant from the Simons Foundation (#SFLife 291812; awarded to L.S. and G.E. Robinson). Additional funding was provided by the Institute for Genomic Biology at the UIUC and by the William F. Milton Fund (awarded to S.C.).
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.214221.116.
- Received August 3, 2016.
- Accepted March 24, 2017.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see http://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








