
TP53 engagement with the genome occurs in distinct local chromatin environments via pioneer factor activity
- Departments of Cell and Developmental Biology, Genetics, and Biology, University of Pennsylvania, Philadelphia, Pennsylvania 19104, USA; Penn Epigenetics Program, University of Pennsylvania, Philadelphia, Pennsylvania 19104, USA
- Corresponding author: bergers{at}upenn.edu
Abstract
Despite overwhelming evidence that transcriptional activation by TP53 is critical for its tumor suppressive activity, the mechanisms by which TP53 engages the genome in the context of chromatin to activate transcription are not well understood. Using a compendium of novel and existing genome-wide data sets, we examined the relationship between TP53 binding and the dynamics of the local chromatin environment. Our analysis revealed three distinct categories of TP53 binding events that differ based on the dynamics of the local chromatin environment. The first class of TP53 binding events occurs near transcriptional start sites (TSS) and is defined by previously characterized promoter-associated chromatin modifications. The second class comprises a large cohort of preestablished, promoter-distal enhancer elements that demonstrates dynamic histone acetylation and transcription upon TP53 binding. The third class of TP53 binding sites is devoid of classic chromatin modifications and, remarkably, fall within regions of inaccessible chromatin, suggesting that TP53 has intrinsic pioneer factor activity and binds within structurally inaccessible regions of chromatin. Intriguingly, these inaccessible TP53 binding sites feature several enhancer-like properties in cell types within the epithelial lineage, indicating that TP53 binding events include a group of “proto-enhancers” that become active enhancers given the appropriate cellular context. These data indicate that TP53, along with TP63, may act as pioneer factors to specify epithelial enhancers. Further, these findings suggest that rather than following a global cell-type invariant stress response program, TP53 may tune its response based on the lineage-specific epigenomic landscape.
TP53 (protein product of TP53, also known as p53) is a DNA-binding transcription factor that acts as a master tumor suppressor to protect organisms from uncontrolled cell proliferation and genotoxic damage (Vousden and Lane 2007; Junttila and Evan 2009). Loss of TP53 activity correlates with increased genome instability, cell proliferation, and higher prevalence of cancer (Zilfou and Lowe 2009; Robles and Harris 2010). TP53 integrates multiple stress-induced signals and enacts a specific transcriptional program to induce factors and regulators involved in DNA repair, cell cycle arrest, and apoptosis. The ultimate functional outcomes of the TP53-dependent transcriptional program are a reversible cell cycle arrest followed by DNA damage repair, permanent arrest (senescence), or apoptosis (Bieging et al. 2014). These varied outcomes are dependent upon the type of stress and type of cell, suggesting that additional factors govern the specific TP53-dependent stress response (Vousden and Prives 2009).
There is intense interest in elucidating mechanisms regulating TP53 activation and genomic localization. Recent TP53 genome-wide data sets have identified numerous new TP53 transcriptional gene targets using combined TP53 binding and gene expression analyses (Smeenk et al. 2008; Li et al. 2012; Nikulenkov et al. 2012; Kenzelmann Broz et al. 2013; Lim et al. 2013; Melo et al. 2013; Menendez et al. 2013; Schlereth et al. 2013; Zeron-Medina et al. 2013; Akdemir et al. 2014; Allen et al. 2014). Importantly, recent studies in human, mouse, and flies indicate that TP53 binding is not restricted to promoter regions, but extends to enhancer elements, where TP53 may also modulate transcriptional activity (Link et al. 2013; Melo et al. 2013; Zeron-Medina et al. 2013). For example, TP53 binds to an enhancer upstream of CDKN1A that correlates with binding the gene-proximal promoter and increased transcriptional output (Melo et al. 2013). There has not yet been a genome-wide investigation of TP53 binding to transcriptional regulatory elements.
The ability of TP53 to modulate cell fate after genotoxic damage or other stress partially depends on specific cofactors that alter TP53 activity and target gene expression (Vousden and Lu 2002). TP53 undergoes abundant posttranslational modification by a diverse group of enzymes, including lysine acetyltransferases, such as CREBBP/EP300 and KAT2A/KAT2B (also known as GCN5/PCAF), and lysine methyltransferases, such as SETD8 and SMYD2, that are believed to modulate DNA binding and cofactor recruitment (Gu and Roeder 1997; Liu et al. 1999; Huang et al. 2006; Shi et al. 2007). TP53 is also acetylated by KAT5 (also known as TIP60) or KAT8 (also known as hMOF) at TP53 lysine 120 (TP53 K120ac) to influence transcriptional activation, specifically in regulation of apoptosis (Sykes et al. 2006; Tang et al. 2006). Many other enzymes and cofactors modify and modulate TP53-dependent transcription altering functional outcomes (Kruse and Gu 2008; Carter and Vousden 2009; Meek and Anderson 2009).
Posttranslational modification of histones directly influences chromatin structure and recruitment of specific transcriptional regulatory proteins. Transcription-associated histone modifications show spatial and temporal localization to specific regulatory regions. Active transcriptional start sites (TSS) are enriched for histone H3 lysine 4 trimethylation (H3K4me3), whereas transcriptional enhancers are enriched for monomethylation of H3K4 (H3K4me1) and acetylation of H3K27 (H3K27ac). The enzyme modifiers are recruited to specific genes via transcription factor association (histone acetylation) or via RNA polymerase II association (histone methylation). H4K16ac was recently shown to associate with enhancers (Taylor et al. 2013), although its specific function and relationship with gene activation has not been reported.
Chromatin modification dynamics at TSS have been linked to changes in TP53 activity. For example, coordinate regulation of H3K4me3 (MLL complexes) and H4K16ac (KAT8) at TSS activates TP53-dependent transcription (Dou et al. 2005). Similarly, coordination of H3K4me3 and H3K27ac by CREBBP/EP300 occurs at TP53-regulated genes (Lauberth et al. 2013; Tang et al. 2013). In contrast, TP53-regulated genes can be down-regulated by SUV39H1-mediated deposition of H3K9me3 and repressive chromatin formation at TP53 binding sites (Mungamuri et al. 2012). Overall, gene-specific and genome-wide approaches have identified TP53 gene targets and gene-proximal TP53 binding sites (Beckerman and Prives 2010). However, a comprehensive understanding of the role of TP53 in gene activation, specifically the function of TP53 at gene-distal regulatory regions and the interaction of TP53 with the underlying chromatin environment in response to stress, remains to be elucidated.
Results
TP53 activation is associated with changes to the local chromatin environment
We identified genome-wide TP53 binding sites and the corresponding changes to the local chromatin environment in basal and TP53-activated conditions in primary IMR90 human lung fibroblasts using ChIP-seq. Regions of TP53 and chromatin modification enrichment were identified by comparison to a condition-specific input using MACS (Supplemental Tables S1, S2; Zhang et al. 2008). Treatment with 5 μM nutlin, an MDM2 inhibitor, led to stabilization of TP53 and a concomitant increase in significantly enriched regions (FDR < 1) of TP53 (Fig. 1A; Supplemental Fig. S1A; Vassilev et al. 2004). The majority of induced TP53 binding events occur >5 kb from the TSS of a protein-coding gene, with the modal group between 5 and 50 kb from the nearest TSS (Fig. 1B).
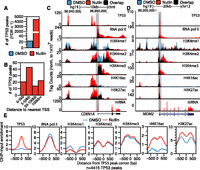
Epigenomic analysis of TP53 activation in primary human fibroblasts. (A) The number of significantly enriched TP53 peaks (versus input, defined by MACS identified by ChIP-seq in IMR90 primary human lung fibroblasts after treatment with DMSO [blue] or nutlin [red]; 5 μM final in DMSO) for 6 h. (B) The percentage of TP53 peaks after nutlin treatment within varying distances to the nearest TSS of a RefSeq gene. (C,D) UCSC Genome Browser track view of TP53, RNA pol II, poly(A)+ selected RNA (mRNA), H3K4me1, H3K4me3, H4K16ac, and H3K27ac at the CDKN1A (C) and MDM2 (D) genes. Tracks for the DMSO and nutlin treatment condition are shown in blue and red, respectively, with regions of overlap depicted in black. The y-axis is scaled to the maximum intensity for each set of data. (E) Enrichment profiles (input subtracted) at TP53 peaks (TP53 peak center ±750 bp) in the DMSO (blue) and nutlin (red) treatment condition for TP53, RNA pol II, H3K4me1, H3K4me2, H3K4me3, H4K16ac, and H3K27ac.
Genome-wide characterization of histone modification dynamics and RNA expression after nutlin treatment allowed definition of TP53-induced genes and potential regulatory regions. Fewer than 5% of transcripts show significant differential expression between DMSO and nutlin treatments (Supplemental Fig. S1C), and importantly, gene ontology (GO) analysis suggests these changes are consistent with well-characterized TP53-dependent processes (Supplemental Fig. S1D). Genome browser views of the canonical TP53 transcriptional targets CDKN1A (Fig. 1C) and MDM2 (Fig. 1D) show induced TP53 binding near the TSS with transcription-linked increases of RNA pol II, H3K27ac, and H4K16ac occupancy. In contrast, RNA pol II occupancy and histone acetylation are strongly reduced at down-regulated genes, such as the cell cycle associated PLK1 (Supplemental Fig. S1E).
Inspection of regions upstream of CDKN1A and MDM2 (Fig. 1C,D, boxed) revealed that TP53 binding distal to genes may correlate with increased histone acetylation. We then assessed the genome-wide correlation of induced TP53 binding sites with changes in chromatin modifications and RNA pol II occupancy (Fig. 1E). H3K27ac and, notably, H4K16ac show increased enrichment after TP53 induction, with H4K16ac showing the most dramatic change (Fig. 1E). Conversely, the average enrichment of histone H3K4 methylation at TP53 binding sites does not change, suggesting that histone methylation may be regulated in a different manner than acetylation at TP53 binding sites.
TP53 binds to three distinct and dynamic local chromatin environments
We explored distinguishing chromatin features between induced TP53 binding sites. Transcriptional regulatory elements can be distinguished by specific patterns of chromatin modification, such as H3K4me3 at TSS and H3K4me1 at enhancers (Bernstein et al. 2002; Santos-Rosa et al. 2002; Koch et al. 2007). Thus, we classified TP53 binding sites via their overlap with enriched regions of H3K4me3 or H3K4me1. H3K4me3-positive TP53 peaks (hereafter called TSS peaks due to gene proximity) (Fig. 2A, green) account for the smallest group of TP53 binding sites (Fig. 2A). TP53 sites lacking H3K4me3 enrichment are split between overlapping enriched regions of H3K4me1 (hereafter called enhancer peaks) (Fig. 2A, orange) or the absence of H3K4me1 (hereafter called distal peaks) (Fig. 2A, pink). The ratio of H3K4me1 to H3K4me3 enrichment is high at the orange enhancer peaks, as expected for putative enhancers (Figs. 2B,C), whereas TSS TP53 peaks are more highly enriched for H3K4me3 compared to either enhancer or distal TP53 peaks (Fig. 2C) (P < 2.2 × 10−16). H3K4me3+ TP53 peaks are located near genes, whereas peaks lacking H3K4me3 are located more gene-distal (Fig. 2D). This distribution of TP53 peaks relative to H3K4me is significantly enriched compared to the expected genome-wide distribution (Supplemental Fig. S1A,B) and suggests that TP53 specifically binds to functional transcriptional regulatory regions.
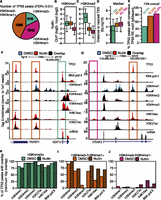
TP53 interacts with the genome at three distinct categories of binding sites defined by a dynamic local chromatin environment. (A) The classification of TP53 peaks based on their overlap with significantly enriched regions of H3K4me3 or H3K4me1. (B,C) Box plot analyses of H3K4me1 (B) and H3K4me3 (C) enrichment at each class of TP53 binding site (TP53 peak center ±750 bp). (D) Distances of each class of TP53 peak to the nearest TSS of a RefSeq gene. (E) Percentage of TP53 motifs within each category of TP53 peak. (F,G) Example UCSC Genome Browser track view of the GDF15 locus (F) and the STEAP3 locus (G), depicting the three classes of TP53 peaks. Tracks for the DMSO and nutlin treatment condition are shown in blue and red, respectively, with regions of overlap depicted in black. The y-axis is scaled to the maximum intensity for each set of data. Different classes of peak types are boxed, corresponding to the class of peak as in Figure 2A. (H–J) The percent of TP53 peaks (nutlin treatment) overlapping with significantly enriched regions of chromatin and RNA pol II in the DMSO (solid) and nutlin (striped) treatment conditions for (H) H3K4me3+ (TSS); (I) H3K4me3−/H3K4me1+ (enhancer); and (J) H3K4me3−/H3K4me1− (distal) TP53 peaks.
A total of 73% of identified TP53 peaks contain a consensus, TP53 response element (RE) motif (Wang et al. 2009); but remarkably, <50% of H3K4me3+ TP53 sites contain the RE (Fig. 2E). Similarly, H3K4me3+ TP53 peaks are poorly represented in a meta-analysis of published TP53 genome-wide binding data sets, especially when compared to the enhancer (orange) or distal (pink) TP53 sites (Supplemental Fig. S2C; Supplemental Tables S3, S4). H3K4me3+ TP53 sites with TP53 RE motifs correlate with increased nutlin-induced expression compared to sites lacking the TP53 motif (Supplemental Fig. S2D). TP53 sites lacking RE motifs display significantly lower (P < 2.2 × 10−16) nutlin-inducible TP53 binding (Supplemental Fig. S2E) and H4K16ac enrichment (Supplemental Fig. S2F) than consensus TP53 motif-containing peaks. Thus, consensus TP53 binding at TP53 RE motifs correlates with higher nutlin-induced TP53 enrichment and dynamic H4K16ac.
We visually inspected a number of TP53 binding sites in order to confirm our classification using H3K4me enrichment. Transcription of GDF15 is strongly up-regulated upon TP53 activation (Fig. 2F) (mRNA) and contains a TP53 peak at the TSS (Fig. 2F, green box) and two TP53 peaks with enhancer signatures upstream (Fig. 2F, orange boxes). The STEAP3 gene contains a putative enhancer TP53 peak within the first intron (Fig. 2G, orange box), but also contains a distal TP53 binding site directly upstream of the TSS (Fig. 2G, pink box). Interestingly, despite the presence of two nutlin-induced TP53 binding sites, no change occurred in the transcription of the STEAP3 gene after TP53 activation (Fig. 2G) (mRNA).
We assessed changes in histone modifications enrichment to gain further insight into the potential functions of different TP53 sites. The majority of green H3K4me3+ TSS peaks co-occupy regions of all surveyed chromatin modification and RNA pol II enrichment before and after TP53 binding (Fig. 2H), consistent with features of promoter-region associated chromatin (Bernstein et al. 2002; Santos-Rosa et al. 2002; Ernst et al. 2011). Orange enhancer TP53 peaks overlap extensively with H3K4me1 and H3K27ac (Fig. 2I). The overlap of TP53 with RNA pol II and H4K16ac increases after nutlin treatment, further suggesting a link between TP53 binding specific changes in the local chromatin environment. Overall, the patterns of chromatin modification enrichment (H3K27ac, high H3K4me1, low H3K4me3) for the enhancer class of TP53 binding sites (Fig. 2A, orange) fit the commonly used chromatin-based definition for active transcriptional enhancers.
Conversely, pink distal TP53 sites show low overlap with histone H3K4 methylation (Fig. 2J). However, a subset of distal TP53 sites overlaps H3K27ac, RNA pol II, and most notably, H4K16ac after nutlin treatment (Fig. 2J). Hence, the three groups of inducible TP53 binding sites have distinct and characteristic profiles of chromatin modification changes correlating with TP53 activation. The observed TP53 intersection with regions of enriched chromatin is significantly higher (P = 0.01) than expected for either locally or genome-wide random peak locations (Supplemental Fig. S2G–I). Strikingly, the most dynamic chromatin changes, like H4K16ac, occur at gene-distal elements, away from classically defined TP53-regulated promoter regions.
TP53 binds to established, but dynamic, enhancers
We explored the enhancer class of inducible TP53 binding sites by examining local chromatin dynamics compared to the TSS class using heatmaps. Enhancer TP53 sites (orange) are enriched for H3K4me1 and depleted of H3K4me3, especially when compared to TSS TP53 sites (Fig. 3A). TP53 enhancer sites are more dynamic than TSS sites, with significantly increased enrichment of RNA pol II, H3K27ac, and H4K16ac after nutlin treatment (Fig. 3A; Supplemental Fig. 3A). Histone H3K4me enrichment did not increase to the extent observed for acetylation at TP53 enhancer binding sites (Fig. 3A; Supplemental Fig. S3A).
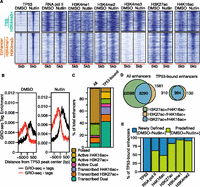
TP53 binds to established H3K4me1-marked enhancers and recruits RNA pol II and H4K16ac. (A) Heatmaps of TP53, RNA pol II, H3K4me3, H3K4me1, H3K27ac, and H4K16ac enrichment within a 5000-bp window (±2500 bp from the TP53 peak center) for TSS (H3K4me3+) and enhancer (H3K4me1+/H3K4me3−) TP53 peak types after DMSO or nutlin treatment. (B) Average GRO-seq profiles (normalized to 1 × 10−7 reads) at TP53 enhancer (H3K4me1+/H3K4me3−) peaks after DMSO or nutlin treatment. (C) Distribution of combinatorial histone modifications and RNA pol II enrichment at TP53-bound H3K4me1+ enhancers compared to the genome-wide complement of H3K4me1+ enhancers. (Poised enhancers) H3K4me1+; (active enhancers) H3K4me1+ and at least one acetylation event (H3K27ac or H4K16ac); (transcribed enhancers) H3K4me1+, RNA pol II+, and at least one acetylation event (H3K27ac or H4K16ac). Dual enhancers are occupied by both H4K16ac and H3K27ac. (D) Venn diagram representation of the overlap between H3K27ac+ and H4K16ac+ enhancers for all genome-wide and TP53-bound enhancers. (E) Analysis of the preestablishment of occupancy for each indicated factor at TP53-bound enhancers. Predefined enhancers are those in which the surveyed factor was significantly enriched (MACS-defined) during the DMSO treatment (no TP53 activation), whereas newly defined enhancers are those in which the enrichment of each factor was dependent on treatment with nutlin.
Recent reports suggest that transcription factor binding to enhancers leads to dynamic chromatin changes in H3K27ac and H3K4me1 and an increase in bidirectional transcription of noncoding RNAs, called eRNA, that reflect enhancer licensing and activity (Wang et al. 2011; Melo et al. 2013; Mousavi et al. 2013). We observed increased enrichment of H3K27ac and H4K16ac, but not H3K4me1 or H3K4me2, at TP53 enhancer binding sites (Fig. 3A; Supplemental Fig. S3A). We then examined TP53-induced eRNA transcription at the TP53-bound enhancers using published GRO-seq data from nutlin-treated HCT116 colon carcinoma cells (Allen et al. 2014). We observe a twofold increase in bidirectional GRO-seq tags overlapping putative TP53 enhancers in the nutlin condition (Fig. 3B; Supplemental Fig. S3C), suggesting that TP53 binding to these regions induces transcription of eRNA. Interestingly, bidirectional eRNA transcription occurs at most TP53-bound enhancers even in the DMSO-treated condition (Fig. 3B). The occupancy of H3K4me1 and H3K27ac and eRNA transcription at future TP53 binding sites (DMSO condition) suggests that these TP53-bound enhancers are preestablished, and that TP53 binding further activates eRNA transcription and H3K27ac/H4K16ac deposition.
Taken together, chromatin modification occupancy and eRNA transcription suggest that the enhancer (H3K4me1+) class of TP53 binding sites are actively transcribed. We then explored if TP53-bound enhancers contained specific features compared to the genome-wide enhancer repertoire. We defined TP53 enhancers as significantly enriched regions of H3K4me1 (MACS-defined) after treatment with nutlin (Fig. 3C) and grouped them as poised (H3K4me1 only), active (H3K4me1 and any combination of histone acetylation), or transcribed (H3K4me1, histone acetylation, and RNA pol II occupancy). Greater than 60% of TP53-bound enhancers are transcribed based on significant RNA pol II enrichment compared to <20% of genome-wide enhancers (Fig. 3C). TP53-bound enhancers are also less likely to be poised than genome-wide enhancers. Further analysis of enrichment of H3K27ac or H4K16ac revealed that almost 70% of TP53-bound enhancers are marked with both modifications, compared to 25% of genome-wide enhancers (Fig. 3D). These data strongly suggest that TP53-bound enhancers are more likely to be actively transcribed and enriched for H4K16ac than the average genome-wide enhancer, potentially reflecting direct TP53-mediated activation of these enhancer elements.
Enhancer licensing is established by the cell lineage-dependent epigenomic landscape defined by chromatin regulators and cell type-specific transcription factors (Ong and Corces 2012; van Oevelen et al. 2013). We investigated whether TP53 might serve as a licensing factor by assessing de novo enrichment of histone modifications reflective of enhancer activation. H3K4me1, H3K4me2, and H3K27ac peaks are present at essentially all TP53-bound enhancers before nutlin induction (Fig. 3E; Supplemental Fig. S3A,C), suggesting that TP53 activation does not commission new enhancers within 6 h of activation (Lee et al. 2013). However, we observe de novo enrichment of RNA pol II and H4K16ac at ∼50% of TP53-bound enhancers (Fig. 3E). Strikingly, new RNA pol II recruitment is more likely to occur at TP53-bound enhancers marked by H4K16ac alone compared to those with only H3K27ac (Supplemental Fig. S3D,E).
TP53 binds to inaccessible chromatin lacking H3K4 methylation
Although H3K4me3 (TSS) and H3K4me1-enriched (enhancer) TP53 binding sites reflect known transcriptional regulatory regions, the largest group of TP53 binding sites (44%) occurred within regions lacking H3K4 methylation (Fig. 4A; Supplemental Fig. S4A). Therefore, we investigated whether distal TP53 binding sites represent uncharacterized regulatory regions. Interestingly, H4K16ac enrichment at distal TP53 sites increases after nutlin treatment (Figs. 4A; Supplemental Fig. S4A) and is significantly enriched compared to random distance and size-matched peaks (2.6-fold induction, P < 2.2 × 10−16) (Supplemental Fig. S4A). RNA pol II and H3K27ac display more modest increases post-nutlin treatment, with H3K4me1, H3K4me2, and H3K4me3 showing little enrichment at distal TP53 binding sites (Supplemental Fig. S4A).
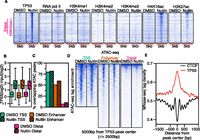
Distal TP53 peaks lie within regions of inaccessible chromatin and display dynamic histone acetylation. (A) Heatmap plots of TP53, RNA pol II, H3K4me3, H3K4me1, H3K27ac, and H4K16ac enrichment (±2500 bp from the TP53 peak center) for the distal (H3K4me1−/H3K4me3−) TP53 peak type for DMSO and nutlin. (B) Boxplot analysis of the input-subtracted TP53 enrichment (±750 bp from TP53 peak center) for TSS (green), enhancer (orange), or distal (pink) TP53 peaks in the DMSO and nutlin conditions. (C) Analysis of the fraction of TSS (green), enhancer (orange), or distal (pink) TP53 peaks overlapping significantly enriched (MACS-defined) ATAC-seq peaks for the DMSO (solid color) and nutlin (striped color) treatment conditions. (D) Heatmap of ATAC-seq enrichment (normalized to 1 × 10−7 reads) over a 5000-bp window (±2500 bp from TP53 peak center) at TP53 peaks of each class (TSS, enhancer, distal) for the DMSO and nutlin-treated conditions. (E) Average MNase-seq tag density (±750 bp from TP53 peak center) from all nutlin-induced TP53 binding sites and CTCF binding sites.
The general lack of histone modifications at distal TP53 binding sites suggested that these regions might be in a more closed conformation compared to promoters or enhancers. We initially hypothesized H4K16ac deposition after TP53 binding might reflect an opening of the chromatin architecture that would support TP53 binding and performed ATAC-seq (Buenrostro et al. 2013) to examine chromatin accessibility at TP53 binding sites (Fig. 4C,D; Supplemental Fig. S4B). Strikingly, we found <10% of distal TP53 peaks (pink) overlap ATAC-seq-defined accessible chromatin regions, compared to 50% and 80% for enhancers (orange) and promoters (green), respectively (Fig. 4C). Interestingly, we did not observe strong increases in ATAC-seq tag enrichment after nutlin treatment at any class of TP53 sites, suggesting that global TP53 binding does not directly induce a more open chromatin conformation (Fig. 4C,D; Supplemental Fig. S4B).
Our ATAC-seq analysis is highly consistent with DNase I hypersensitive site (DHSs) data from proliferating IMR90 cells from the Roadmap Epigenomics Project (Supplemental Fig. S4C,D; Bernstein et al. 2010) and are consistent with previous observations that promoters and enhancers occur within open chromatin regions (Biddie et al. 2011; Thurman et al. 2012; Buenrostro et al. 2013). Surprisingly, more than half of TP53 binding sites in nutlin-induced IMR90 fibroblasts, including almost all distal (pink) sites, are in “closed/inaccessible” regions.
The majority of characterized transcription factors bind within DHSs, which is thought to allow the factor access to DNA (Thurman et al. 2012). Our data demonstrate that 57% of TP53 binding sites occur within closed or compact chromatin (Fig. 4C; Supplemental Fig. S4B). This suggests that TP53 may act as a pioneer factor, binding directly to its response element in the context of a nucleosome, as was previously suggested for TP53 acting at individual binding sites (Lidor Nili et al. 2010; Sahu et al. 2010; Laptenko et al. 2011; Cui and Zhurkin 2014). We therefore examined nucleosome enrichment using published genome-wide mononucleosome (MNase) data from IMR90 fibroblasts (Kelly et al. 2012). Indeed, we observe enrichment of MNase density over all TP53 binding sites (Fig. 4E). Conversely, we observe a large nucleosome-depleted region over the center of IMR90 CTCF binding sites (Fig. 4E; Shah et al. 2013), as has been previously observed for other transcription factors like CTCF (Thurman et al. 2012). Similar mononucleosome enrichment is observed over all three classes of TP53 binding sites (Supplemental Fig. S4E), suggesting that even in the context of “open” chromatin at TSS and enhancers, TP53 binding occurs mainly over nucleosomes.
TP53-bound enhancers are cell type-specific, and proto-enhancers are active in epithelial cell types
Enhancers are established by cell- and lineage-specific transcription factors (Ong and Corces 2012; van Oevelen et al. 2013). TP53 is widely expressed, but its activity is temporally restricted during development and normal cellular proliferation. We observed that TP53-bound enhancers in IMR90 are established before TP53 activation (Fig. 3A,E), whereas binding of TP53 to distal regions does not lead to enhancer licensing within 6 h of TP53 activation (Fig. 4A). We predicted that distal TP53 sites might lack particular co-occupying transcription factor binding sites present in the TSS or enhancer class of TP53 binding sites, which would regulate TP53 enhancer licensing. Using HOMER (Heinz et al. 2010), we determined that enhancer and distal TP53 sites are highly enriched for consensus TP53 motifs over background regions, as expected (Fig. 5A; Supplemental Tables S8, S9). JUN/FOS motifs are significantly enriched in TP53 enhancer peaks and absent from the distal class (Fig. 5A). Similarly, JUN/FOS family members overlap TSS and enhancer TP53 binding sites significantly more than distal sites (Fig. 5B). The JUN/FOS family can mediate chromatin accessibility for other transcription factors (Biddie et al. 2011) and thus may function to license a set of TP53-bound enhancer elements in IMR90 fibroblasts.
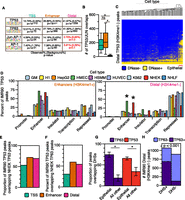
Distal (H3K4me1−) TP53 peaks bind to accessible chromatin in a cell-specific manner. (A) Transcription factor motif analysis for enhancer and distal TP53 peaks using HOMER. (B) Boxplot of JUN/FOS/TP53 colocalization at the three categories of TP53 binding sites. Asterisks denote that the P-value for the pairwise comparison is less than 0.05 (Wilcoxon rank sum test). (C) Hierarchal clustering (Euclidean distance) of the overlap between TP53 distal (H3K4me1−) peaks and DHSs from the ENCODE Project (minimum of 1-bp overlap between TP53 and DHSs; additional information in Supplemental Table S5). (D) Analysis of the overlap between TP53 enhancer (orange) and distal (pink) peaks with ChromHMM-defined genomic regulatory regions from nine human cell types. Similar functional ChromHMM categories were grouped into single categories (i.e., strong and weak enhancers grouped together). TP53 peaks were allowed to overlap multiple ChromHMM regulatory regions, and each region was reported (minimum of 1-bp overlap). (E) The proportion of IMR90 TP53 peaks overlapping TP53 binding sites from cisplatin-treated normal human epithelial keratinocytes (NHEK). (F) The proportion of IMR90 TP53 peaks overlapping TP63 binding sites from cisplatin-treated NHEK. (G) The proportion of TP63 (NHEK) and TP53 (IMR90) binding sites overlapping regions of DHSs from epithelial cell types and all other cell types with available data. P-values generated using a two-tailed t-test (P-values < 0.001). (H) Dependency of TP53 distal (H3K4me1−) peak overlap with DHSs (normal human epithelial keratinocytes) on co-occupancy with TP63: P-value < 0.001 (χ2 test).
We explored whether the distal TP53 binding sites in IMR90 fibroblasts might have context-dependent enhancer activity by assessing whether these TP53 sites display chromatin signature of enhancers in other cell types. Using genome-wide DHSs data from the ENCODE Project (The ENCODE Project Consortium 2011; Thurman et al. 2012), we observe that distal TP53 binding sites show strong and specific overlap with DHSs in epithelial cell types (Fig. 5C; Supplemental Fig. S5B). We observe similar specificity for epithelial cells when intersecting distal TP53 sites in IMR90 with H3K4me1 and H3K27ac-enriched regions from other ENCODE cell types (Supplemental Fig. S5C,D). In contrast, TP53-bound enhancers in IMR90 fibroblasts display broad overlap with DHSs in other cell types (Supplemental Fig. S5A), suggesting a shared mechanism of licensing of TP53-bound enhancers in diverse lineages.
We used ChromHMM chromatin state classifications (Ernst et al. 2011; Ernst and Kellis 2012) to further explore potential enhancer licensing of distal TP53 binding sites in epithelial cell types. TP53-bound TSS and enhancer sites in IMR90, as well as CTCF, overlap the expected ChromHMM classifications (Fig. 5D; Supplemental Fig. S5E,F). The percentage of TP53-bound enhancers scoring as “enhancer” or “repressed” is consistent with our analysis of DHSs enrichment (Supplemental Fig. S5B). Strikingly, nearly 50% of the distal TP53 peaks (H3K4me1−) in IMR90 are predicted to be active enhancers in two epithelial cell types and inactive/repressed in the remaining seven cell types (Fig. 5D, right panel) (HMEC and NHEK), again consistent with our DHSs clustering (Fig. 5C).
Thus, we propose that the H3K4me3−/H3K4me1− class of distal TP53 peaks represents “proto-enhancers” that are inactive in IMR90 fibroblasts but are active enhancers in epithelial cells, and that these enhancers are specified by an epithelial-specific factor. We reasoned that TP63, sharing a nearly identical DNA binding motif and serving as a master regulator of epithelial cell identity (Mills et al. 1999; Yang et al. 1999), was a likely candidate. We used genome-wide TP63 and TP53 binding data from epithelial keratinocytes to determine if TP53-bound proto-enhancers in IMR90 are co-occupied by TP53/TP63 (McDade et al. 2014). Almost 70% of TP53-bound proto-enhancer peaks in IMR90 are also found in cisplatin-treated keratinocytes (NHEK) (Fig. 5E). Consistent with our hypothesis, a subset of TP53-bound proto-enhancers is co-occupied by TP63 in keratinocytes (Fig. 5F). Overall, TP63 binding sites from NHEK cells strongly overlap DHSs present only in epithelial cell types (Fig. 5G); TP53 proto-enhancer binding sites are also significantly more likely to be DNase-accessible if TP63 is cobound than if TP63 is absent (Fig. 5H; Supplemental Fig. S5G). These data strongly suggest that binding of TP63 at the TP53-bound proto-enhancers defined in IMR90 cells acts to preestablish active, open enhancers in epithelial cells and also suggest that cell type-specific transcription factors may regulate TP53-bound enhancers and TP53-mediated transcriptional responses.
Discussion
Three classes of genomic TP53 binding sites defined by the local chromatin environment
Genome-wide analyses of TP53 occupancy have revealed tens of thousands of TP53 binding sites, but the functional role of most sites remains unclear. In this report, we focus on the chromatin context of TP53 binding sites, thus identifying three distinct classes of TP53 binding sites. These sites comprise (1) H3K4me3-enriched, gene-proximal sites; (2) transcriptional enhancer sites; and (3) distal proto-enhancers, which had not been previously defined. This study is the first to characterize TP53 binding sites with respect to dynamic chromatin environment change and to define a complete set of TP53-bound enhancers in fibroblasts.
Our data also demonstrate that <50% of TP53 binding sites near TSS and enriched for H3K4me3 contain a TP53 response element motif in the underlying DNA sequence, whereas between 80% and 95% of enhancer and distal TP53 binding sites contain these motifs (Supplemental Fig. S2A). Although the reason for this is unclear, it is possible that many TP53 TSS binding sites identified by ChIP-seq result from promoter:enhancer looping interactions. Indeed, recent reports suggest that TP53-regulated genes are controlled in part by long-range chromatin interactions (Link et al. 2013; Melo et al. 2013). Hence, it is striking that TP53 binding sites adjacent to activated genes comprise the smallest overall proportion of sites and have lower correspondence to consensus binding motifs, suggesting a crucial role for previously underappreciated TP53-bound enhancer elements.
Dynamic chromatin modifications at TP53-bound TSS and enhancers
Our observations suggest that preestablishment of H3K4 methylation at TP53 binding sites may be controlled by the activity of other sequence-specific transcription factors recruited to establish the enhancers, such as JUN/FOS (Fig. 5A). Conversely, pulsatile TP53 activity during the cell cycle, in response to intrinsic cellular stress, could account for preestablishment of TP53-bound enhancers (Batchelor et al. 2008; Loewer et al. 2010). Further investigation into this epigenetic memory at TP53 binding sites is warranted and likely will reveal critical mechanisms underlying TP53 activity.
In contrast to methylation, regulation of histone acetylation at TP53-bound TSS and enhancer sites is more dynamic. H4K16ac enrichment at TP53-bound enhancers supports a recent report of H4K16ac marking active enhancers in mouse embryonic stem cells (mES) and suggests that classes of enhancers may be differentially marked with histone acetylation (Taylor et al. 2013). Dynamic deposition of H3K27ac after transcription factor binding has been observed at SPI1 (also known as PU.1) binding sites and VEGF-stimulated enhancers (Kaikkonen et al. 2013; Zhang et al. 2013), but has not yet been described for H4K16ac. This is particularly interesting as both known H4K16 acetyltransferases, KAT8 and KAT5, directly modify TP53 to regulate its activity (Sykes et al. 2006; Tang et al. 2006; Li et al. 2009). KAT8 and KAT5 may have complementary or redundant roles in the regulation of H4K16ac at TP53 binding sites, which suggests that H4K16ac likely serves a critical but previously unrecognized role at nonpromoter TP53 binding sites.
We propose that differential modification of chromatin structure at TP53 binding sites may be one potential mechanism regulating the timing of a TP53 transcriptional response. Remodeling and modification of nucleosomes at TF binding sites is a time-dependent process. Although our data suggest that TP53 does not license new enhancers during the early TP53 response, TP53 activity might be necessary for the establishment and activation of enhancers required for late TP53-mediated responses, an idea supported by increases in transcription from TP53 bound enhancers observed using GRO-seq (Allen et al. 2014).
Alternatively, TP53 binding to preestablished enhancers may be a mechanism to generate cell- or lineage-specific TP53 responses. We observed that preestablishment of enhancers at TP53 binding sites is cell type-specific, as has been extensively observed for cell- and lineage-specific enhancers (Ong and Corces 2012; van Oevelen et al. 2013). Our data suggest that members of the JUN/FOS family are putative candidates to regulate TP53-bound enhancer establishment (Fig. 5A), as suggested for other transcription factors (Biddie et al. 2011; Andersson et al. 2014). JUN/FOS regulates the cellular stress response and can mediate TP53-dependent transcription (Shaulian and Karin 2001). Other transcription factors, like SP1 and STAT family members, have been implicated in modulation of TP53-mediated responses through co-occupation of binding sites (Nikulenkov et al. 2012; Li et al. 2014). It is interesting to speculate that global establishment of enhancers by co-occupancy of various transcription factors before TP53 activation is a potentially powerful mechanism to generate rapid cell type-specific, TP53-mediated stress responses, like TP63 in epithelial cell types.
TP53 family pioneer activity at enhancer elements
The TP53 proto-enhancers we identified in IMR90 fibroblasts are likely to be active enhancers in epithelial cell types. Importantly, our data indicate that TP53-bound epithelial enhancers are co-occupied, and potentially licensed, by TP63 (Fig. 5E), whose expression is restricted to epithelial cell types (Mills et al. 1999; Yang et al. 1999; Collavin et al. 2010; Su et al. 2010, 2013). TP53 response element motifs are specifically and highly enriched in epithelial-specific enhancers, further supporting this notion (Andersson et al. 2014). Our data strongly suggest that coregulation of epithelial-specific enhancers is a shared function of TP53 and TP63, and we speculate that TP53 may function to regulate critical epithelial genes.
A main observation from our analysis is that TP53 can bind to DNA within closed chromatin lacking other chromatin features of enhancers (Fig. 4A), thus strongly suggesting that TP53 can function as a pioneer factor. Furthermore, TP63 likely also functions as a pioneer factor and likely establishes open chromatin at epithelial-specific, TP53-bound enhancers (Fig. 5G). Taken together, our data thus provide a genome-wide view of TP53 and TP63 as pioneer factors, as suggested by previous observations that some TP53 binding sites are nucleosomal (Lidor Nili et al. 2010; Sahu et al. 2010; Laptenko et al. 2011; Cui and Zhurkin 2014). We speculate that the pioneer activity of TP53 is a crucial, intrinsic property of the TP53 protein and contributes to its tumor suppressive function by allowing for discrimination of its response elements in many chromatin contexts. Together, our results demonstrate that TP53 binding to the genome occurs within varied chromatin contexts that likely reflect cell type-specific enhancer usage and suggest unique chromatin-based regulatory mechanisms for controlling TP53-mediated responses in a lineage-specific manner.
Methods
Chromatin immunoprecipitation
IMR90 fetal lung fibroblasts (population doubling 30–40) were cultured in complete DMEM media (10% FBS + 1% Penn/Strep) at physiological oxygen (3.5% O2) at 37°C. Cells were treated with DMSO or 5 μM nutlin (in DMSO) for 6 h to induce TP53 activity and then either crosslinked with formaldehyde (1% final) for 10 min at room temperature or snap-frozen for RNA isolation. Crosslinking reactions were quenched with glycine (125 mM final) for 5 min, followed by 2× washes in cold PBS. Nuclei were isolated from 25 million cells as previously described (Shah et al. 2013), and chromatin was sheared to 250 bp average size (Diagenode Bioruptor, high setting, 30 s on/off, 30 min).
Immunoprecipitation reactions were performed using 1 mg sheared chromatin and 5 μg antibodies preconjugated to Protein G beads (Invitrogen): TP53 (10 μγ/rxn, Abcam #ab80645), H3 (Abcam, #ab1791), H3K4me3 (Abcam, #ab8580), H3K4me2 (Millipore, #07-030), H3K27ac (Active Motif, #39133), H4K16ac (Millipore, #07-329), POLR2A (RNA pol II, Santa Cruz, #sc-56767), or H3K4me1 (Abcam, #ab8895). ChIP reactions were incubated overnight at 4°C with rotation and washed 4× in wash buffer (50 mM HEPES-HCl pH 8, 100 mM NaCl, 1 mM EDTA, 0.5 mM EGTA, 0.1% sodium deoxycholate, 0.5% N-laurylsarcosine) and 1× in ChIP final wash buffer (1X TE, 50mM NaCl). Immunoprecipitated DNA was eluted from the washed beads, purified, and used to construct sequencing libraries.
ChIP/RNA sequencing and data analysis
Sequencing libraries for ChIP experiments were prepared using NEBNext Ultra reagents (New England Biolabs). All ChIP samples and input were single-end sequenced on an Illumina HiSeq 2000 or NextSeq 500. Uniquely aligned reads (up to one mismatch) were retained and aligned to NCBI37/hg19 using Bowtie 2 (Langmead and Salzberg 2012).
Poly(A)+ RNA was isolated using double selection with poly-dT beads, followed by first- and second-strand synthesis. RNA-seq reads (unique, concordant paired) were mapped via STAR (Dobin et al. 2013) to Ensembl v.75/hg19. Expression values were determined using Cufflinks (Trapnell et al. 2010). Numbers for total sequenced and aligned reads for all experiments are in Supplemental Table S1. Significantly enriched peaks (false discovery rate <1) were called with MACS (v1.4, default parameters) (Zhang et al. 2008), with treatment-matched input DNA as a control. bigWig Genome Browser tracks (hg19) were generated using HOMER.
Assay for transposase-accessible chromatin (ATAC-seq)
ATAC-seq experiments were performed as previously described (Buenrostro et al. 2013), except that 100,000 cells and 5 μL Tn5 transposase (Nextera XT Kit, Illumina) were used to tagment intact nuclei. Reads were aligned to NCBI37/hg19 using Bowtie 2, and bigWig files were generated using HOMER. Significant regions of enrichment were identified using MACS (v1.4), with default local enrichment settings.
Analysis of ENCODE DHS, MNase, and ChromHMM-predicted regulatory regions
BED files for DHSs and transcription factor binding data from ENCODE were downloaded from http://hgdownload.cse.ucsc.edu/goldenPath/hg19/encodeDCC/. IMR90 TP53 peaks were intersected with DHSs using BEDTools (intersectBed, min. 1-bp overlap) (Quinlan and Hall 2010), and hierarchal clustering (Euclidean distance) was performed using Partek Genome Analysis software. MNase-seq data from proliferating IMR90 were obtained from GEO Accession GSE21823 (Kelly et al. 2012), aligned to NCBI37/hg19 using Bowtie 2, and processed with HOMER. ChromHMM chromatin state segmentation was obtained from http://hgdownload.cse.ucsc.edu/goldenPath/hg19/encodeDCC/. All ChromHMM regions overlapping a TP53 site were included. The 15 chromatin states were combined into five broad categories as previously described (Ernst and Kellis 2012).
Intersection of TP53 peak types with chromatin features and genome-wide enhancer prediction
TP53 peaks were intersected with MACs-defined chromatin features using BEDTools (intersectBed, minimum 1-bp overlap). Genome-wide enhancer subtype predictions for Figure 3C were made by intersecting MACS-defined nutlin H3K4me1 peaks with MACS-defined chromatin features using BEDTools (intersectBed, minimum 1-bp overlap, -u). The order of intersection was H3K4me1, H3K4me3, H3K27ac, RNA pol II, and H4K16ac. Randomization and statistical testing of TP53 peak type intersection was performed using poverlap with local peak shuffling (https://github.com/brentp/poverlap).
Generation of tag enrichment statistics, histogram plots, and heatmaps
Tag enrichment TP53 peak histograms were generated using HOMER (annotatePeaks.pl). Tag enrichments for given regions were computed with HOMER (annotatePeaks.pl, -size given). A Wilcoxon rank sum test was used to compute P-values (R Core Team 2014, wilcox.test). Tag count heatmaps were generated using HOMER (annotatePeaks.pl, -hist 10, -size 5000, -ghist) and visualized using JavaTreeView.
Analysis of TP53 and TP63 binding in human primary keratinocytes
Raw data for TP53 and TP63 ChIP-seq in keratinocytes in Figure 5 were obtained from GEO series GSE56640 (McDade et al. 2014). Raw reads from duplicate TP53 (GSM1366691 and GSM1366697) and TP63 (GSM1366688 and GSM1366694) ChIP-seq experiments were individually aligned to NCBI37/hg19 using Bowtie 2, and significant regions of enrichment were identified using MACS. A high-confidence set of TP53 and TP63 peaks were constructed using peaks identified in replicate data sets.
Data access
Sequencing data from this study have been submitted to the NCBI Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE58740.
Acknowledgments
We thank Piotr Kopinski for assistance with ChIP experiments; and Daniel Simola, Parisha Shah, and Daniel Bose for insightful discussion. M.A.S. was supported by a Postdoctoral Fellowship from the American Cancer Society. This research was supported by a grant from the National Institutes of Health (CA078831 to S.L.B.).
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.181883.114.
- Received July 21, 2014.
- Accepted November 12, 2014.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see http://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








