
The landscape of antisense gene expression in human cancers
- O. Alejandro Balbin1,2,3,
- Rohit Malik1,2,7,
- Saravana M. Dhanasekaran1,2,7,
- John R. Prensner1,2,
- Xuhong Cao1,2,
- Yi-Mi Wu1,2,
- Dan Robinson1,2,
- Rui Wang1,2,
- Guoan Chen4,
- David G. Beer4,
- Alexey I. Nesvizhskii1,2,3,8 and
- Arul M. Chinnaiyan1,2,3,5,6,8
- 1Michigan Center for Translational Pathology, University of Michigan, Ann Arbor, Michigan 48109, USA;
- 2Department of Pathology, University of Michigan, Ann Arbor, Michigan 48109, USA;
- 3Department of Computational Medicine and Bioinformatics, University of Michigan, Ann Arbor, Michigan 48109, USA;
- 4Department of Surgery, Section of Thoracic Surgery, University of Michigan, Ann Arbor, Michigan 48109, USA;
- 5Department of Urology, University of Michigan, Ann Arbor, Michigan 48109, USA;
- 6Comprehensive Cancer Center, University of Michigan, Ann Arbor, Michigan 48109, USA
- Corresponding authors: arul{at}umich.edu, nesvi{at}med.umich.edu
-
↵7 These authors contributed equally to this work.
Abstract
High-throughput RNA sequencing has revealed more pervasive transcription of the human genome than previously anticipated. However, the extent of natural antisense transcripts’ (NATs) expression, their regulation of cognate sense genes, and the role of NATs in cancer remain poorly understood. Here, we use strand-specific paired-end RNA sequencing (ssRNA-seq) data from 376 cancer samples covering nine tissue types to comprehensively characterize the landscape of antisense expression. We found consistent antisense expression in at least 38% of annotated transcripts, which in general is positively correlated with sense gene expression. Investigation of sense/antisense pair expressions across tissue types revealed lineage-specific, ubiquitous and cancer-specific antisense loci transcription. Comparisons between tumor and normal samples identified both concordant (same direction) and discordant (opposite direction) sense/antisense expression patterns. Finally, we provide OncoNAT, a catalog of cancer-related genes with significant antisense transcription, which will enable future investigations of sense/antisense regulation in cancer. Using OncoNAT we identified several functional NATs, including NKX2-1-AS1 that regulates the NKX2-1 oncogene and cell proliferation in lung cancer cells. Overall, this study provides a comprehensive account of NATs and supports a role for NATs' regulation of tumor suppressors and oncogenes in cancer biology.
The human genome is widely transcribed (Kapranov et al. 2002; 2007; Okazaki et al. 2002; Carninci et al. 2005; Cheng et al. 2005; Djebali et al. 2012); however, the extent to which both strands of DNA are transcribed at any given locus needs further characterization. Natural antisense transcripts (NATs) are transcribed RNA products from the DNA strand complementary to a region harboring a sense transcript of either protein-coding or noncoding genes (Katayama et al. 2005; Guil and Esteller 2012; Pelechano and Steinmetz 2013). NATs may arise from independent transcriptional units containing cryptic promoters situated within genes, typically in intronic regions, or near transcriptional start sites of neighboring genes. Depending on the orientation of the transcripts involved, overlapping pairs (cis-NAT pairs) are classified as follows: head-to-head (5′-regions overlap) (HTH), tail-to-tail (3′-regions overlap) (TTT), embedded (one transcript is fully contained within the other) (EMB), or intronic (INT) pairs.
NATs can function locally (in the nucleus) or distally (in the cytoplasm) and in a cis or trans manner (Pelechano and Steinmetz 2013) to regulate the expression of their cognate genes. Trans-acting NATs can also regulate the expression and/or function of other genes. Local cis-regulation involves epigenetic changes proximal to a target gene, whereas distal cis-regulation involves RNA-RNA interactions between transcripts originating from the same locus. Cis-regulation of NATs can lead to activation (Sessa et al. 2007; Zhang et al. 2009) or silencing (Modarresi et al. 2012) of the corresponding sense mRNAs via transcriptional activation (Sessa et al. 2007; Zhang et al. 2009), silencing (Yu et al. 2008; Congrains et al. 2013), mRNA stabilization (Mahmoudi et al. 2009; Su et al. 2012), alternative splicing (Morrissy et al. 2011), or post-translational regulation among others. The role of dysregulated antisense transcript expression has been investigated in neurological illnesses such as Alzheimer's disease (Faghihi et al. 2008), schizophrenia (Millar et al. 2000), Parkinson's disease (Scheele et al. 2007), and in multiple cancers (Luo et al. 2006; Huarte et al. 2010; Geng et al. 2011; Kogo et al. 2011; Prensner et al. 2011; Schmidt et al. 2011; Silva et al. 2011; Niinuma et al. 2012; Han et al. 2013; Kim et al. 2013b).
Previous studies of antisense expression often assessed only a small fraction of the transcriptome largely missing low-expressed transcripts, primarily due to methodological limitations including low accuracy and transcriptome coverage (Katayama et al. 2005; He et al. 2008). Antisense transcript detection and assessment, especially in cancer, is hampered by the small data sets (<20 samples) that were characterized. RNA sequencing (RNA-seq) studies allow an unbiased genome-wide analysis of the human transcriptome to elucidate novel disease biology. The Cancer Genome Atlas (TCGA) project has generated data from more than 6000 RNA-seq analyses (http://cancergenome.nih.gov/). However, the conventional methodology utilized in generating these RNA-seq libraries does not preserve transcript strandedness information. Although computational methods relying on splice-site orientation can be used a posteriori to infer the transcript orientation in eukaryotic genomes, accurate resolution of expressed genes with an overlap in their genomic location is challenging. Strand-specific RNA sequencing (ssRNA-seq) (Parkhomchuk et al. 2009) resolves these issues by facilitating precise template strand mapping of the boundaries of antisense transcripts, thereby enabling accurate expression measurements. Here we present the results of the largest ssRNA-seq study to date, profiling 376 samples representing nine different cancer and normal tissue types to comprehensively characterize the landscape of antisense expression.
Results
Pervasive antisense expression across the human transcriptome
We generated strand-specific RNA paired-end sequencing (ssRNA-seq) data from the Michigan Center for Translational Pathology (MCTP) compendium of 376 samples representing cancer and benign conditions from nine different tissue types (303 tissues and 69 cell lines) (Supplemental Table S1; Supplemental Data S1). On average, 101 million read pairs were obtained per library across all cohorts (Supplemental Fig. S1; Supplemental Data S2) for a total of 38.2 billion read pairs, and the data set quality metrics are provided in Supplemental Data S3 and Supplemental Figures S2 and S3. Using this data set, we developed a bioinformatics workflow (Fig. 1A) to characterize the sense/antisense expression of 42,124 gene models (Supplemental Data S4) across human cancers (Supplemental Data S5, S6) (see Methods). This constitutes the most comprehensive assessment of transcription originating from the antisense strand of DNA in cancer genomes.
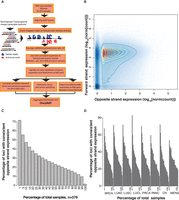
Antisense expression is pervasive across the human transcriptome. (A) Bioinformatics workflow for characterization of expressed antisense loci. The summarized transcriptome was built by reconstructing the longest high quality annotation for each gene, using transcript and exon information provided in the Ensembl v69 assembly. This procedure generated 42,124 gene models (Supplemental Data S4) whose expressions on forward and opposite strands were calculated (Supplemental Data S5, S6). The protocol error rate of a library (pei) corresponds to the fraction of reads mistakenly mapped to the opposite strand but generated by the forward gene (Methods). Bona fide antisense transcripts are loci that have an OPSratio > pei_th in at least 30% of the cohort samples and have a statistically significant antisense expression according to NASTI-seq (Methods). This bioinformatics workflow nominates expressed antisense loci across cancer subtypes, establishes their pattern of expression, and generates a catalog of tumor suppressor and oncogenes with significant antisense expression, OncoNAT (Supplemental Data S11–S15). (B) Density plot of forward and opposite strand expression. Expression over forward and opposite strands is calculated for each locus per sample. Expression in the opposite strand is in general two to three orders of magnitude lower (median of opposite/forward = 0.001) than the expression of the forward strand. (C) The percentage of loci with consistent expression in the opposite strand. A loci is considered to have consistent opposite strand expression if the OPSratio > pei_th in at least 30% of the samples in the cohort. (D) The percentage of loci consistently expressing the opposite strand by tissue type: (BRCA) breast cancer; (LUAD) lung adenocarcinoma; (LUSC) lung squamous carcinoma; (LUCL) lung cancer cell lines; (PRCA) prostate cancer; (PANC) pancreatic cancer; (OV) ovarian cancer; (MENG) meningioma (see Supplemental Fig. S6 for extended version).
In general, at any given locus the magnitude of expression from the opposite (antisense) strand was two to three orders of magnitude lower than the forward (sense) strand (median of opposite/forward = 0.001) (Fig. 1B). To identify experimental variations in the strandedness among the libraries, we first calculated the ssRNA-seq strand specificity (Yassour et al. 2010) for each sample and then determined the fraction of the transcriptome with measurable opposite strand expression (Methods). Typically a library strand specificity ranges from 0.5% to 12%, depending on the quality of the ssRNA-seq libraries (Yassour et al. 2010). The average strand specificity of 0.64% (min = 0.17%; max = 0.69%; SD = 0.0055) that we obtained for the 376 samples sequenced, indicates a high library strand specificity (Supplemental Data S7) supporting the data set's utility in evaluating antisense expression. On average, 8% of total reads originated from the opposite DNA strand (Supplemental Fig. S4A), with minor variation noted across tissue types (Supplemental Fig. S4B,C) or disease stages (Supplemental Fig. S4D).
Various factors, including biological variation, library size differences, strand-specific-protocol efficiency, and more importantly the overall weak opposite strand expression, can collectively influence antisense measurements (Fig. 1B). Here we used a sequential set of filtering criteria to identify loci with significant antisense expression in order to overcome these limitations. First, loci with consistent opposite strand expression were identified by defining a sample-specific confidence interval for the protocol error rate (pei_th) (Methods). Based on simulation studies to define the appropriate pei_th (Supplemental Fig. S5), a locus is considered to have consistent antisense expression if its opposite strand (OPS) expression ratio [OPSratio = Opposite read counts/(Forward read counts + Opposite read counts)] is greater than pei_th in at least 30% (n = 113 for the full data set) of the cohort samples (Methods). On average, we noted consistent opposite strand expression from at least 38% (mean = 11,135; SD = 865) (Table 1; Fig. 1C) of annotated genes. This pattern had minimal variance regardless of the tissue of origin (Fig. 1D; Supplemental Fig. S6; Supplemental Data S8). Altogether, these results reveal prevalent genome-wide transcription from both strands in humans. To further refine our nominations, we used a probabilistic method for natural antisense transcript identification (NASTI-seq) (Li et al. 2013). This second filter uses a model comparison framework to identify loci with statistically significant antisense expression by calculating the probability of the observed read count data under a sense only or a sense/antisense model. In this approach, an antisense locus is defined as a region of DNA wherein the antisense model fits better than the sense plus protocol error rate only model, based on the read count data observed over that region (Methods).
Average number of bona fide antisense loci across the different cancer subtypes
Our bioinformatics workflow applied these filters to identify 11,054 unique antisense loci in the cancer transcriptome that are henceforth referred to as bona fide antisense loci. The number of bona fide antisense loci ranged from 7405 to 11,377 (mean = 9051; SD = 1021) across different cancer subtypes (Table 1; Supplemental Data S8). Out of those, 7241–9259 (98%–81%) genes are involved in annotated cis-NAT pairs (mean = 8422; SD = 699), whereas only 164–1001 (2%–10%) of loci are unannotated to form overlapping gene pairs based on the reference transcriptome (mean = 628; SD = 344) (Supplemental Data S8; Methods). Finally, of all bona fide antisense loci, 17% correspond to HTH, 18% to TTT, 20% to EMB, and 45% to INT cis-NAT pairs; Supplemental Table S2 further classifies these pairs by gene ontology.
Widespread correlation between sense and antisense transcript expression
A systematic characterization of all bona fide antisense loci expression forming cis-NAT pair types (Fig. 2A) revealed an overall positive correlation between sense/antisense genes, with a median Spearman correlation coefficient of 0.28 (Fig. 2B). This correlation is greater than what would be expected by chance. Among cis-NAT gene pairs, expression of cis-NATs with HTH orientation showed the strongest positive correlation (median R = 0.41) (Fig. 2C); whereas the other types—namely, TTT, EMB, and INT configurations—displayed comparable weak correlations (median R = 0.23, 0.22, and 0.26, respectively) (Fig. 2C). These patterns persist across various tissue types (Fig. 2D), suggesting common coordinated regulatory mechanisms controlling the cis-NAT transcription. Our results recapitulate well-studied and experimentally proven cases of coexpressed sense/antisense gene pairs such as WT1/WT1-AS and BDNF/BDNF-AS (Dallosso et al. 2007; Modarresi et al. 2012), validating our bioinformatics approach (Fig. 2E).
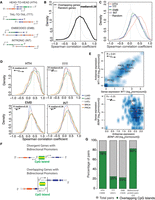
Correlation of cis-NAT pair expression and regulation by bidirectional promoters. (A) Schematic representation of cis-NAT pairs, according to the orientation of the overlapping genes. The arrow indicates transcriptional direction: (dark blue) overlapping exons; (gray) untranslated regions; (green or red) unique exons. (B) Distribution of Spearman's correlation coefficient between sense and antisense expression for all overlapping types (median R = 0.28). Correlation between random pairs of genes is represented by a gray dashed line (n = 14,166). (C) Distribution of Spearman's correlation coefficient between sense and antisense expression by overlapping type. Head-to-Head cis-NAT gene pairs show the highest positive correlation among all overlapping types (median R = 0.41). Correlation between random pairs of genes is depicted by a gray dashed line. HTH = 2485; TTT = 2515; EMB = 2788; and INT = 6378. (D) Distribution of Spearman's correlation coefficient between sense and antisense expression by tissue type. HTH = 2485; TTT = 2515; EMB = 2788; and INT = 6378. (E) Examples of coexpressed cis-NAT gene pairs previously reported. Density scatter plots for WT1/WT1-AS (Dallosso et al. 2007) (top) and BDNF/BDNF-AS (Modarresi et al. 2012) (bottom). The units of the x- and y-axis are log10[norm(count)], and red dots indicate the average value for the each cohort. (F) Schematic representation of CpG islands and bidirectional promoters: (top) CpG islands (green) are present in 80%–95% of bidirectional promoters (yellow) controlling nonoverlapping protein-coding genes in opposite directions; (bottom) model representation of a HTH cis-NAT gene pair sharing a bidirectional promoter located within the overlapping region. Arrows indicate the direction of transcription. (G) Percentage of cis-NAT gene pairs with CpG islands found in their overlapping regions. Seventy-eight percent of HTH cis-NAT contained CpG islands within their overlapping region, similar to the 83% observed for known bidirectional protein-coding gene pairs. Less than 25% of TTT and EMB pairs contained CpG islands in their overlapping regions.
Inspection of the HTH cis-NAT pairs revealed that 87% of them involved overlapping regions within their corresponding 5′UTR (5UTR-5UTR) or the 5′UTR and the first exon (5UTR-exon). This pattern is consistent with the usage of bidirectional promoters that are shared by ∼10% of protein-coding genes (Liu et al. 2011) and are known to produce divergent transcription. Because paired transcripts from bidirectional promoters often exhibit similar expression patterns (Trinklein et al. 2004), we hypothesized that HTH cis-NAT pairs might share bidirectional-like promoters directing their concerted expression.
To assess bidirectional promoters, we searched for loci that harbored two nonoverlapping transcriptional start sites with opposite orientation and separated by <1000 bp (Trinklein et al. 2004). Because bidirectional promoters are rich in CpG islands (CGI), present in >80% compared to only 30% of unidirectional promoters (Liu et al. 2011), we reasoned that if a bidirectional promoter exists between the genes of a HTH cis-NAT pair, it will likely be associated with a CGI in the overlapping region between the two genes (Fig. 2F). We found that 78% of the HTH cis-NAT pairs have CGIs in the region of overlap between the two genes (Fig. 2G). Similarly, we found that 83% of bidirectional but not overlapping protein-coding genes had CGIs in their promoters. In contrast, we observed CGIs only in 25% of the overlapping regions of cis-NAT pairs with TTT or EMB configurations (Fig. 2G). Taken together, these results support our hypothesis that shared bidirectional promoters may tightly regulate the coexpression of HTH cis-NAT pairs.
Cognate sense gene regulation by antisense transcripts
Recent studies with gene pairs from bidirectional promoters have shown the influence of noncoding transcript expression on the cognate protein-coding mRNA (Xu et al. 2009; Wei et al. 2011; Guil and Esteller 2012). In particular, regulation of sense gene expression by its antisense counterparts are reported for highly coexpressed HTH cis-NAT pairs involving HOXA11/HOXA11-AS (Chr 7), HOXA1/HOTAIRM1 (Chr 7), and WT1/WT1-AS (Chr 11) (Sessa et al. 2007; Zhang et al. 2009). These cis-NAT pair expressions were positively correlated in our data, consistent with previous reports (Fig. 2; Supplemental Fig. S7A). Our analysis nominated additional cases of potential cis-NAT pair regulation, and representative cases in the HOXD (Chr 2), HOXC (Chr 12), and HOXB (Chr 17) gene clusters are shown in Figure 3A and Supplemental Figure S7A. This suggests the existence of similar regulatory mechanisms between sense and antisense transcripts in other homeobox gene clusters. The positive correlation between sense and antisense transcript expression for representative cis-NAT pairs (HOXD1/HAGLR and HOXC10/HOXC10-AS3) was experimentally verified using qRT-PCR in a panel of cell lines (Fig. 3B; Supplemental Fig. S7B), which also credentialed our bioinformatics approach. A detailed description of the genomic arrangement of the HOXD1/HAGLR pair is shown in Figure 3C. Moreover, we showed that similarly to HOXA1/HOTAIRM1, knocking down of HAGLR (also known as HOXD-AS1) using two specific and independent siRNAs (Supplemental Fig. S8A) decreased HOXD1 (cognate sense gene) expression in H1792, a lung cancer cell line that expresses both transcripts (Fig. 3D).
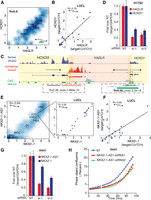
Antisense regulation of cognate sense genes. (A) Density scatter plot for HOXD1/HAGLR expression across the compendium. The units of the x- and y-axis are log10[norm(count)], and red dots indicate the average value for each cohort. (B) HOXD1 and HAGLR expression measured by quantitative PCR across a cohort of 29 lung cell line samples. (C) Representative example of tight coexpression of HTH cis-NAT gene pairs. Loci containing HOXD1/HAGLR/HOXD3 in PRCA sample (mctp_sample_337) is illustrated. Read support for expression in the sense and antisense strands is shown as tracks in blue and red, respectively. Transcript structure, CpG islands (green boxes), and H3K27ac (light blue) tracks are obtained from the UCSC Genome Browser (https://genome.ucsc.edu/) (Karolchik et al. 2014). Yellow and light red shadows represent the limits of sense and antisense genes, respectively. HOXD1/HAGLR HTH gene pair is tightly coexpressed: (right inset) R = 0.90; P-value ≤ 2.2 × 10−10. These transcripts share an overlapping region containing CpG islands, suggesting a bidirectional promoter. HOXD3/HAGLR, on the other hand, form a TTT gene pair with a lower coexpression pattern ([left inset] R = 0.36; P-value = 1.9 × 10−10) and no CpG islands in the overlapping region. (D) Knockdown of HOXD1-AS1 in the H1792 cell line, which expresses HOXD1 sense/antisense transcripts with two independent specific siRNAs. Knockdown decreases the expression levels of both the antisense (HOXD1-AS1) and the cognate sense genes (HOXD1). (E) Density scatter plot for the expression of NK2 homeobox 1 (NKX2-1), master regulator essential for lung development, and NKX2-1-AS1 (R = 0.94). In the inset, a density scatter plot showing only the sense and antisense expression for the samples in the ssRNA-seq lung cell line cohort (R = 0.96). The units of the x- and y-axis are log10[norm(count)], and red dots indicate the average value for each cohort. (F) NKX2-1 and NKX2-1-AS1 expression measured by quantitative PCR across a cohort of 29 lung cell line samples. (G) Knockdown of the antisense gene NKX2-1-AS1 with two independent and specific siRNAs decreases the expression level of their cognate sense gene NKX2-1 in H441, a cell line that expresses both transcripts (see also Supplemental Fig. S8). (H) Knockdown of the antisense gene NKX2-1-AS1 with two independent siRNAs impairs proliferation of the H441 cell line. The y-axis, “Phase object confluence,” is a label-free measure of cell confluence used for IncuCyteZOOM live-cell imaging platform to assess the cell growth.
Intriguingly, these regulatory patterns are not restricted to homeotic genes because our analyses also nominated cis-NAT pairs associated with functions as diverse as cell adhesion and migration (BVES), ras guanine nucleotide-releasing factors (RASGRF2), transmembrane proteins (TMEM220, TMEM176B, TMEM176A), and transcription factors (NKX2-1, WT1, TBX5, HAND2, FOXD3), among others (Fig. 3E; Supplemental Fig. S7C,D). Among these candidates, the cis-NAT pair formed by NKX2-1 (also known as thyroid transcription factor 1, TTF-1) and NKX2-1-AS1 has one of the highest gene expression correlations in our data set (R = 0.94) (Fig. 3E). The expressions of NKX2-1 cis-NAT pair transcripts were highest in lung lineage (both adeno- and squamous carcinomas) in the combined cohort, and their positive correlation was also clearly recapitulated in our lung cell line ssRNA-seq data (Fig. 3E, inset). These results reveal additional regulatory mechanisms of NKX2-1, a master regulator essential for lung development and terminal respiratory units, with a known “lineage-survival” oncogene function in lung cancers (Yamaguchi et al. 2013).
The strong coexpression of NKX2-1 and NKX2-1-AS1 suggests a cis-NAT regulation in lung cancer; hence, we validated this result using qRT-PCR in a panel of 29 lung cell lines (Fig. 3F). To investigate the regulation of this cis-NAT gene pair expression, we designed two independent siRNAs that are highly specific to NKX2-1-AS1 (as revealed by BLAST analysis) (Supplemental Fig. S8B; Supplemental Data S9). Our results showed that knockdown of NKX2-1-AS1 decreases NKX2-1 expression level (Fig. 3G). Likewise, knockdown of NKX2-1 with specific siRNAs also reduced expression of NKX2-1-AS1 (Supplemental Fig. S8C), suggesting for the first time a reciprocal regulation of this cis-NAT pair. We next assessed the location of this cis-NAT pair RNAs in cells and noticed equal distribution between nuclear and cytoplasmic compartments, further supporting their mutual regulation (Supplemental Fig. S8D). More importantly, knockdown of NKX2-1-AS1 impaired cell proliferation of the lung cell line H441, which expresses both transcripts (Fig. 3H). Taken together, these results demonstrate the utility of our antisense compendium for uncovering potentially new aspects of tumor biology.
Patterns of antisense expression in human cancer tissues
We performed a robust characterization of global expression patterns of antisense loci in our ssRNA-seq data compendium by using tissue cohorts with 10 or more samples (Supplemental Table S1). We observed three broad groups of antisense loci expression, including ubiquitously expressed antisense loci (n = 4828), tissue-enriched/nonspecific antisense loci that were expressed in several but not all tissues (n = 7942), and lineage-specific antisense expression that was present in only one tissue type (n = 894) (Methods). Of 4828 ubiquitously expressed antisense loci, 3273 involved protein-coding genes, including 178 well-known cancer-related genes (tumor suppressors/oncogenes) such as FLI1, NF1, BAD, MDM2, KRAS, PIK3CA, and RAF1 among others (Fig. 4A). Notably, although HTH orientation is commonly observed, KRAS, PIK3CA, and RAF1 correspond to TTT cis-NAT loci, where transcription of the neighboring protein-coding gene runs into the 3′UTR and body of those oncogenes (Supplemental Fig. S9A). Similarly, 4798 of 7942 nonspecific but tissue-enriched antisense loci involved protein-coding genes, including 282 known cancer genes such as AXL, E2F2, CTNNB1, STK11, WNT1, EZH2, and CCND1 (Fig. 4A). Moreover, among the 894 lineage-specific cis-NAT loci, 44 involved tumor suppressors or oncogenes (Fig. 4B). Finally, because our compendium contains a large number of lung cancer cases, we then focused on lung cancer-specific antisense loci. We found 1772 cancer-specific antisense loci that were expressed in LUAD and LUSC but not in the six matched normal samples (Fig. 5A). Among these, 1226 were also represented in our cohort of lung cell lines, and 154 antisense loci were exclusively expressed in lung cancer samples (Fig. 5B). Of the cancer-specific antisense loci, 44 involved known cancer-related genes (Fig. 5C).
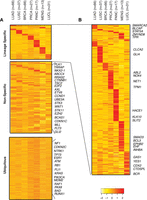
Expression of antisense loci across cancer subtypes. (A) Expression measured in the opposite strand of ubiquitous (n = 3273), tissue nonspecific (n = 4798), and lineage-specific (n = 653) antisense loci involving protein-coding genes. Only loci involving protein-coding genes are depicted in the heatmap, and the gene names displayed are therefore for the cognate genes. For each locus, the value represented in the heatmap corresponds to the average read count data across each cancer subtype. For clarity, the values have been centered and scaled by the mean and SD along the rows. Therefore, the color scale ranges from yellow (−2) to red (2), in units of SD from the mean. (B) Zoom in of lineage-specific antisense loci. Loci involving tumor suppressor or oncogenes in different cancer tissue types are labeled in the heatmap. Color scale as in A.
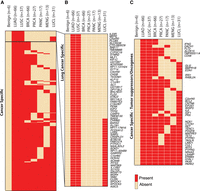
Cancer-specific antisense loci. (A) Presence/absence heatmap of significantly expressed antisense loci found in lung cancer tissues (LUAD, LUSC) but not in benign samples. Presence in other cancer tissues, breast cancer (BRCA), pancreatic cancer (PANC), prostate cancer (PRCA), meningioma (MENG), and the lung cancer cell lines (LUCL) is also shown. All loci depicted in the heatmap involve protein-coding genes, and the gene names displayed are therefore for the cognate genes. Loci present in a particular cancer subtype are indicated by red, whereas absent loci are indicated by beige. (B) Lung cancer-specific antisense loci. Presence/absence heatmap of antisense loci significantly expressed in LUAD and LUSC but not in lung normal samples (n = 1772). Of these, 1212 were also present in in the LUCL cohort. Color scale as in A. (C) Cancer-specific antisense loci involving tumor suppressors or oncogenes (n = 44). Color scale as in A.
We also investigated antisense expression dysregulations in lung cancer samples relative to normal lung counterparts. Loci-specific cis-NAT transcription could vary between tumor and normal samples in a concordant (i.e., sense and antisense are both overexpressed or underexpressed) or discordant (i.e., sense is overexpressed, whereas antisense is underexpressed or vice versa) manner (Faghihi and Wahlestedt 2009; Wahlestedt 2013). We used DESeq (Anders and Huber 2010) normalized locus’ forward and opposite strand read count data to identify cancer-specific dysregulated loci (Methods). A negative binomial test was utilized (with adjusted P-value ≤ 0.1), and a change in expression was considered significant only if the absolute log-fold change (lfc) was >1 (Methods).
A proof-of-concept analysis on three pairs of matched LUAD tumor and normal samples (Supplemental Data S10), using their average sense and antisense log-fold expression change, revealed the presence of both concordant and discordant pairs (Fig. 6A). Expanding this analysis across all LUAD tumor samples (n = 66), we observed similar results (Fig. 6B). We further verified that the average behavior (concordance/discordance) is not driven by outliers but rather uniformly observed across all individual samples (Fig. 6C,D). These analyses identified 831 concordant and 251 discordant candidates in LUAD (Supplemental Table S3), and hypoxia inducible factor 1 alpha (HIF1A) and the tubulin polymerization promoting protein (TPPP) are examples of concordant and discordant loci, respectively (Fig. 6E,F). Our data show that HIF1A and TPPP sense/antisense expression changes were consistent across both matched tumor-normal pairs (Supplemental Fig. S10A–C) and all other tumor samples (Fig. 6G,H).
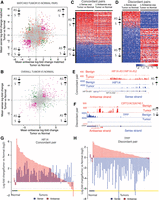
Antisense loci dysregulation in cancer. The expression of the two genes in a cis-NAT pair can both change in a concordant (same direction) or discordant (in opposite direction) manner, when comparing tumor versus normal samples (see Methods). (A) Mean sense expression of log-fold change versus mean antisense expression of log-fold change between matched pairs of lung tumor-normal samples (n = 6 samples, three pairs). Gray dots represent unchanged pairs; green dots, discordant loci; purple dots, concordant loci. A dot is colored orange if the relationship is observed in only one of the tumor-normal matched pairs. (B) Mean sense expression of log-fold change versus mean antisense expression of log-fold change between LUAD tumor (n = 66) and normal samples (n = 3). Color code as in A. (C) Heatmap of cancer-specific concordant cis-NAT pairs. The log-fold change expression between tumor (n = 66) and normal samples (n = 3) for the sense and antisense gene is displayed. Loci are represented in the rows and samples in the columns. Loci are sorted in decreasing order according to average log-fold change across all samples. The color scale ranges from blue (−2) to red (2), in units of log-fold change expression. (D) Heatmap of cancer-specific discordant cis-NAT pairs. The log-fold change expression between tumor (n = 66) and normal samples (n = 3) for the sense and antisense gene is displayed. Loci are represented in the rows and samples in the columns. Loci are sorted in decreasing order according to average log-fold change across all samples. The color scale ranges from blue (−2) to red (2) in units of log-fold change expression. (E) Coverage map for a representative example of a concordant cis-NAT pair (HIF1A). Coverage maps in benign and tumor samples in both sense and antisense strands in the HIF1A locus are presented as individual tracks above the gene schematic. Values on the left indicate track heights. (F) Coverage map for a representative example of a discordant cis-NAT pair (TPPP/CEP72). Coverage maps in benign and tumor samples in both sense and antisense strands in TPPP/CEP72 loci are presented as individual tracks above the gene schematic. Values on the left indicate track heights. (G) Bar plot of the ssRNA-seq expression log-fold change between tumor and normal samples for the concordant pair HIF1A/HIF1A-AS. Red bars indicate antisense expression and blue bars indicate sense transcript expression. (H) Bar plot of the ssRNA-seq expression log-fold change between tumor and normal samples for the discordant pair TPPP/CEP72. Red bars indicate antisense expression and blue bars indicate sense transcript expression.
Overall, there were approximately three times more concordant than discordant loci in both LUAD and LUSC (Supplemental Table S3); and we observed enrichment for HTH pairs in the concordant group, whereas EMB and TTT configurations were overrepresented in the discordant group (Fisher's exact test P-value = 1 × 10−5) (Supplemental Table S4). This is particularly interesting given the possible role for bidirectional promoters in HTH antisense loci transcription. Altogether, these data suggest that HTH cis-NAT gene pairs change in a coordinated fashion during cancer progression.
OncoNAT: a catalog of antisense loci involving cancer genes
We developed OncoNAT, the first catalog of cis-NAT pairs involving cancer-related genes to further support the increasing evidence of antisense dysregulation in cancer (Luo et al. 2006; Huarte et al. 2010; Geng et al. 2011; Kogo et al. 2011; Schmidt et al. 2011; Silva et al. 2011; Niinuma et al. 2012; Han et al. 2013; Kim et al. 2013b; Takayama et al. 2013). OncoNAT is provided as a set of tables for the use by the research community (Supplemental Data S11–S15 and https://github.com/oabalbin/OncoNAT). OncoNAT aggregates all cis-NAT pairs involving at least one known tumor suppressor or oncogene and classifies them according to their orientation into HTH, TTT, and EMB pairs. For each cis-NAT pair, the gene expression correlation between sense/antisense transcripts across our combined cohort of 376 cancer samples, CGI evidence, gene biotypes, overlapping type and length, and the NASTI-seq score are provided. We found that 51% of tumor suppressors and 46% of oncogenes produced transcripts from the opposite strand (Supplemental Table S5; Supplemental Data S16). Given that 46% of all other protein-coding genes harbor overlapping transcripts, the slight enrichment for overlapping antisense transcripts among tumor suppressors (Fisher's exact test P-value = 0.0027) might imply a greater role for antisense expression in their transcriptional regulation.
Searching OncoNAT for HTH cis-NAT pairs involving tumor suppressors or oncogenes that had evidence of bidirectional promoters, high gene expression correlation and statistically significant expression of the antisense strand (high NASTI-seq score and OPSratio) (Supplemental Data S11; Supplemental Table S5), we captured a majority of cancer-related genes with previously known antisense transcript regulation, demonstrating the utility of our catalog (Supplemental Table S6). Moreover, our approach also nominated other HTH cis-NAT pairs involving tumor suppressors and oncogenes such as CCND2, MYCN, TP73, ATM, and ETV7 among others. Similarly, searching OncoNAT for TTT cis-NAT pairs, we found oncogenes such as KRAS, PIK3CA, and RAF1, where the transcription of a neighboring protein-coding gene runs into the 3′UTR and body of those oncogenes (Supplemental Data S12; Supplemental Table S7). Furthermore, searching OncoNAT EMB cis-NAT pairs also identified HIF1A and NF1 (Supplemental Data S13). Finally, we used our ssRNA-seq data to directly examine the antisense expression of cancer-related genes that did not have annotated overlapping transcripts. We found additional examples of oncogenes and tumor suppressors with significant expression of the antisense strand, suggesting potential unannotated overlapping transcripts that may regulate genes such as PLK4, RET, CDKN2C, CDKN1C, AXL, SPRY2, and E2F2 among others (Supplemental Data S15; Supplemental Table S8; Supplemental Fig. S11).
Discussion
In this study, we used strand-specific RNA sequencing on a cohort of 376 human cancer samples to determine the global landscape of antisense expression. On average, at least 38% of all human loci exhibit consistent transcription from the opposite DNA strand, and 31% correspond to bona fide expressed cis-NAT pairs (Table 1). Our estimate suggests that antisense transcription is a widespread genomic phenomenon and updates previous assessments (Katayama et al. 2005; He et al. 2008).
Among overlapping cis-NAT pairs, candidates with HTH configuration showed the highest positive correlation (Fig. 2). This is likely due to the coordinated regulation of the associated bidirectional promoters, whose presence is further supported by the detection of CGIs (78% HTH cis-NATs have CGIs in their overlapping region) (Fig. 2 and 3). This notion is further strengthened by our detailed analyses of known examples (Figs. 2 and 3) and the experimental validation of the coexpression patterns of novel cis-NAT pairs candidates (Fig. 3).
A compendium-wide analysis of antisense expression revealed three broad antisense loci groups: ubiquitous, tissue-enriched, and lineage-specific (Fig. 4); whereas tumor versus benign comparisons identified lung cancer-specific cis-NAT loci (Fig. 5) that are dysregulated in either concordant or discordant fashion (Fig. 6). Remarkably, HTH cis-NAT pairs are enriched in the cancer-specific concordant group (Supplemental Table S4), indicating common regulatory mechanisms.
Notably, cancer-related genes populated all subgroups: For example, FLI1/SENCR (also known as FLI1-AS1) forms a ubiquitously expressed HTH cis-NAT pair, whereas KRAS, PIK3CA, and RAF1 oncogenes form ubiquitously expressed TTT cis-NAT pairs with neighboring protein-coding genes. Noteworthy examples of concordantly regulated cancer-related gene loci in LUAD are zinc finger E-box binding homeobox 2 (ZEB2) and polo-like kinase 4 (PLK4). ZEB2 and ZEB2-AS1 form a bidirectional HTH cis-NAT pair that is essential for down-regulation of E-cadherin during epithelial-mesenchymal transition. Beltran et al. (2008) elegantly showed that ZEB2 and ZEB2-AS1 are transcribed from a bidirectional promoter, and more importantly, ZEB2-AS1 up-regulates ZEB2 protein expression, which in turn down-regulates E-cadherin expression. PLK4 is essential for centriole duplication and when overexpressed, induces centrosome aberrations during tumorigenesis. Notably, the antisense transcript overlapping PLK4 has not yet been annotated, but according to our ssRNA-seq data, it displays a HTH configuration overlapping the 5′ region of PLK4 (Supplemental Fig. S11D).
Several mechanisms have been proposed for concordant regulation of sense/antisense transcripts, including stabilization of the sense transcript by its antisense counterpart. For example, in the case of beta-site APP-cleaving enzyme 1 (BACE1), the cis-NAT pair forms a transient RNA duplex with secondary or tertiary structures contributing to BACE1 mRNA stability (Faghihi et al. 2010). Similarly, HAS2-AS1 and HAS2 are simultaneously transcribed in renal proximal tubular epithelial cells in kidney fibrosis, and transcription of the antisense RNA stabilizes or augments HAS2 mRNA expression in these cells via RNA/mRNA heteroduplex formation (Michael et al. 2011). In this study, we showed that NKX2-1-AS1/NKX2-1 concordantly regulates one another's expression, and even more importantly, NKX2-1-AS1 knockdown impaired cell proliferation. Both transcripts were also equally distributed between nuclear and cytoplasmic compartments (Supplemental Fig. S8D), suggesting a coregulation, possibly through modulation of the RNA stability. The exact mechanism by which NKX2-1-AS1 regulates its cognate gene remains to be uncovered.
Finally, we created OncoNAT, a catalog of cancer-related genes with significant antisense expression (Supplemental Data S11–S15 and https://github.com/oabalbin/OncoNAT). Our catalog provides a comprehensive examination of the expression patterns of known and new cis-NAT pairs and will assist in nominating promising candidates (e.g., NKX2-1/NKX2-1-AS1, HOXD1/HAGLR) that merit further investigation in cancer. OncoNAT raises the exciting possibility of novel regulatory mechanisms for well-known oncogenes and tumor suppressors; elucidating the biological implications of these very different expression patterns will deepen our understanding of antisense regulation and their role in cancer.
Overall, our study contributes to a growing body of literature supporting a role for NAT regulation of well-studied cancer genes and the increasing evidence of antisense dysregulation in cancer (Luo et al. 2006; Huarte et al. 2010; Geng et al. 2011; Kogo et al. 2011; Schmidt et al. 2011; Silva et al. 2011; Niinuma et al. 2012; Han et al. 2013; Kim et al. 2013b). The proposed molecular mechanisms of this regulation are multiple and poorly understood. Nevertheless, utilization of natural antisense transcripts to modify the expression of their cognate sense genes could be a promising technology (Modarresi et al. 2012) for gene-specific precision therapies.
Taken together, this study characterizes the landscape of antisense expression in human cancers and provides a resource (OncoNAT) that will enable researchers to elucidate the mechanisms of sense/antisense regulation in cancer.
Methods
Biorepository description
The Michigan Center for Translational Pathology (MCTP) strand-specific RNA-seq repository included in this study has 376 samples. Most of the samples correspond to cancer tissues, and the largest tissue cohorts are breast, lung adenocarcinoma, lung squamous carcinoma, prostate cancer, ovarian cancer, pancreatic cancer, meningioma, rare cancers, and lung cell lines. A breakdown of major and minor cohorts included in this study is presented in Supplemental Table S1.
Preparation of strand-specific RNA-seq libraries
Transcriptome libraries were prepared following a modified protocol previously described for generating strand-specific RNA-seq-libraries (see Supplemental Information) (Yassour et al. 2010).
Bioinformatics workflow for antisense expression analysis
First, sequencing reads were mapped to the human genome (hg19, GRCh37) using TopHat 2 (TopHat/2.0.4) (Kim et al. 2013a). Then, a summarized transcriptome was built by reconstructing the longest annotation for each gene, using transcript and exon information provided in the Ensembl v69 assembly (http://www.ensembl.org) (Flicek et al. 2013). Only high quality transcript isoforms were included, while problematic and misannotated transcripts were filtered out (see Supplemental Information). This procedure generated 42,124 gene models. Second, these gene models were used as reference loci to compute the number of strand-specific pair-end reads mapping to the forward or opposite strand of each locus and then to calculate the expression level of each strand in that locus (see Supplemental Information). Loci expression was then normalized using DESeq (Anders and Huber 2010). Third, strand specificity was calculated for each library in order to determine the protocol error (pei) or background noise affecting our estimation of the expression coming from the opposite strand (see Supplemental Information). Fourth, loci consistently expressing both forward and opposite strands across our cohort were identified. Moreover, a locus that has OPSratio > pei_th in at least 30% of the cohort samples (see Supplemental Information) was considered as a locus with measurable antisense expression. Fifth, a probabilistic method, NASTI-seq, was used for refining natural antisense transcripts identification (Li et al. 2013). This method accounts for the variable protocol error in order to identify loci with significant antisense expression (see Supplemental Information).
Finally, we calculated the correlation between sense and antisense transcripts forming cis-NAT pairs and determined tissue-specific, tissue-enriched/nonspecific, ubiquitous and cancer-specific antisense loci. This bioinformatics pipeline nominates expressed antisense loci across different cancer subtypes and establishes their pattern of expression. The pipeline aggregates tumor suppressor and oncogenes with significant antisense expression into a single catalog, OncoNAT (Supplemental Data S4–S6, S11–S15 and https://github.com/oabalbin/OncoNAT).
Gene expression by quantitative PCR
H441 and H1793 cells were seeded onto six-well plates and were transfected with either nontargeting siRNA or siRNAs targeting NKX-2-1-AS1 and HOXD1-AS1 (HAGLR), respectively. Seventy-two hours post transfection, total RNA was isolated using TRIzol (Invitrogen) and an RNeasy kit (Qiagen) according to the manufacturer's instructions. Total RNA was reverse transcribed into cDNA using SuperScript III (Invitrogen) and random primers (Invitrogen). qPCR was performed using SYBR Green Master Mix (Applied Biosystems) on an Applied Biosystems 7900HT Real-Time System. The relative quantity of the target gene was computed for each sample using the ΔΔCt method by comparing mean Ct of the gene to the mean Ct of the housekeeping gene, glyceraldehyde-3-phospate dehydrogenase (GAPDH). All the primers were obtained from Integrated DNA Technologies (IDT). Sequences of all the siRNAs and primers used are listed in Supplemental Tables S9, S10.
Cell proliferation assays
H441 cells were seeded onto six-well plates and were transfected with two independent siRNAs targeting NKX-2-1-AS1. Seventy-two hours post transfection, cells were plated on 24-well plates in triplicate, and cell proliferation was monitored using IncuCyte live-cell imaging system (http://www.essenbioscience.com). Sequences of all the siRNAs used are listed in Supplemental Table S10.
Data access
The raw sequencing data from this study have been submitted to the NCBI Sequence Read Archive (SRA; http://www.ncbi.nlm.nih.gov/sra) and dbGaP (http://www.ncbi.nlm.nih.gov/gap) under accession numbers SRP048484 and phs000937.v1.p1. The raw count data for the sense and antisense gene expression for all samples in the cohort have been submitted to the NCBI Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE66729. The source code for the analyses presented in this study and the OncoNAT catalog are available in the Supplemental Material and at https://github.com/oabalbin/OncoNAT, and https://github.com/oabalbin/OncoNAT/tree/master/onconat, respectively.
Acknowledgments
We thank K. Giles, D. Banka, J. Athanikar, and Christine Betts for administrative support; T. Barrette for IT support; Shruthi Subramanian for technical assistance; and B. Veeneman and D. Mellacheruvu for useful scientific discussion about the manuscript. O.A.B. dedicates this work to his family. This work was supported in part by US National Institutes of Health Prostate Specialized Program of Research Excellence grant P50CA69568 and Early Detection Research Network grant UO1 CA111275. A.M.C. is supported by the Prostate Cancer Foundation and the Howard Hughes Medical Institute. A.M.C. is an American Cancer Society Research Professor and a Taubman Scholar of the University of Michigan. A.I.N. is supported by US National Institutes of Health grant R01-GM-094231. O.A.B. is supported by the F31 National Institutes of Health Ruth L. Kirschstein National Research Service Award for Individual Predoctoral Fellowships to Promote Diversity in Health-Related Research (F31-CA-165866) and by T32 Proteome Informatics of Cancer Training Program at the University of Michigan (T32-CA-140044). R.M. is supported by a US Department of Defense Post-doctoral award (W81XWH-13-1-0284) and a Prostate Cancer Foundation Young Investigator Award. J.R.P. is supported by a Prostate Cancer Foundation Young Investigator award. D.G.B. and A.M.C. are supported by US National Institutes of Health grant RO1 CA154365.
Author contributions: O.A.B., A.I.N., and A.M.C. conceived and design the study. O.A.B. performed data assembly, sense/antisense bioinformatics analysis pipeline, ssRNA-seq data processing, data integration and statistical analysis, and designed and generated all figures. R.M. and J.R.P. conducted experimental validation of sense/antisense gene pairs and functional characterization. S.M.D., D.R., and Y.-M.W. prepared ssRNA-seq libraries that were sequenced by X.C. and R.W. D.G.B. and G.C. provided RNA for the LUAD and LUSC samples. A.I.N. oversaw data analysis and methods development. A.M.C. provided overall scientific oversight. A.O.B., R.M., S.M.D., J.R.P., A.I.N., and A.M.C. wrote the manuscript, which was reviewed by all authors.
Footnotes
-
↵8 These authors share senior authorship.
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.180596.114.
- Received June 27, 2014.
- Accepted May 11, 2015.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see http://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








