
Genetic and phenotypic intra-species variation in Candida albicans
- Matthew P. Hirakawa1,
- Diego A. Martinez2,5,
- Sharadha Sakthikumar2,5,
- Matthew Z. Anderson1,3,
- Aaron Berlin2,
- Sharvari Gujja2,
- Qiandong Zeng2,
- Ethan Zisson1,
- Joshua M. Wang1,
- Joshua M. Greenberg1,
- Judith Berman3,4,
- Richard J. Bennett1,5 and
- Christina A. Cuomo2,5
- 1Department of Molecular Microbiology and Immunology, Brown University, Providence, Rhode Island 02912, USA;
- 2Broad Institute of MIT and Harvard, Cambridge, Massachusetts 02142, USA;
- 3Department of Molecular, Cellular Biology and Genetics, University of Minnesota, Minneapolis, Minnesota 55455, USA;
- 4Department of Molecular Microbiology and Biotechnology, Tel Aviv University, Ramat Aviv 69978, Israel
- Corresponding authors: richard_bennett{at}brown.edu, cuomo{at}broadinstitute.org
-
↵5 These authors contributed equally to this work.
Abstract
Candida albicans is a commensal fungus of the human gastrointestinal tract and a prevalent opportunistic pathogen. To examine diversity within this species, extensive genomic and phenotypic analyses were performed on 21 clinical C. albicans isolates. Genomic variation was evident in the form of polymorphisms, copy number variations, chromosomal inversions, subtelomeric hypervariation, loss of heterozygosity (LOH), and whole or partial chromosome aneuploidies. All 21 strains were diploid, although karyotypic changes were present in eight of the 21 isolates, with multiple strains being trisomic for Chromosome 4 or Chromosome 7. Aneuploid strains exhibited a general fitness defect relative to euploid strains when grown under replete conditions. All strains were also heterozygous, yet multiple, distinct LOH tracts were present in each isolate. Higher overall levels of genome heterozygosity correlated with faster growth rates, consistent with increased overall fitness. Genes with the highest rates of amino acid substitutions included many cell wall proteins, implicating fast evolving changes in cell adhesion and host interactions. One clinical isolate, P94015, presented several striking properties including a novel cellular phenotype, an inability to filament, drug resistance, and decreased virulence. Several of these properties were shown to be due to a homozygous nonsense mutation in the EFG1 gene. Furthermore, loss of EFG1 function resulted in increased fitness of P94015 in a commensal model of infection. Our analysis therefore reveals intra-species genetic and phenotypic differences in C. albicans and delineates a natural mutation that alters the balance between commensalism and pathogenicity.
Candida albicans is an important human fungal pathogen, responsible for both debilitating mucosal infections and life-threatening systemic infections (Wey et al. 1988; Gudlaugsson et al. 2003; Pfaller and Diekema 2007). In addition to being an opportunistic pathogen, C. albicans is a frequent and benign component of the human gastrointestinal microbiota (Kleinegger et al. 1996). Many attributes have been linked to the ability of C. albicans to successfully colonize and infect the human host, including its ability to rapidly switch between different cell states in response to environmental cues (Slutsky et al. 1987; Lo et al. 1997; Kumamoto and Vinces 2005; Phan et al. 2007; Zhu and Filler 2009).
The majority (97%) of C. albicans isolates are diploid strains assigned to one of 17 clades, as defined by multilocus sequence typing (MLST). These clades are enriched for different geographical areas, with the five largest clades accounting for three-quarters of clinical isolates (Bougnoux et al. 2004; Tavanti et al. 2005; Odds et al. 2007). The population structure is largely clonal, although recombination events have led to isolates with mixed evolutionary histories, consistent with a sexually or parasexually reproducing species (Tibayrenc 1997; Odds et al. 2007). C. albicans mating has been shown to occur between MTLa and MTLα diploids, as well as within unisexual populations (Hull and Johnson 1999; Hull et al. 2000; Magee and Magee 2000; Alby et al. 2009). Mating is regulated by a phenotypic switch, in which cells must transition from the default “white” state to the mating-competent “opaque” state to undergo efficient conjugation (Miller and Johnson 2002). Despite efficient mating, a conventional meiosis has not been observed in C. albicans; instead, cells shift from the tetraploid state to the diploid state via a program of random chromosome loss (Bennett and Johnson 2003; Forche et al. 2008; Berman and Hadany 2012). A viable haploid state was also recently uncovered, although C. albicans haploids are unstable, rapidly reverting to diploid, and also exhibit decreased fitness relative to diploid forms (Hickman et al. 2013).
Natural isolates of C. albicans exhibit a broad spectrum of phenotypic properties. For example, clinical isolates display striking differences in virulence when compared in the murine model of systemic infection (Wu et al. 2007; MacCallum et al. 2009). Clinical isolates also show differences in their ability to form biofilms (Li et al. 2003); these are cellular communities that promote infection arising from implanted medical devices (Nobile and Mitchell 2006; Finkel and Mitchell 2011). Phenotypic variation is further evident in the form of morphological plasticity, such as the white-opaque switch and the yeast to hyphal transition, which involve developmental shifts between distinct cellular states (Slutsky et al. 1985, 1987; Soll 1993; Lohse and Johnson 2009; Sudbery 2011).
Genomic variation is also well established for C. albicans and includes large-scale chromosomal rearrangements such as translocations, chromosome truncations, and aneuploidy (Rustchenko 2007; Selmecki et al. 2010; Bennett et al. 2014). A number of studies have demonstrated that karyotypic differences exist between clinical isolates, including evidence for both monosomic and trisomic chromosomes (Rustchenko-Bulgac et al. 1990; Rustchenko-Bulgac 1991; Barton and Gull 1992; Perepnikhatka et al. 1999; Chibana et al. 2000; Chen et al. 2004). In one example, the presence of an extra isochromosome composed of the two left arms of Chromosome 5 mediates fluconazole resistance in a subset of clinical strains (Selmecki et al. 2006, 2008). C. albicans strains also exhibit genetic diversity due to variable tracts of loss of heterozygosity (LOH). Most C. albicans strains are considered to be heterozygous diploids, and yet different strains often contain different LOH tracts, some of which can provide phenotypic advantages. For example, homozygosis of hyperactive alleles of ERG11, TAC1, and MRR1 (encoding the target of azole drugs or transcription factors regulating drug efflux pumps) can result in strains with increased azole resistance (White 1997; Coste et al. 2006; Dunkel et al. 2008). Both LOH and karyotypic changes are induced in response to environmental stimuli including drug treatment, heat shock, nutrient utilization, and oxidative stress (Rustchenko et al. 1997; Janbon et al. 1998; Forche et al. 2011; Harrison et al. 2014).
Sequencing of C. albicans isolates has provided additional insights into the genome structure of C. albicans. Two isolates have been sequenced to date: SC5314, the standard laboratory strain of C. albicans; and WO-1, in which white-opaque switching was first observed (Jones et al. 2004; van het Hoog et al. 2007; Butler et al. 2009). The genomes of these strains consisted of eight diploid chromosomes, although WO-1 had lost one >300-kb region on Chromosome 3. Both SC5314 and WO-1 contained ∼6100 genes and were generally heterozygous, although extended LOH regions were observed in both isolates (Butler et al. 2009). Recent phasing of the single nucleotide polymorphisms (SNPs) in SC5314 included nearly all of the 69,668 SNPs identified in this strain (Muzzey et al. 2013).
Here, we sequenced the genomes of 21 clinical isolates of C. albicans selected to represent different clades, different sites of anatomical origin, and differences in virulence (Wu et al. 2007). We find that genomic variation in C. albicans occurs at the level of SNPs, inversions, copy number variation, loss of heterozygosity (LOH), and aneuploidy. Extensive phenotypic analysis was also performed on the 21 isolates to characterize properties associated with pathogenesis. We discuss our findings with respect to intra-species variation in C. albicans and reveal how a natural isolate has evolved a mutation that promotes the commensal lifestyle at the expense of a more pathogenic one.
Results
Sequencing of 21 clinical isolates of C. albicans
We selected 21 C. albicans strains for phenotypic and genotypic characterization (Supplemental Table S1). This is a geographically diverse set of strains that originated from bloodstream infections (14 isolates) or superficial infections of oral or vaginal tissues (seven isolates) and includes the previously sequenced laboratory strain SC5314 for comparison (Jones et al. 2004). With the exception of SC5314, the virulence of each clinical isolate was previously compared using a murine model of disseminated candidiasis (Supplemental Table S1; Wu et al. 2007).
The set of 21 strains was typed by fingerprint analysis to clades I, II, III, SA, and E of C. albicans (Lockhart et al. 1996; Blignaut et al. 2002; Pujol et al. 2002). Using variants identified from whole-genome sequence data (Supplemental Table S2; Methods), we evaluated the phylogenetic relationship of these 21 strains (Fig. 1). The major groups corresponded to the previously described fingerprint-based clades, with the exception of P94015, which is distantly positioned relative to other clade I strains. MLST analysis was carried out and compared to a previously typed population (Tavanti et al. 2005). The sequenced isolates represent seven of the 17 previously identified clades, including representatives of the five largest clades (1, 2, 3, 4, and 11), as well as two less common clades (6 and 8) (Supplemental Table S3; Supplemental Fig. S1; Odds et al. 2007).
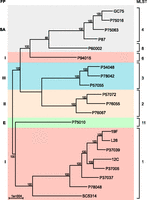
Phylogenetic relationship and clade assignment of studied isolates. The phylogenetic relationship of the strains was inferred based on 112,223 informative SNP positions using maximum parsimony in PAUP*; node labels indicate the support frequency of 1000 bootstrap replicates. Clades were assigned based on multilocus sequence type (MLST) analysis of seven loci or prior work reporting fingerprint clades (FP).
Analysis of the genome-wide variation for these C. albicans isolates revealed they contain, on average, a heterozygous SNP every 267 bases. Furthermore, when compared to SC5314, other clade I strains displayed a homozygous variant every 1404 bases, whereas non-clade 1 isolates contained a homozygous variant every 235 bases. Using these variants, the average genome-wide nucleotide diversity between any two C. albicans isolates is estimated at 0.37%. In contrast, the average diversity at neutrally evolving fourfold degenerate codon positions is 1.78%; the 4.8-fold higher rate at fourfold degenerate sites suggests that many sites in the genome are under selective constraint. Overall, the gene content of C. albicans isolates was highly conserved across the different isolates and clades. An average of 6069 (98.1%) of the 6189 genes in SC5314 were present in each of the assemblies of the other isolates (Supplemental Table S4).
Phenotypic analysis of C. albicans isolates
Phenotypic analyses were performed on the 21 clinical isolates, revealing marked differences in virulence, growth rates, filamentation, biofilm formation, stress resistance, and resistance to antifungal drugs. To determine the virulence of these strains, isolates were tested in the Galleria mellonella insect model of infection (Fuchs et al. 2010b; Jacobsen 2014) and compared to that in the murine model of systemic infection (Wu et al. 2007). Isolates showed widely differing virulence levels, with some strains causing death of the larvae within 48 h, whereas others caused only limited mortality even after 7 d infection (Supplemental Fig. S2). Virulence data in insects showed a significant correlation with that in the murine infection model (P < 0.04, Spearman’s rs = 0.46) (Supplemental Table S5).
To compare in vitro fitness, doubling times were measured in different culture media. Doubling times of clinical isolates ranged from ∼70 to ∼170 min (Fig. 2A; Supplemental Table S6). Relative growth rates were similar across the conditions tested, with P34048 being the fastest growing and P76067 and P37037 among the slowest growing.
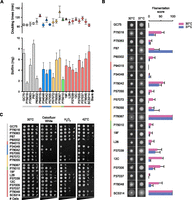
Phenotypic profiling of C. albicans isolates. (A) Doubling times and biofilm formation. Doubling times were measured in liquid YPD medium at 30°C. Circles represent euploid isolates and triangles represent aneuploid isolates. Biofilm dryweight biomasses ± standard error. Colors represent clade designation as follows: (SA) gray; (I) red; (II) yellow; (III) blue; (E) green. (B) Filamentation of clinical isolates grown on Spider medium for 7 d at 30°C or 37°C. Bar graphs represent filamentation scores ± standard error for strains at 30°C (pink) or 37°C (blue). (C) Assays of clinical isolates grown in the presence of stressors. Tenfold dilutions of cells were plated under regular growth conditions (SCD, 30°C), cell wall stress (SCD + 100 µg/mL calcofluor white, 30°C), oxidative stress (SCD + 2 mM hydrogen peroxide, 30°C), and thermal stress (SCD medium, 42°C).
Clinical isolates varied widely in their ability to undergo filamentous growth. Filamentation on solid media was scored using a modified morphology score (M score) (Noble et al. 2010). On Spider medium, the standard laboratory strain SC5314 showed the highest filamentation score (100), whereas P94015 had the lowest score (0) (Fig. 2B). Filamentation in liquid RPMI at 37°C also revealed variable filamentous phenotypes, with SC5314 being one of the most filamentous (Supplemental Fig. S3). Despite being associated with pathogenesis, we detected no significant correlation between filamentation scores and virulence under any of the tested conditions (Supplemental Table S5).
Biofilm formation was evaluated by measuring the total biomass accumulated on silicone elastomer, a common component of medical devices (Nobile et al. 2012). SC5314 scored as one of the most efficient biofilm producers (7.5 mg), whereas P94015 formed the weakest biofilms (0.61 mg) (Fig. 2A). There were no correlations between biofilm formation and clade designation or between biofilm formation and virulence. However, significant positive correlations were observed between biofilm efficiency and filamentation in both liquid RPMI medium (P < 0.011, Spearman’s rs = 0.54) and on solid Spider medium at 37°C (P < 0.05, Spearman’s rs = 0.43) (Supplemental Table S5). This is consistent with filamentation being essential for efficient biofilm formation in C. albicans (Baillie and Douglas 1999; Nobile and Mitchell 2006).
We also compared resistance to different in vitro stressors, including calcofluor white (CFW), which interferes with chitin in the cell wall (Ram and Klis 2006), as well as oxidative stress (Fig. 2C). We observed several clade-specific trends, including that strains from clades II and III were the most sensitive to CFW (with the exception of P78042), whereas strains from clade SA were all resistant to CFW. Many of the strains that were sensitive to CFW also were sensitive to oxidative stress. Thus, clade III isolates were the most sensitive to hydrogen peroxide, whereas clade SA isolates again were the most resistant to this stress (Fig. 2C). However, there was no overall correlation between a strain’s susceptibility to CFW stress, oxidative stress or heat shock stress, and its virulence in the host (Supplemental Table S5).
Copy number and structural variation in C. albicans isolates
Comparing sequencing read depth across chromosomes identified genomic regions showing copy number variation. We identified regions showing statistically significant coverage variation and focused on genes present in amplified regions of at least two strains (Supplemental Table S11). The genes with the most variable copy number were ORFs encoding retroelements. Zorro elements are retrotransposons that lack long terminal repeats (Goodwin et al. 2001), and Zorro2 and Zorro3 ORFs were amplified at least twofold in several strains relative to SC5314. Tca2 elements, representing Ty1/copia-type retrotransposons (Holton et al. 2001), were present at sixfold higher copy number in several strains from each clade except clade II. We also detected a highly elevated copy number (35× above diploid) of the TAR1 gene present in the rDNA array. The genome assembly of SC5314 only contains one copy of the ribosomal DNA locus (5S-18S-5.8-25S), and our data therefore suggest this locus is, on average, an array of 35 copies. Aside from these regions, the most commonly encountered gene of variable copy number was CYP5, encoding a cytoplasmic cyclophilin; this gene was duplicated in 15 strains, providing an example of a widely occurring amplification not found in the reference strain.
Targeted Sanger sequencing of the subtelomeric regions showed that these regions exhibited hypervariation between isolates. The telomere-associated (TLO) gene family resides at subtelomeric regions, and this family is greatly expanded in C. albicans relative to other Candida species (Butler et al. 2009). Analysis of TLO genes revealed that both the number and position of TLO genes varied widely, with the total number of TLOs varying between 10 and 15 between isolates. This indicates significant plasticity in this gene family with potential consequences for fitness of the organism (see Supplemental Material; Supplemental Fig. S4; Supplemental Table S12).
We also assessed the genomes of the 21 isolates for inversions and translocations. Each assembly was aligned to SC5314 to identify candidate inversions and read support for the breakpoints examined (see Methods). We found that 18 of the 21 isolates have local sequence inversions. Each isolate contained between one and eight total inversions ranging in size from 705 bases to 144 kb; many of the inverted regions were identified across multiple isolates (Supplemental Table S13). We did not find evidence for chromosomal translocations supported by both the assemblies and read mapping analyses.
Substitution rates and evolutionary selection in clinical isolates
To examine the regional variation in evolutionary rates, we plotted the rates of nucleotide substitution in coding regions across the C. albicans genome. One region in particular, the subtelomeric region on the right arm of Chromosome R, exhibited a uniformly high omega value, though also a lower dS value (Supplemental Fig. S5). Notably, this chromosomal region is also the only region in the genome that is homozygous in all 21 strains; genome-wide, dS positively correlated with heterozygosity (P < 1 × 10−7, R = 0.19, Pearson’s correlation). Across the genome, we observed high variability in dS (Supplemental Fig. S5), and although dS significantly correlated with omega values (P < 2 × 10−16, R = −0.34), dN did not show a clear correlation (R = 0.03). This suggests that regional variation in dS, but not dN, is a major driver of differences in omega values.
To identify the most rapidly changing proteins, we focused on the top 5% of dN values, as the local variation in dS is likely to skew omega values. Estimates of dN provide a view of protein divergence, particularly in cases of extremely low or high dS (Luo and Hughes 2012), and can correlate well with omega values even when dS measurements are accurate (Drosophila 12 Genomes Consortium 2007). This set of 299 genes (dN > 0.087) included a number of genes encoding cell wall proteins, such as HWP1, IFF5, IHD1, MP65, PGA41, PGA44, PGA46, PGA49, PGA60, and PGA61 (Supplemental Table S14). Several of these encode predicted GPI (glucosylphosphatidylinositol)-anchored proteins implicated in cell wall function, adhesion, or host interactions (Richard and Plaine 2007). Genes producing cell wall modifying activities also displayed high dN values, such as the chitinase CHT1 gene as well as the mannosyltransferase MNT1 gene. This presumably underscores the importance of the cell wall in interactions with the mammalian host. Other genes with high dN values include those implicated in filamentous growth (FGR18 and FGR43), a gene encoding an oligopeptide transporter (OPT9), and the peptidase-encoding genes SAP5 and PEP3. Genes with the highest dN included many with unknown function (173 with no GO function), which prevented the detection of GO term enrichment overall.
To characterize variants resulting in loss-of-function mutations, we identified homozygous nonsense mutations as well as insertions and deletions (indels) predicted to prematurely terminate protein-coding genes. Across the 21 genomes, nonsense mutations truncated 175 genes and indels resulted in frameshift mutations that disrupted 391 genes, with many of these mutations shared by multiple strains (Supplemental Tables S15, S16). Interestingly, nonsense mutations and indels targeted the same genes in many cases (Supplemental Fig. S6) (P < 2 × 10−16, χ2), suggesting strong evolutionary pressure on these loci. For isolate P94015, we scanned the loss-of-function mutations to identify candidates that might be responsible for the extreme phenotypes exhibited by this strain and explored in more detail below.
Multiple aneuploidies are present in C. albicans isolates
By examining sequence read depth across the genome, large-scale copy number variations were identified in more than one-third of isolates (8 of 21 strains) (Fig. 3; Supplemental Figs. S7, S8). Whole chromosome gains were commonly observed, with five strains containing one trisomic chromosome and one strain, P60002, containing two trisomic chromosomes. Whole chromosome gains were observed for each of the four smallest chromosomes, 4–7, with extra copies of Chr 4 and 7 present in multiple strains (Fig. 3; Supplemental Fig. S8). In addition, segmental regions of two chromosomes were found at trisomic and tetrasomic levels. In P60002, an 805-kb region on Chr R spanning from 263 kb to the left of CENR up to 70 kb to the right of the rDNA repeat was trisomic for a 487-kb region and the terminal 318-kb region distal to the rDNA was tetrasomic (Fig. 3; Supplemental Fig. S8). For P94015, a 340-kb internal region of Chr 1 was tetrasomic, with an adjacent 74-kb region being trisomic. One strain, P76067, was also hemizygous (only one copy of one allele present) for the final 200 kb on the right arm of Chr 3; interestingly, a larger hemizygous region encompassing 433 kb of this chromosome was previously noted in strain WO-1 (Butler et al. 2009). Because P76067 is a clade II strain, whereas WO-1 maps to a different clade (Odds et al. 2007), this loss is best explained by independent losses of these subtelomeric regions.
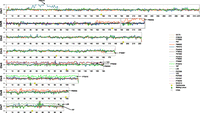
Aneuploid regions of clinical isolates. For each sequenced isolate, normalized read depth in 1-kb windows is shown along each chromosome relative to the SC5314 reference. Chromosome position is shown for every 100 kb along the x-axis; average normalized depth on the y-axis is relative to diploid levels. Regions exhibiting partial or full chromosome aneuploidies are noted with the corresponding isolate name. See also Supplemental Figure S8 for chromosome copy number in each strain.
As a group, aneuploid strains (with partial, whole, or multiple aneuploidy regions) exhibited significantly slower growth than euploid strains in several growth media (SCD, Spider, and YPD media at 30°C; P < 0.05, Student’s t-test) (Supplemental Table S5). These results are consistent with aneuploidy being associated with a general fitness defect in yeast species when grown in replete media (Torres et al. 2007; Forche et al. 2008; Pavelka et al. 2010; Siegel and Amon 2012). In contrast, aneuploidy did not correlate with any other strain attributes including virulence (Supplemental Table S5).
Whole-genome heterozygosity and LOH events
Previous sequencing of isolates SC5314 and WO-1 revealed high levels of heterozygosity, with 86% and 70% of the genome, respectively, containing SNPs (Jones et al. 2004; Butler et al. 2009). Genome heterozygosity in the 21 isolates analyzed here ranged from 48% in P94015% to 89% in P34048, with LOH of whole chromosomes observed for Chr 3, 5, 6, 7, and R (Fig. 4). The transition points between homozygous and heterozygous regions along each chromosome differed between strains; however, the centromeres were excluded from having undergone LOH except for cases of whole chromosome LOH. As discussed earlier, only one region, extending from the rDNA locus to the end of the right arm of Chr R, was homozygous in all strains. Three regions, encompassing the centromeres of Chr 1, 2, and 4, were heterozygous across all strains (Fig. 4). Overall genome heterozygosity correlated with faster growth rates in YPD medium at 30°C (P < 0.027, Spearman’s rs = 0.48) (Supplemental Table S5). Genome heterozygosity did not, however, correlate with increased virulence in either the murine or insect model of infection (Supplemental Table S5).

Loss of heterozygosity in sequenced isolates. For each isolate, the frequency of SNPs, including both heterozygous and homozygous variants relative to SC5314, is plotted for 5-kb windows across the genome (gray bars). Homozygous regions are shaded blue; this includes regions that are homozygous and shared with the reference SC5314 and regions containing a different homozygous haplotype. Additional features shown on the chromosome plot (bottom profile) include centromeres (green circles), major repeat sequences (blue rectangles), and the MTL locus on Chromosome 5 (orange rectangle).
We more closely examined heterozygosity on Chr 5 that contains the mating type-like (MTL) locus. More than 90% of clinical isolates have been shown to be heterozygous at the MTL locus, containing both MTLa and MTLα idiomorphs (Lockhart et al. 2002; Legrand et al. 2004; Odds et al. 2007). We found that all a/α isolates were heterozygous surrounding the MTL locus as expected, but were often homozygous for other regions of Chr 5. For example, five strains showed LOH on the right arm of Chr 5 that extended to the end of the chromosome (Fig. 5A). Similarly, four a/α strains showed LOH on the left arm of Chr 5 that extended to the telomere. It is likely that these LOH events are due to break-induced replication or single crossover recombination events. Overall, a/α strains were heterozygous for 37%–100% of SNPs on Chromosome 5 (Fig. 5B).
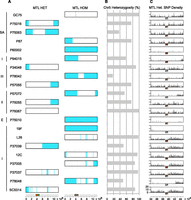
Pattern of heterozygosity on Chromosome 5 containing the MTL locus. (A) Isolates were typed as either MTL heterozygous (left column) or MTL homozygous (right column). Homozygous regions (shaded blue) of Chromosome 5 were identified from SNP data. The lower axis indicates the position of the MTL locus (orange rectangle) and centromere (green circle) on Chromosome 5, with scale shown in bases. (B) Percentage of Chromosome 5 that is heterozygous in each isolate (fraction of 5-kb windows typed as heterozygous; see Methods). (C) Pattern of SNP heterozygosity immediately flanking the MTL locus. Note that SNPs within the MTL loci were not mapped due to the high sequence divergence of the two loci. MTLa loci are marked by orange triangles and MTLα loci are marked by white triangles. Axis indicates position on Chromosome 5, with scale shown in bases.
Strains that were MTL homozygous also exhibited a wide range of Chr 5 configurations. In three isolates, the entire Chr 5 was homozygous; whereas in nine isolates, partial LOH events had occurred (Fig. 5A). For example, the entire left arm of Chr 5 was homozygous in four isolates. In contrast, the regions flanking the MTL locus were heterozygous in five isolates with only a small tract at the MTL locus itself being homozygous. In these strains, the length of the LOH tract varied from 11 kb to 20 kb (Fig. 5C). Given that the MTL locus is ∼9 kb, these results demonstrate how recombination events close to the MTL can drive homozygosity of this locus.
We also examined whether overall heterozygosity of Chr 5 correlated with virulence, as previously proposed (Wu et al. 2007). Chromosome 5 heterozygosity varied from 0% (19F) to 100% (GC75), but these levels did not show a significant correlation with either growth rates or virulence (Supplemental Table S5). Thus, unlike genome-wide heterozygosity, heterozygosity on Chr 5 does not appear to affect fitness either in vitro or in vivo.
Characterization of avirulent isolate P94015
Isolate P94015 was chosen for further investigation, as this strain was nonfilamentous (Fig. 2; Supplemental Fig. S3), resistant to azole drugs (Supplemental Material), exhibited the lowest virulence of the clinical isolates (Wu et al. 2007), and exhibited a phenotypic state distinct from conventional “white” or “opaque” cells. C. albicans white cells are round and smooth and form dome-shaped colonies, while cells in the opaque state are elongated and pimpled and form flatter, darker colonies (Slutsky et al. 1987). The cellular phenotype of P94015 included a mixture of round white-like cells and elongated opaque-like cells, as well as cells intermediate in shape to both states (Supplemental Fig. S9A). Fluorescent microscopy and cell cytometric analysis of cells expressing pOP4-GFP or pWH11-GFP reporters established that P94015 cells exhibited differential expression of these reporters to those in control white or opaque cells (Supplemental Fig. S9B,C).
Genome-wide transcriptional profiling of P94015 was also performed, and compared to the profiles of white and opaque cells, as well as those of “GUT” and “gray” cells. The latter two phenotypes have been shown to be related to, but distinct from, those of white and opaque cells (Pande et al. 2013; Tao et al. 2014). Comparative analysis revealed that gene expression in P94015 showed a significant correlation with that in opaque cells, GUT cells, and gray cells relative to white cells, but that the P94015 profile was also distinct from each of these three cell types (see Supplemental Material; Supplemental Fig. S10).
Genome analysis of P94015 revealed that the genes regulating white-opaque switching were intact with the exception of EFG1, which contained a homozygous nonsense mutation at amino acid 80 (full-length Efg1 is 525 amino acids). None of the other clinical isolates contained a premature stop mutation in EFG1. In addition to controlling white-opaque switching (Sonneborn et al. 1999; Srikantha et al. 2000; Zordan et al. 2007), EFG1 is a key regulator of filamentous growth and virulence (Lo et al. 1997; Stoldt et al. 1997; Braun and Johnson 2000; Korting et al. 2003; Noffz et al. 2008). To test whether the truncated EFG1 gene product in P94015 is functional, we introduced the nonsense mutation into the full-length SC5314 EFG1 allele, generating efg1STOP. The efg1STOP allele did not restore filamentation when introduced into an SC5314 efg1Δ/efg1Δ strain, whereas reintegration of intact EFG1 successfully restored filamentation (Supplemental Fig. S11). Furthermore, introduction of one copy of efg1STOP into wild-type SC5314 did not inhibit filamentation (Supplemental Fig. S11). This establishes that the truncated EFG1 gene in P94015 represents a recessive, loss-of-function allele. Complementation of P94015 with full-length EFG1 partially restored filamentous growth (Fig. 6A, B) and also yielded colonies that resembled the conventional white state (data not shown). These results indicate that complementation of P94015 with full-length EFG1 promotes formation of the white phenotypic state as well as the ability of this strain to undergo filamentous growth.
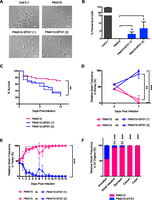
Analysis of native and EFG1-complemented P94015 strains in commensal and pathogenic models of infection. (A,B) Induction of filamentation in SC5314 and P94015 strains in YPD medium supplemented with 10% serum at 37°C. Two independent EFG1-complemented strains are shown: P94015 + EFG1(1) and P94015 + EFG1(2). (C) Survival curves for Galleria mellonella infected with strain P94015 and EFG1-complemented P94015. n = 30 worm per strain, P < 0.01, log-rank Mantel-Cox test. (D) Competition between native P94015 and EFG1-complemented P94015 in a murine model of systemic infection. C. albicans cells were recovered from kidneys after 7 d infection and compared to the inoculum. Data are two independent competition experiments with four mice each. (E) Testing of P94015 or derived strains in a murine model of commensal fitness. C. albicans cells were recovered from fecal pellets each day, and the relative levels of native P94015 or EFG1-complemented P94015 were determined. Error bars, ±SD. n = 3 mice for each competition. (F) Relative levels of P94015 or EFG1-complemented P94015 in gastrointestinal tissues after 2 wk of commensal growth. Error bars, ±SD.
To determine if loss of EFG1 function contributes to the low virulence of P94015, native and EFG1-complemented strains were compared in an invertebrate model of virulence. Over the course of 10 d, wild-type P94015 killed 30% of Galleria larvae, whereas P94015 isolates expressing full-length EFG1 exhibited significantly increased killing (P < 0.01, log-rank test) (Fig. 6C). We also directly competed native P94015 against EFG1-complemented P94015 strains in the murine model of systemic infection. P94015 strains containing full-length EFG1 showed significantly higher fungal burdens than the native P94015 strain after 7 d co-infection (P < 0.005, Student’s t-test) (Fig. 6D). We conclude that reintroduction of full-length EFG1 increases the virulence of P94015 in systemic infection.
Previous studies demonstrated that commensal fitness in SC5314 was enhanced by deletion of EFG1 (Pierce and Kumamoto 2012; Pande et al. 2013; Pierce et al. 2013). We therefore hypothesized that loss of EFG1 function in P94015 could promote commensal growth. To test this, we performed a 1:1 competition between P94015 and P94015 + EFG1 isolates in a murine model of gastrointestinal colonization. By 3 d post infection, P94015 had significantly outcompeted P94015 + EFG1 in two independent competition experiments, and this competitive advantage was maintained for 14 d post infection (P < 0.05, day 3; P < 0.001, day 14, Tukey’s pairwise test) (Fig. 6E; Supplemental Fig. S12). Similar fitness differences were observed when C. albicans cells were recovered from GI organs 14 d post infection; P94015 had outcompeted P94015 + EFG1 in the stomach, small intestine, cecum, and colon (P < 0.0005, Tukey’s test) (Fig. 6F; Supplemental Fig. S12). These results demonstrate that native P94015 has a significant fitness advantage in commensal growth over isolates in which EFG1 function has been restored. This raises the possibility that P94015 evolved a mutated form of this gene to promote commensal colonization within the mammalian host at the expense of decreased virulence.
Discussion
Here, intra-species analyses of C. albicans isolates revealed extensive variation between strains, both at the genotypic and phenotypic level. Substantial genomic differences were observed between the set of 21 clinical strains and included single nucleotide polymorphisms, inversions, copy number changes, LOH events, and whole or partial chromosomal aneuploidies. It is expected that each of these genomic differences contribute to the range of phenotypes exhibited by clinical isolates of C. albicans. This work provides the first step toward the identification of novel variants associated with phenotypes important for virulence in C. albicans.
Frequent LOH in C. albicans isolates
Overall genome heterozygosity varied from 48% to 89%, establishing LOH as an important mechanism contributing to C. albicans diversity. LOH events included segmental LOH on one or more of each of the eight chromosomes, as well as whole chromosome LOH events for Chr 3, 5, 6, 7, and R. This indicates that many chromosome homologs do not harbor recessive lethal alleles, as these homologs would be unable to exist as homozygous chromosomes in viable cells. Only one region of the genome was homozygous in all 21 strains; this region showed high omega (dN/dS) values, although an extremely low dS value was found to be the driver of this signal. While dN is relatively constant across the genome, dS exhibits strong local variation that will need to be taken into account in future studies of C. albicans genome evolution.
In general, we observed few correlations between genotypic and phenotypic characteristics, although overall genome heterozygosity showed a significant correlation with faster growth rates. This is consistent with heterozygosity contributing to strain fitness and with recent studies showing that completely homozygous strains of SC5314 exhibit slower growth, presumably reflecting the loss of alleles that influence fitness (Hickman et al. 2013).
LOH is also the primary mechanism by which homozygosity of the MTL locus is achieved, a necessary prelude to mating (Wu et al. 2005, 2007). Analysis of homozygous MTL alleles showed that LOH could include small regions of homozygosity at the MTL locus itself or LOH of the entire Chr 5 harboring the MTL locus. The frequent observation of LOH tracts that encompass only the MTL locus suggests that there may be recombination hotspot(s) close to the MTL. This is similar to what has been observed in Cryptococcus neoformans, where recombination hotspots flank the mating-type locus (Hsueh et al. 2006). However, in contrast to a previous analysis (Wu et al. 2007), we did not find a correlation between Chr 5 heterozygosity and virulence, or indeed between overall genome heterozygosity and virulence.
Aneuploidy in C. albicans
Variation between C. albicans genomes included extensive karyotypic differences, with more than one-third of the clinical isolates being aneuploid. This is consistent with earlier studies that revealed karyotypic variation in multiple C. albicans isolates (Suzuki et al. 1982; Rustchenko-Bulgac 1991; Perepnikhatka et al. 1999; Selmecki et al. 2006; Rustchenko 2007; Bouchonville et al. 2009; Yang et al. 2011). In the current study, seven of the 21 strains contained trisomic chromosomes, with Chr 4 and Chr 7 being trisomic in multiple isolates. This suggests that extra copies of these chromosomes may be particularly well tolerated by C. albicans or may confer a fitness benefit under some growth conditions. Interestingly, whole chromosome aneuploidies involved only the four smallest chromosomes (Chr 4, 5, 6, and 7); aneuploidies of the larger chromosomes may be less prevalent due to the higher number of genes impacted, particularly if changes in ploidy cause corresponding changes in transcript levels. Previous studies found that fluconazole-treated C. albicans strains were also biased toward aneuploidies of the smallest chromosomes (Selmecki et al. 2006; Bouchonville et al. 2009).
Consistent with experiments in both C. albicans and S. cerevisiae (Chen et al. 2004; Torres et al. 2007; Forche et al. 2008; Pavelka et al. 2010; Abbey et al. 2011; Siegel and Amon 2012), a significant correlation was observed between aneuploidy and longer doubling times, although not all aneuploid strains exhibited slow growth. A previous study noted a correlation between faster growth rates and increased virulence (MacCallum et al. 2009), but this was not observed in the analysis of the 21 strains described here.
Copy number variation and positive selection
Analysis of the 21 C. albicans genomes revealed gene copy number variation, including transposons that have previously been shown to be active and to have differential representation between strains (Goodwin et al. 2001; Holton et al. 2001; Yang et al. 2011). Variable copy number was also observed for the CYP5 gene that encodes a cytoplasmic cyclophilin. Cyclophilins are implicated in a variety of processes, including the response to stress, and have been shown to be necessary for pathogenesis in some fungal species (Wang et al. 2001; Viaud et al. 2002). This suggests that duplication of CYP5 could similarly contribute to increased fitness or virulence in C. albicans. Overall, the protein-coding gene duplications identified here did not overlap with the five duplications detected in a study of multiple C. parapsilosis isolates (Pryszcz et al. 2013).
Genes with high rates of nonsynonymous substitution (dN) included many structural cell wall proteins, including GPI-anchored proteins, as well as cell wall modifying enzymes. A previous study by Butler et al. (2009) showed that rapidly evolving gene families in the most pathogenic Candida species were also highly enriched for cell wall proteins. This is consistent with cell surface proteins being important for host-pathogen interactions, and there is perhaps ongoing selection for faster evolution of these factors. For example, our study found high variability in the FGR38-like Leucine-rich repeat family, previously noted to be fast evolving and to be unique to C. albicans and C. tropicalis (Butler et al. 2009).
Further evidence of genomic diversity was apparent in hypervariation of the subtelomeric TLO gene family in C. albicans. This gene family displays the largest increase in gene copy number between C. albicans and related, less pathogenic Candida species (Butler et al. 2009). We found that the C. albicans TLO gene family is continuing to rapidly evolve, including changes both in the number and relative positions of TLO genes within the genome. TLO genes encode proteins with similarity to Med2, a component of Mediator that regulates transcription by RNA polymerase II; and some Tlo proteins have been confirmed as Med2-like Mediator components (Zhang et al. 2012). TLO gene expression is also highly variable due primarily to Sir2-dependent telomere associated silencing (Anderson et al. 2014). The challenge now is to determine if expansion and diversification of TLO genes represents a mechanism to alter global transcription without extensive rewiring of transcriptional networks.
Mutations affecting commensalism and pathogenesis
One clinical isolate, P94015, was chosen for more detailed analysis as it exhibits an altered phenotypic state, an inability to filament, and limited systemic virulence. Each of these attributes was shown to be the result, at least in part, of a premature stop codon in the EFG1 transcription factor gene. It is striking that a clinical strain lacks this key transcriptional regulator given its established role in C. albicans filamentation and virulence (Lo et al. 1997; Stoldt et al. 1997; Braun and Johnson 2000; Korting et al. 2003; Noffz et al. 2008). However, loss of EFG1 in P94015 provides a fitness advantage in commensal growth. Thus, we identify a natural mutation that alters the balance between commensal and pathogenic lifestyles.
Our results raise additional questions about C. albicans commensalism. If loss of EFG1 can improve commensal fitness, the predominant state of C. albicans, why is there not a similar loss of function in more natural isolates? One intriguing possibility is that loss of EFG1 was selected for in an isolate infecting an immunocompromised host. Pierce and Kumamoto (2012) showed that efg1 mutants in SC5314 initially show increased fitness in gut colonization, but that in mice with an intact immune system, these mutants then decline in fitness relative to a wild-type strain. In contrast, in nude mice lacking T cells, the efg1 mutants do not show the same decline in fitness. It is therefore possible that loss of EFG1 function has a greater benefit in an immunocompromised host than in a healthy host. It should be noted, however, that in both our experiments and in another study using SC5314 (Pande et al. 2013), loss of EFG1 function consistently improved commensal fitness in immunocompetent mice. It is therefore unclear under which conditions, or by what mechanisms, the immune system impacts commensal colonization by C. albicans.
It is also striking that the P94015 isolate was recovered from a bloodstream infection despite lacking functional EFG1. This is surprising considering that loss of EFG1 compromises virulence. However, if the host was immunocompromised, this could explain the ability of an isolate to mount a successful bloodstream infection despite its reduced virulence. In fact, Sampaio et al. (2010) recently showed that successive isolates from a recurring bloodstream infection in a chemotherapy patient exhibited a progressive reduction in virulence. The authors speculated that C. albicans underwent microevolution and this favored adaptation toward increased commensal fitness and attenuated virulence (Sampaio et al. 2010). Our current study further supports this hypothesis and demonstrates that mutation of EFG1 is one mechanism by which C. albicans may limit killing of the host while enhancing its commensal lifestyle.
Conclusions
The analysis described here provides the most detailed examination of genomic and phenotypic intra-species variation in C. albicans to date. The phenotypic plasticity of this species has long been recognized, and our studies reveal the genetic differences underlying phenotypic differences are due to a variety of mechanisms, of which LOH and aneuploidy are major contributors. Furthermore, we uncover a genetic polymorphism responsible for altered phenotypic behavior, including a change in the balance between commensalism and pathogenesis. Uncovering these links will undoubtedly provide greater insights into the commensal-pathogen dichotomy and will present novel opportunities for clinical interventions against this opportunistic pathogen.
Methods
C. albicans strains, plasmids, and media
The set of 20 clinical isolates of C. albicans (Supplemental Table S1) were a gift of Dr. David Soll (University of Iowa) and have previously been tested in a murine model of systemic virulence (Wu et al. 2007). This set of strains is available at http://www.beiresources.org/. Additional strains used in this study are listed in Supplemental Table S17. Standard culture media were prepared as previously described (Guthrie and Fink 1991). For detailed strain construction and microscopy, see Supplemental Material.
Whole-genome sequencing, variant identification, and de novo assembly
To extract genomic DNA, ∼109 cells grown overnight in YPD at 30°C were processed using the Qiagen Genomic Buffer Set and the Qiagen Genomic-tip 100/G. Lyticase (Sigma, L2524) was used to enzymatically digest the fungal cell wall. Two libraries were constructed with average insert sizes of 197 bases and 2.5 kilobases (Supplemental Table S2) as previously described (Fisher et al. 2011; Grad et al. 2012) and were sequenced on an Illumina HiSeq to generate 101 base paired-end reads.
For SNP calling, reads were aligned to the SC5314 genome (version A21-s02-m01-r01; http://www.candidagenome.org) using BWA 0.5.9 (Li and Durbin 2010) and variants identified with GATK (McKenna et al. 2010). Genomic regions exhibiting copy number variation were identified from GC normalized read depth. Potential inversions and translocations identified from alignments of the de novo assemblies were validated by evaluating read support using BreakDancer (Chen et al. 2009).
The Illumina reads for each strain were assembled using ALLPATHS-LG (Gnerre et al. 2011), and genes were predicted for each assembly. Both the assemblies and SNPs were used to evaluate variation in gene content and sequence. For detailed DNA sequence analysis methods, see Supplemental Material.
RNA extraction, sequencing, and differential expression analysis
C. albicans cells for selected strains were grown overnight in YPD at 30°C, and RNA was isolated. Differentially expressed genes were identified using EdgeR (version 3.4.2) (Wolf et al. 2009) by comparing P94015 or P60002 transcript levels to those of SC5314, P87 and GC75 (Supplemental Table S18). For detailed methods see Supplemental Material.
Galleria mellonella infections
C. albicans cells were grown overnight in YPD medium at 30°C, washed, and injected into the terminal pro-leg of G. mellonella larvae, as previously described (Fuchs et al. 2010a). Inocula were ∼2.5 × 105 CFUs for analysis of the full set of clinical isolates and ∼5 × 105 CFUs for analysis of P94015 versus P94015 + EFG1. G. mellonella larvae were kept for 10 d at 37°C and scored daily for signs of death (see Supplemental Material).
Murine competition experiments
For systemic infection, cells were grown overnight in liquid YPD medium at 30°C, washed, and mice infected via the tail-vein with a total inoculum of ∼6.0 × 105 colony forming units (CFUs). Competition experiments involved a 1:1 mixture of wild-type P94015 (NAT-sensitive) and P94015 + EFG1 (NAT-resistant) isolates. Mice were euthanized 7 d post-infection, and the number of CFUs from the kidney determined.
For in vivo commensal experiments, an antibiotic-treated murine model of commensalism was used (Pande et al. 2013). To prepare C. albicans cells for infection, strains were grown overnight in 3 mL YPD at 30°C, washed, and ∼1 × 108 CFUs of C. albicans introduced by oral gavage. For each competition mixture, one strain was NAT sensitive and one strain was NAT resistant. Fecal pellets were collected daily and fungal CFUs were determined. For complete details see Supplemental Material.
Data access
The sequence data from this study have been submitted to NCBI under BioProject ID PRJNA193498 (http://www.ncbi.nlm.nih.gov/bioproject). SNP data have been submitted to dbSNP (http://www.ncbi.nlm.nih.gov/projects/SNP/) under ss# 1456786277 to 1457237021 (noninclusive). Differential expression analysis of RNA-seq data was submitted to the NCBI Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE55158.
Acknowledgments
We would like to thank David Soll for providing the C. albicans isolates, Felipe Riberio for assistance with Haploidify, and Darren Abbey for the Matlab script used to calculate doubling times. We acknowledge the Broad Institute Sequencing Platform for generating all DNA and RNA sequences described here. This work was supported by National Institutes of Health grants AI081704 and AI112363 (to R.J.B.) and AI075096 and AI0624273 (to J.B.), by EU Career Integration Grant 303635 and ERCAdv Award 340087 (to J.B.), and by the Investigators in the Pathogenesis of Infectious Disease award from the Burroughs Wellcome Fund (to R.J.B.). M.P.H. was supported by a training grant from the USDOE Graduate Assistance in Areas of National Need program (P200A100100) and an NIH F31 fellowship (F31DE023726). M.Z.A. was funded by an NIH Research Supplement to Promote Diversity in Health-Related Research (AI075096-S1). This project has been supported in part with federal funds from the National Institute of Allergy and Infectious Diseases, National Institutes of Health, Department of Health and Human Services under contract no. HHSN272200900018C.
Author contributions: R.J.B. and C.A.C. designed experiments and wrote the manuscript with editorial input from D.A.M., M.P.H., M.Z.A., and J.B. M.P.H. performed genetic and phenotypic analysis on the set of 21 isolates and tested strains in murine models of commensalism and virulence. D.A.M. performed transcriptional analysis. D.A.M and J.W. performed selection analysis. S.S. identified DNA variants and performed phylogenetic and heterozygosity analysis. J.G. mapped nonsense mutations and indels. M.Z.A. and J.B. designed and M.Z.A. performed the telomere analysis. M.Z.A. performed the G. mellonella infection experiments. E.Z. performed phenotypic analysis. A.B. assembled the genomes. S.G. and Q.Z. annotated the assemblies.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.174623.114.
- Received February 27, 2014.
- Accepted December 9, 2014.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see http://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








