
DIS3 shapes the RNA polymerase II transcriptome in humans by degrading a variety of unwanted transcripts
- Teresa Szczepińska1,2,
- Katarzyna Kalisiak1,2,4,
- Rafal Tomecki1,2,4,
- Anna Labno1,2,4,
- Lukasz S. Borowski1,2,
- Tomasz M. Kulinski1,2,
- Dorota Adamska1,2,
- Joanna Kosinska3 and
- Andrzej Dziembowski1,2
- 1Institute of Biochemistry and Biophysics, Polish Academy of Sciences, 02-106 Warsaw, Poland;
- 2Institute of Genetics and Biotechnology, Faculty of Biology, University of Warsaw, 02-106 Warsaw, Poland;
- 3Department of Medical Genetics, Center for Biostructure Research, Medical University of Warsaw, 02-106 Warsaw, Poland
- Corresponding author: andrzejd{at}ibb.waw.pl
-
↵4 These authors contributed equally to this work.
Abstract
Human DIS3, the nuclear catalytic subunit of the exosome complex, contains exonucleolytic and endonucleolytic active domains. To identify DIS3 targets genome-wide, we combined comprehensive transcriptomic analyses of engineered HEK293 cells that expressed mutant DIS3, with Photoactivatable Ribonucleoside-Enhanced Cross-Linking and Immunoprecipitation (PAR-CLIP) experiments. In cells expressing DIS3 with both catalytic sites mutated, RNAs originating from unannotated genomic regions increased ∼2.5-fold, covering ∼70% of the genome and allowing for thousands of novel transcripts to be discovered. Previously described pervasive transcription products, such as Promoter Upstream Transcripts (PROMPTs), accumulated robustly upon DIS3 dysfunction, representing a significant fraction of PAR-CLIP reads. We have also detected relatively long putative premature RNA polymerase II termination products of protein-coding genes whose levels in DIS3 mutant cells can exceed the mature mRNAs, indicating that production of such truncated RNA is a common phenomenon. In addition, we found DIS3 to be involved in controlling the formation of paraspeckles, nuclear bodies that are organized around NEAT1 lncRNA, whose short form was overexpressed in cells with mutated DIS3. Moreover, the DIS3 mutations resulted in misregulation of expression of ∼50% of transcribed protein-coding genes, probably as a secondary effect of accumulation of various noncoding RNA species. Finally, cells expressing mutant DIS3 accumulated snoRNA precursors, which correlated with a strong PAR-CLIP signal, indicating that DIS3 is the main snoRNA-processing enzyme. EXOSC10 (RRP6) instead controls the levels of the mature snoRNAs. Overall, we show that DIS3 has a major nucleoplasmic function in shaping the human RNA polymerase II transcriptome.
Eukaryotic genomes are pervasively transcribed, and many transcripts without protein-coding potential arise from previously unexpected parts of these genomes (Jensen et al. 2013).
The exosome complex is a primary eukaryotic 3′-to-5′ exonuclease involved in the processing of stable RNA species (5.8S rRNA and snoRNA), RNA quality control, and mRNA decay (Chlebowski et al. 2013; Schneider and Tollervey 2013). It is also implicated in the decay of the products of pervasive transcription. Exosome dysfunction causes the accumulation of unstable transcripts, many of which originate as a result of bidirectional transcription and give rise to so-called Cryptic Unstable Transcripts (CUTs) in yeast (Wyers et al. 2005) and Promoter Upstream Transcripts (PROMPTs) in humans (Preker et al. 2008). Such classes of long noncoding RNA (lncRNA) are barely detectable in normal cells. More recently, it has also been shown that bidirectional transcripts that arise from enhancer regions accumulate upon exosome depletion in humans (Andersson et al. 2014). The exosome is also involved in transcriptional termination in Schizosaccharomyces pombe (Lemay et al. 2014) and targeting activation-induced cytidine deaminase (AID) to transcribed DNA, which is essential for immunoglobulin class switch recombination and somatic hypermutation, two phenomena that can generate antibody diversity in mammalian B cells (Basu et al. 2011; Pefanis et al. 2014).
The exosome is composed of a catalytically inactive, ring-shaped nine-subunit core and associated catalytic subunits that belong to the DIS3 and RRP6 families (Liu et al. 2006; Dziembowski et al. 2007; Makino et al. 2013; Wasmuth et al. 2014). DIS3 proteins can be endowed with both processive exonucleolytic and endonucleolytic activities that originate from the RNB and PIN domains, respectively (Lebreton et al. 2008; Schaeffer et al. 2009; Schneider et al. 2009). RRP6 (EXOSC10 in humans) is in turn a distributive exonuclease (Mitchell et al. 1997; Chlebowski et al. 2010). In yeast, Dis3 is present in both the nucleus and cytoplasm, whereas Rrp6 is restricted to the nucleus. The functions of nuclear Dis3 and Rrp6 overlap, but only DIS3 is essential (Gudipati et al. 2012; Schneider et al. 2012). In humans, the situation is more complex because the genome encodes three DIS3 proteins—DIS3, DIS3L, and DIS3L2 (Staals et al. 2010; Tomecki et al. 2010; Lubas et al. 2013) and one RRP6 (EXOSC10) protein. Both DIS3 and DIS3L possess all DIS3 domains that are typical for that family, and they associate with the exosome core, whereas DIS3L2 lacks an N-terminal PIN domain and is not a constituent of any known stable macromolecular assembly (Chang et al. 2013; Lubas et al. 2013; Ustianenko et al. 2013). The DIS3 PIN domain has endoribonucleolytic activity; but in the case of DIS3L, the two catalytic residues within the PIN domain are missing, and the protein has no detectable endoribonucleolytic activity (Tomecki et al. 2010). Human DIS3 proteins are differently localized, as DIS3 is mainly nuclear but is restricted from the nucleolus, whereas DIS3L and DIS3L2 are exclusively cytoplasmic. EXOSC10 is mainly a nucleolar protein, but a small fraction is also present in the nucleoplasm and perhaps in the cytoplasm (Tomecki et al. 2010). Thus, in the nucleus, which is the subject of the present study, there are three potential forms of exosome complexes: a nucleoplasmic 10-subunit assembly with DIS3 as a catalytic subunit, a nucleolar 10-subunit assembly with EXOSC10, and a nucleoplasmic 11-subunit complex with DIS3 and EXOSC10 (Lykke-Andersen et al. 2011). The interplay and dynamics between these assemblies in humans remains to be elucidated. Human DIS3 attracted our attention because it is essential for the survival of vertebrate cells (Tomecki et al. 2014). Interestingly, DIS3 is frequently mutated in multiple myeloma and other cancers (Chapman et al. 2011; Walker et al. 2012; Weißbach et al. 2014). Approximately 10%–15% of multiple myeloma cases have specific hemizygous or homozygous mutations in the DIS3 RNB domain, which leads to DIS3 dysfunction, but not complete inactivation (Tomecki et al. 2014).
To thoroughly understand the impact of nuclear exosome dysfunction on RNA metabolism, we applied an effective model based on the HEK293 cell line in which an endogenous DIS3 is silenced and replaced with exogenous wild-type or defective DIS3 proteins (Tomecki et al. 2014). This strategy overcame the caveats of, so far utilized, systems which did not abolish the nuclear activity nor could distinguish the nuclear and cytoplasmic functions (Preker et al. 2008; Tomecki et al. 2010; Ntini et al. 2013). A comprehensive genome-wide analysis of human DIS3 targets presented herein shows the essential role of this nuclease in maintaining RNA polymerase II transcriptome homeostasis, indicating that the levels of pervasive transcription initiation and premature termination are much higher than previously anticipated.
Results
To identify nucleoplasmic exosome substrates, we conducted RNA-seq analysis of HEK293 cells with DIS3 dysfunction. These analyses were supplemented by the direct identification of DIS3 targets using Photoactivatable Ribonucleoside-Enhanced Cross-Linking and Immunoprecipitation (PAR-CLIP) assays (Hafner et al. 2010).
RNA-seq experiments were performed using HEK293 cell lines, which upon tetracycline-mediated induction simultaneously express sh-miRNAs that silence endogenous DIS3 and one of the following sh-miRNA-insensitive exogenous DIS3 variants: DIS3WT (WT), PIN domain catalytic mutant DIS3D146N (PIN), RNB catalytic mutant DIS3D487N (RNB), or PIN and RNB domain double mutant DIS3D146N D487N (PIN RNB) (Tomecki et al. 2014). The transgenic exogenous DIS3 is overexpressed ∼fivefold with regard to the endogenous protein in the analyzed cell lines (Tomecki et al. 2014). Experiments were carried out in triplicate; RNA was isolated three days after induction and rRNA-depleted before strand-specific total RNA libraries were prepared and sequenced in a paired-end protocol.
In parallel, PAR-CLIP experiments were performed. The in vivo cross-linking and immunoprecipitation efficiency of the RNA was first examined in HEK293 cells that stably expressed C-terminal eGFP-tagged WT or RNB mutant DIS3 proteins, as well as in parental, nontransfected control cells. Initial experiments showed that the DIS3 RNB mutant produced a much stronger PAR-CLIP signal (Supplemental Fig. 1) than did DIS3 WT. Therefore, further analyses were conducted using cells that synthesized a DIS3 RNB mutant allele. The entire immunoprecipitation procedure was performed using high-salt conditions in which DIS3 could no longer interact with the remainder of exosome core (Tomecki et al. 2010). 4-Thiouridine-enhanced cross-linking often results in thymidine to cytidine transitions; and in our case, nearly 30% of the reads contained such changes (27.34% in data set 1, 32.54% in data set 2), pointing to high specificity of the experiments because this value is well above the typical background level (Hafner et al. 2010). Since DIS3 protein is an exoribonuclease, degrading the transcript throughout its length without any strong sequence preferences, we have not identified any specific motives in PAR-CLIP data sets; therefore, to allow for higher coverage, we have not narrowed down our further analysis to reads containing the T-C transitions.
In CLIP experiments performed for yeast Dis3, a substantial fraction of sequencing reads contained untemplated adenosines at their 3′-ends (Schneider et al. 2012), so we checked whether this was also the case for human DIS3. Surprisingly, untemplated poly(A) sequences (from two to nine in a row) represented only 3.98% of all partially untemplated mapped reads in one replicate, and 6.31% in the second one, which represents 0.25% and 0.39% of all mapped reads, respectively. This strongly suggests that oligoadenylation is not a main targeting signal for human DIS3; therefore, it was not analyzed further.
DIS3 dysfunction causes the global accumulation of RNAs from unannotated parts of the genome, allowing for the identification of thousands of novel transcripts
To provide a general overview of the DIS3 RNA-seq and PAR-CLIP data, the sequencing reads were mapped to the reference human genome and divided into different categories of transcripts (Table 1). Importantly, in the case of PAR-CLIP, distribution of reads mapped to the genome between RNA classes was essentially the same for all reads and the ones containing T-C transitions (Supplemental Table 1), suggesting that reads without transitions are also specific. Several molecular phenotypes of DIS3 dysfunction were immediately visible.
The distribution of sequence reads over different classes of transcripts
The well-known exosome substrates accumulated robustly in DIS3 RNB and PIN RNB double mutants, but not in the PIN domain single-mutants (Table 1; Supplemental File 1). The accumulation of such RNAs usually correlated with a strong PAR-CLIP signal. Reads that mapped to the PROMPTs class of lncRNAs grew from 0.16% in control (WT) cells to 0.88% of all mapped reads in PIN RNB mutants. Moreover, PROMPTs represented as much as 8.29% of PAR-CLIP reads.
One of the prominent effects of DIS3 dysfunction was the accumulation of reads that mapped to regions of the human genome that lack any known annotation, increasing from ∼4% in wild-type controls to ∼9% in DIS3 double mutants. They also represented as much as 8% of all PAR-CLIP reads. Because the reads from unannotated parts of the genome were rather dispersed, to assemble these reads into novel transcripts, we increased the depth of sequencing of the RNA libraries prepared from HEK293 cells that expressed WT DIS3 and DIS3 PIN RNB double mutants. We identified hundreds of thousands of novel transcripts that did not overlap with any known transcripts (Supplemental File 2), some of which were validated using quantitative PCR (qPCR) (Fig. 1A,B; Supplemental Fig. 2). These transcripts cover as much as 25.2% of the genome, have median length of 1334 bp (80% have length between 649 bp and 3656 bp), and 92% of them are intronless.
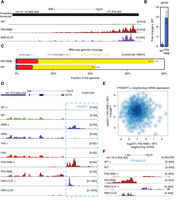
DIS3 inactivation leads to the accumulation of RNAs from unannotated parts of the genome and uncovers novel transcripts. (A) A genome browser screenshot of the genomic region encompassing one of the novel transcripts predicted by Cufflinks software with mapped RNA-seq reads for WT, PIN RNB double mutant, and DIS3 PAR-CLIP reads. The uniquely mapped reads are shown to indicate the minus strand. (B) Quantitative PCR validation of novel transcripts. Bars represent the standard deviation for three biological replicates. (C) The fraction of the genome covered by RNA-seq reads in WT and cell lines expressing the DIS3 PIN RNB double mutant. Coverage was measured by normalized read counts over each nucleotide. The sequencing depth for RNA-seq was ∼100 × 106 read pairs per sample. Bars represent the standard deviation for three biological replicates. (D) A genome browser screenshot of previously detected PROMPTs. RNA-seq reads mapped for WT, RNB, PIN, PIN RNB double mutants, and DIS3 PAR-CLIP. In D and F, uniquely mapped reads are visualized separately for the plus and minus strands. (E) The accumulation of PROMPTs does not correlate with the expression of neighboring genes. The relationship of expression fold changes between DIS3 WT and DIS3 PIN RNB double mutants for PROMPT and neighboring protein-coding transcripts, as shown on a logarithmic scale. (F) A genome browser screenshot of one of the enhancers accumulating eRNA in DIS3 double mutants. RNA-seq reads mapped for WT, PIN RNB double mutants, and DIS3 PAR-CLIP.
Finally, we calculated the percentage of nucleotides in the genome that were covered by at least one read in deeply sequenced libraries (Fig. 1C). The fraction of the genome covered increased from 43% (SD 1.99%) in WT to 74% (SD 2.52%) in the DIS3 double mutants, suggesting that the level of pervasive transcription initiation is much higher than previously anticipated, and DIS3 effectively degrades such unstable RNAs.
Impairment of DIS3 functions leads to global accumulation of PROMPTs and enhancer RNAs
The levels of several PROMPTs increased >50-fold in DIS3 mutant cells compared with WT cells (Supplemental Files 1, 3). We detected the accumulation of 66% of previously detected PROMPTs in DIS3 PIN RNB double mutants (68% in deeply sequenced libraries). Generally, the effect was not strongly enhanced in comparison to RNB domain single mutants, in which 60% of previously detected PROMPTs accumulated (Fig. 1D; Supplemental Figs. 2, 3). Interestingly, although PROMPTs accumulated robustly, there was no indication of their direct involvement in the regulation of the expression of neighboring genes because we observed no negative or positive correlation between the accumulation of PROMPTs and sense mRNA (Spearman correlation coefficient = 0.0431, P-value = 0.144) (Fig. 1E).
The exosome plays a prominent role in the decay of enhancer RNA (eRNA) molecules (Andersson et al. 2014). Unfortunately, the expression levels of eRNAs from a list of 35,265 possible enhancers were low in our system. We could detect statistically significant changes in the expression level of only 85 enhancers from that list in our deeply sequenced libraries (Fig. 1F; Supplemental Fig. 3), which probably reflects high cell-type specificity of enhancer RNA expression since Andersson and colleagues (Andersson et al. 2014) did not analyze HEK293 cells. In all cases, respective eRNAs accumulated in DIS3 double mutants compared with WT enhancers (Supplemental File 4; Andersson et al. 2014). The increase in the expression of those enhancers did not entail changes in the expression of the genes located in close proximity because 80% of neighboring genes showed no or very little expression. We observed increased or decreased expression for only 3.5% and 16.5% such genes, respectively. When the analysis was extended up to the end of chromatin domains marked by CTCF in case of six enhancers (7.06%), there was at least one gene with increased expression; whereas for 18 eRNAs (21.18%), at least one gene with decreased expression was identified. Three enhancers (3.53%) were linked to genes, which were either up-regulated or down-regulated.
DIS3 dysfunction results in the accumulation of NEAT1 lncRNA and paraspeckles
No evidence for the general accumulation of lncRNAs was observed, and only a few lncRNAs accumulated robustly (Supplemental Files 1, 3). One of the most interesting examples was NEAT1. There are two forms of this RNA species in human cells: a shorter one (NEAT1.1 of 3.7 kb) and a longer one (NEAT1.2 of ∼23 kb) (Naganuma et al. 2012). The latter transcript has a triple helix and the tRNA-like structures at the 3′-end, which are essential for its stability and processing respectively (Wilusz et al. 2008, 2012). We noted up to ∼eightfold accumulation of NEAT1.1 transcripts in the DIS3 mutants, whereas the steady state level of NEAT1.2 did not change significantly (Fig. 2A; Supplemental File 3). A prominent PAR-CLIP signal covering NEAT1.1 indicated that DIS3 is directly involved in promoting its degradation.
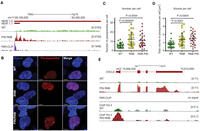
DIS3 dysfunction leads to NEAT1 lncRNA overexpression and the accumulation of paraspeckles. (A) A genome browser screenshot of the genomic region encompassing the NEAT1 locus with reads for mapped RNA-seq and DIS3 PAR-CLIP. (B) Fluorescent images of HEK293 cells transfected with constructs encoding the paraspeckle component NONO fused with mCherry. Nuclei were stained with Hoechst. White bars represent 5 µm. (C) The distribution of number of paraspeckles per cell in WT, RNB, and PIN RNB mutant cell populations. (D) Total volume of paraspeckles in a single cell, in WT, RNB, and PIN RNB mutant cell populations. (E) A genome browser screenshot of the genomic region encompassing the CXCL8 locus with mapped RNA-seq, RNA polymerase II ChIP-seq, and DIS3 PAR-CLIP reads (no signal). The mock control signals for Pol II ChIP are overlaid in scale on the respective tracks in gray color. Bars (C,D) represent the standard deviation from 28 measurements. P-values obtained using a t-test are shown. The uniquely mapped reads (A,E) are shown for the plus strand.
NEAT1.1 and NEAT1.2 form nuclear substructures termed paraspeckles (Fox and Lamond 2010). To analyze the influence of NEAT1 overexpression in DIS3 mutant cells on paraspeckle formation, we visualized these structures in cells by transfections with a construct expressing mCherry-labeled paraspeckle component, NONO (Fig. 2B). Indeed, both the number and volume of paraspeckles increased ∼twofold upon DIS3 dysfunction (Fig. 2C,D). The cellular role of paraspeckles remains to be well defined, but there are indications of their involvement in the regulation of gene expression through the sequestration of positive or negative transcription regulators. The enhanced formation of paraspeckles leads to the sequestration of SFPQ protein, which negatively regulates CXCL8 (formerly known as IL8) mRNA, resulting in its increased expression (Imamura et al. 2014). Interestingly, CXCL8 mRNA was overexpressed up to eightfold in our HEK293 mutant cells (Fig. 2E; Supplemental File 3). We performed chromatin immunoprecipitation (ChIP) for RNA polymerase II (Pol II) combined with massively parallel DNA sequencing experiments in WT and DIS3 PIN RNB double mutants cells, followed by analysis of signal distribution over the CXCL8 gene. This analysis revealed that the Pol II levels are 2.14 higher (P-value = 1.9 × 10−12) in the DIS3 mutant (Fig. 2E), strongly suggesting that CXCL8 up-regulation is a result of increased transcription.
DIS3 mutations abolishing its nucleolytic activity lead to global dysregulation of the expression of protein-coding genes
The DIS3 double mutant exhibited general deregulation of mRNA levels: 53.35% of protein-coding transcripts were down-regulated and 4.09% were up-regulated; and in deeply sequenced libraries, these values increased to 61.47% and 5.59%, respectively (Fig. 3A; Supplemental Files 1, 3). Such general down-regulation of mRNA levels in DIS3 double mutants might be considered as an artifact of normalization, resulting from the general up-regulation of unstable RNAs, because the data were normalized to all mapped reads. If normalization was performed to consider reads that mapped exclusively to annotated genes, the fractions of down-regulated and up-regulated genes were comparable (29.65% and 25.28%, respectively) but still represented ∼50% of protein-coding genes expressed. Notably, individual catalytic mutations within the RNB or PIN domains led to less pronounced changes in expression of mRNA (16.43% or 6.44%, respectively). For the majority of up-regulated or down-regulated genes in DIS3 mutant cells, the expression level changed less than two- to fourfold, but there are examples, like TNFRSF9 (Fig. 3B,C) or CXCL8, which are highly overexpressed. Importantly, very little correlation between the up-regulation of mRNAs and the corresponding DIS3 PAR-CLIP signal (Spearman correlation coefficient = 0.291, P-value = 1.110 × 10−83) suggests that the observed effects were secondary.
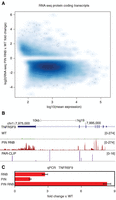
Global deregulation of the expression of protein-coding genes caused by DIS3 inactivation. (A) A volcano plot showing a large number of up-regulated and down-regulated transcripts. (B) A genome browser screenshot of TNFRSF9 mRNA transcripts, which are up-regulated in a cell line expressing the DIS3 PIN RNB double mutant. The RNA-seq signal was not supported by the PAR-CLIP signal. The uniquely mapped reads are shown for the minus strand. (C) Quantitative PCR validation of TNFRSF9 up-regulation in double mutants. Bars represent the standard deviation of three biological replicates.
Premature termination of protein-coding genes is a widespread phenomenon
An analysis of reads that mapped to known introns of protein-coding genes that did not contain snoRNA or any other genomic feature (e.g., an exon of another transcript) showed that there was no general trend for intron accumulation, indicating that DIS3 (and probably the exosome) is not a major intron-degrading enzyme (Table 1). However, a more careful inspection of intronic reads showed that in multiple cases, there was an overrepresentation of reads that mapped to the first intron of a gene (but not to a second intron) in DIS3 mutants (Fig. 4A; Supplemental Fig. 4). For each deeply sequenced library, we counted reads that overlapped exon1/intron1 junction and intron1/exon2 junction in protein-coding transcripts, and then the fold changes between those counts were calculated. For 11.88% of transcripts, this fold change was larger in DIS3 double mutants than in wild-type cells, whereas only 1.54% cases showed a change in the opposite direction. For the remaining 86.58% of transcripts, there was no statistically significant difference between the two cell lines (t-test, Benjamini–Hochberg corrected P-value <0.05) (Fig. 4B; Benjamini and Hochberg 1995). These results suggest that such accumulation of the first intron represents unprocessed transcripts that most likely arise from premature transcription termination events within the gene body. Such transcripts accumulated in DIS3 double mutants in ∼12% of transcripts that contained at least two exons. We analyzed whether the intron length has an effect on premature termination frequency. Indeed, we observed that the incidence of this phenomenon was slightly more pronounced for longer first introns, nevertheless the effect was relatively mild. Namely, for 5% of the longest first introns, the ∼17% were increased in comparison to ∼11.5% for the remaining ones. It is possible that the premature termination reported herein is not restricted to introns and may also take place within the intronless protein-coding genes; but due to high background of reads originating form mature mRNAs, such cases were more difficult to detect.
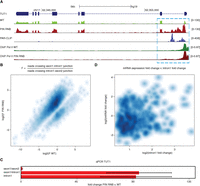
DIS3 degrades transcripts that arise from the premature termination of pre-mRNA transcripts. (A) A genome browser screenshot of the TUT1 gene, which is an example of the accumulation of prematurely terminated transcripts. The RNA-seq signal is supported by the PAR-CLIP and RNA polymerase II ChIP-seq signal. The mock control signals for Pol II ChIP, barely visible due to relatively low signal, are overlaid in scale on the respective tracks in gray color. (B) A higher number of fragments that overlap with the exon1/intron1 junction than the intron1/exon2 junction in DIS3 double mutant by RNA-seq. Transcripts that showed no change, defined here as the quotient between PIN RNB double mutants and WT, are not included in this plot. (C) Quantitative PCR validation of increases in levels of transcripts that cross the exon1/intron1 junction for the TUT1 gene. Bars represent the standard deviation for three biological replicates. (D) Lack of correlation between mRNA and intron 1 expression fold changes in DIS3 double mutants. The Spearman correlation coefficient = 0.0879, and the P-value = 0.0207. Only those mRNA transcripts that showed an increase in intron 1, but not in intron 2, were included in the analysis.
Next, we performed qPCR validation for several selected examples, which showed that the unspliced, prematurely terminated RNAs indeed accumulated in cells that expressed DIS3 mutant variants (Fig. 4C; Supplemental Fig. 4). Finally, we looked at the Pol II ChIP signal over relevant genes with a high level of putative premature termination. The most significant ChIP peaks corresponded to the promotor regions, but the drop of polymerase occupancy was visible in the regions of putative premature termination, where the RNA-seq signal decreases in DIS3 mutant cells (Fig. 4A; Supplemental Fig. 4). There was basically no difference in the ChIP signal from WT and mutant cells, indicating that indeed RNA species observed by us arise from premature termination. Nevertheless, there was no apparent correlation between the accumulation of putative premature termination products and the expression levels of the mature mRNAs (Fig. 4D).
DIS3, but not EXOSC10, is involved in snoRNA processing
Most snoRNA in human cells are encoded within introns of abundant mRNAs. In the RNA-seq analysis of DIS3 mutant cells, there was an accumulation of snoRNA precursors (2.12% of reads in double mutant versus 1.18% in WT controls) (Table 1), and they represented a significant fraction of the PAR-CLIP signal (∼2%) (Fig. 5A). Although the levels of mature snoRNAs in cells are orders of magnitude higher than for the precursors, they represented a much smaller fraction of the PAR-CLIP reads (0.17% and 0.29% in the first and the second replicate, respectively), which indicates that DIS3 is involved in snoRNA processing rather than decay. Indeed, by Northern blot analyses, we observed a significant accumulation of snoRNA precursors in DIS3 mutant cells (Fig. 5B). In contrast to PROMPTs, the accumulation of snoRNA precursors increased significantly from DIS3 single to double mutants (from 1.35% in RNB mutant and 1.34% in PIN mutants to 2.12% in PIN RNB double mutants), indicating that endonucleolytic DIS3 activity plays a role in snoRNA processing (Fig. 5B). In yeast, in addition to Dis3, Rrp6 also plays a prominent role in snoRNA processing (Allmang et al. 1999). To assess the relative contributions of human EXOSC10 and DIS3 to this process, we generated HEK293 cell lines with silenced endogenous EXOSC10 and exogenously produced EXOSC10 WT or catalytic mutant, similarly to the DIS3 model cell lines. This experimental system worked efficiently, because endogenous protein expression was reduced by 80% (data not shown), and we were able to note the accumulation of 21S precursors of 18S rRNA in cells that synthesized the mutant variant of this protein, which is a typical phenotype for EXOSC10 dysfunction (Supplemental Fig. 5; Preti et al. 2013; Sloan et al. 2013). We compared the effects of EXOSC10 and DIS3 mutations on snoRNA processing (Fig. 5B). Interestingly, unlike in the case of DIS3, we were unable to observe the accumulation of snoRNA precursors upon EXOSC10 inactivation (Fig. 5B). The only effect that we could observe was a slight increase in the levels of mature transcripts.
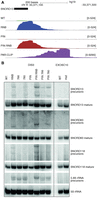
DIS3, but not EXOSC10, is involved in snoRNA processing. (A) A genome browser screenshot of the genomic region encompassing SNORD13 snoRNA with mapped RNA-seq and DIS3 PAR-CLIP reads. The uniquely mapped reads are shown for the plus strand. Note that mature snoRNA are underrepresented in the long read RNA-seq libraries. (B) Northern blot analysis of selected snoRNAs, using cell lines with DIS3 and EXOSC10 mutations. Multiple myeloma-associated mutations are represented by 766 and 780 for DIS3 G766R and DIS3 R780G, respectively (Tomecki et al. 2014).
Discussion
Our analysis of the effects of DIS3 dysfunction showed a generally high level of transcription in non-protein-coding regions of the human genome and revealed the robustness of the exosome-mediated nuclear quality control pathways. Moreover, the accumulation of dispersed RNA fragments strongly suggests that pervasively occurring transcriptional initiation is a common phenomenon in human cells, which was previously questioned (van Bakel et al. 2010; Clark et al. 2011).
The putative premature termination products of protein-coding genes and PROMPTs represent the most prominent targets of DIS3 (Fig. 6), indicating that there are basically no alternative pathways for their decay. The absence of a direct correlation between the accumulation of noncoding RNA species that originate from bidirectional transcription in the direction opposite to protein-coding transcripts with levels of respective mRNAs strongly suggests that the majority of PROMPTs lack a general regulatory role. Likewise, the accumulation of products of widespread premature termination for protein-coding genes, which we detected in this present study, does not have a large influence on mRNA accumulation. All these data suggest that in the majority of cases, such unwanted transcripts do not act in cis to regulate gene expression. In case of enhancer RNAs, for which we also did not observe a strong effect on neighboring gene expression, it is possible that transcription itself, rather than the resulting RNA products, has an impact on gene activation; but due to the relatively low number of enhancer RNA analyzed in this study, we cannot exclude that in specific cases such RNAs play a regulatory role, as was suggested previously (Melo et al. 2013; Schaukowitch et al. 2014).
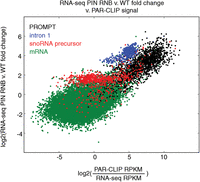
The relationship between PAR-CLIP signals and mRNA, PROMPTs, snoRNA precursors, and intron 1 expression changes in DIS3 double mutants. The PAR-CLIP signal was defined as PAR-CLIP RPKM divided by RNA-seq RPKM in WT cells. Transcripts with no PAR-CLIP signal were not included in this plot. Data for the first intron are only from transcripts that show up-regulation of the first intron in double mutants but not for the second intron.
The global deregulation of gene expression in DIS3 mutant cells most likely represents a secondary effect that results from the accumulation of nuclear RNAs, which might sequester factors that are involved in mRNA biogenesis, like NEAT1 lncRNA and CXCL8 mRNA. Such a regulatory mechanism might be more common, however difficult to identify, because of the complexity of the molecular phenotypes in DIS3 mutant cells.
DIS3 targets snoRNA precursors; however, the fact that the level of mature snoRNA is not decreased in DIS3 mutant cells suggests that there are also other nucleases that participate in snoRNA processing. In contrast to DIS3, nucleolus-enriched EXOSC10 appears to degrade mature snoRNA. In some cases, the level of mature snoRNA in DIS3 mutants is also slightly increased (Fig. 5B), suggesting that DIS3 could be involved in the degradation of mature species, but very low DIS3 PAR-CLIP signal over mature snoRNAs argues against such a possibility. The different roles of DIS3 and EXOSC10 in snoRNA metabolism underscore the strong functional specialization between different catalytic subunits of the human nuclear exosome, which appears to be less prevalent in yeast.
RNA polymerase I and III products represent more than half of Dis3 substrates in yeast; whereas in humans, they are a minority of DIS3 targets (Table 1; Gudipati et al. 2012; Schneider et al. 2012). However, there are exceptions, such as 5.8S rRNA 3′-extended precursors that are processed by DIS3 proteins both in humans and yeast (Dziembowski et al. 2007; Tomecki et al. 2010, 2014). Interestingly, although the precursors are transcribed by RNA polymerase I as a part of 47S pre-rRNAs, they are processed by DIS3 rather than EXOSC10, most likely because this late rRNA processing step also occurs in the nucleoplasm rather than in the nucleolus (Thomson and Tollervey 2010). Our PAR-CLIP data also suggest that—in contrast to yeast in which Dis3 controls tRNA levels—tRNAs are not the major targets of human DIS3. Again, this finding might reflect differences in localization patterns because in yeast, Dis3 is present in the cytoplasm where the turnover of tRNAs occurs.
An outstanding question that needs to be answered is how DIS3 and the exosome can distinguish between its targets and stable, mature RNAs, because both species have a similar architecture in humans because both PROMPTs and mRNAs contain cap and poly(A) tails (Preker et al. 2011). Premature termination products are also expected to contain poly(A) tails, since they most probably represent molecules that evaded protection by U1 snRNP and are generated by a canonical termination pathway (Kaida et al. 2010; Berg et al. 2012; Almada et al. 2013). In contrast to mRNAs, direct DIS3 RNA targets are generally short intronless molecules, and we hypothesize that this might be one of the main distinctions between stable and unstable RNA polymerase II transcripts in human cells. In the case of mRNA molecules or cytoplasmic lncRNAs, splicing can enhance RNP formation and nuclear RNA export, which would help to evade the DIS3 activity. Notably, however, we could not detect a statistically significant difference in the sensitivity of multiexonic or intronless mRNA and lncRNA to DIS3-mediated degradation. This phenomenon might be related to the existence of a stabilization mechanism specific for single-exon short functional RNAs, as suggested previously (Andersen et al. 2012). The mechanism of recognition of unstable RNAs in human cells is different from the one in yeast, in which the specific transcription termination mechanism is linked to noncanonical polyadenylation (Vasiljeva et al. 2008; Wlotzka et al. 2011; Tudek et al. 2014). A dedicated exosome activation complex, TRAMP, oligoadenylates exosome substrates, thereby promoting their decay (LaCava et al. 2005; Vanácová et al. 2005; Wyers et al. 2005). A similar complex found in humans is enriched in nucleoli, and thus plays a minor (if any) role in exosome-mediated decay in the nucleoplasm (Lubas et al. 2011). The cofactor of the human nucleoplasmic exosome is the NEXT complex, which is composed of the RNA helicase SKIV2L2 and two RNA-binding proteins, and interacts with newly synthetized RNAs (Lubas et al. 2011, 2015). How this complex distinguishes between stable and unstable RNAs remains to be determined, although there are indications for its cooperation with the cap-binding complex, which suggest that the distance from the RNA 5′-end could play a role in directing exosome-mediated 3′-5′ RNA decay (Andersen et al. 2013; Hallais et al. 2013). It has also been suggested that in humans, nuclear poly(A)-binding protein promotes the hyperadenylation and decay of unstable transcripts, including the short form of NEAT1 (Bresson and Conrad 2013). Whether and how hyperadenylation can induce exosome-mediated decay is also unknown and will require further study.
In conclusion, our present study describes the repertoire of various DIS3-dependent human nucleoplasmic exosome substrates and underscores the importance of efficient quality control mechanisms that ensure that all pervasive transcription products and other unwanted RNA molecules, which were detected at surprisingly high levels, are quickly degraded.
Methods
RNA-seq
HEK293 Flp-In T-REx cell lines were grown, and RNA was isolated as described previously (Tomecki et al. 2014). RNA was ribodepleted, and strand-specific libraries were prepared using a slightly modified standard protocol (see Supplemental Data for details; Supplemental Fig. 6). RNA libraries were sequenced using an Illumina HiSeq 1500 sequencing system in paired-end mode. Sequencing reads were preprocessed by cutadapt-1.2.1 (Martin 2011) to remove reads shorter than 12 nt, those with quality lower than a score of 20, and to cut adapters. Reads were then mapped to the human genome (hg19) using RUMv2.05_05 (Grant et al. 2011), which was the most recent assembly at the time the analyses were initiated. Since most of the conclusions are of general genome-wide nature, it is highly unlikely that they should be affected by an analysis performed on a different genome assembly. The program requires left and right reads to be the same length, so the reads had been stripped adequately. Information about the known transcripts was supplied to the program. It contained: (1) GENCODE v18 transcript data; (2) a supplement to GENCODE v18 data tRNA coordinates from the tRNA scan program; (3) previously published PROMPT data, which is a set of 3-kb upstream regions from the selected 2471 genes; and (4) intronic snoRNA precursors defined as the regions from the end of the snoRNA to the end of the intron. RNA-seq experiments were performed in triplicate. Following sequencing and quality filtering for RNA-seq samples, we obtained an average of 16.34 (SD 1.27) million read pairs per library that map to the human reference genome. Deeply sequenced libraries for WT and PIN RNB double mutant cells had an average of 101.74 (SD 9.64) million read pairs per library. Read counts from the samples were normalized to the average number of all mapped read pairs for the first sequenced libraries and deeper sequenced libraries separately. Only uniquely mapped reads were counted for further analyses.
PAR-CLIP
The PAR-CLIP procedure was performed using HEK293 Flp-In T-REx cell lines producing DIS3-eGFP fusion protein similarly to Hafner et al. (2010), with some modifications. Briefly, cells were grown in the presence of thiouridine (4-sU) and irradiated with 365 nm UV light. Proteins cross-linked to RNA were then immunoprecipitated with anti-GFP resin, followed by RNA labeling, SDS-PAGE, and transfer to the nitrocellulose membrane. Finally, the isolated RNA was converted into a cDNA library and subjected to deep sequencing. For a more detailed procedure, please see the Supplemental Data and Supplemental Figure 7. PAR-CLIP was carried out in two biological replicates. We obtained 8.89 and 5.79 million read pairs that map to the human reference genome. The sequencing data analysis was the same as for RNA-seq data. All uniquely mapped reads were counted for further analyses. Moreover, the percentage of reads with T-C transitions was investigated. We counted reads with T-C transitions together with those that are pairs of such reads.
ChIP analysis
ChIP experiments for RNA polymerase II were performed with MNase treatment and anti-RNA Polymerase II antibody (clone 8WG16, BioLegend). The protocol was based on X-ChIP protocol from Abcam (http://www.abcam.com/protocols/). For detailed procedure, please refer to Supplemental Data. ChIP-seq libraries were prepared with KAPA Hyper Prep Kit (Kapa Biosystems) in three biological replicates for WT and DIS3 double mutant with their respective controls, consisted of fragments that were on average 160 bp in length and were sequenced to the mean depth of 110.5 million single-end 75-bp-long reads. Reads were mapped to the human reference genome hg19 (GRCh37) using the short read aligner program t-test 2 (version 2.2.5) using default settings (Langmead and Salzberg 2012). On average, 69.2% of ChIP reads and 60.9% of mock control reads mapped uniquely, and only those were further analyzed using custom scripts implementing elements of the SAMtools (version 0.1.19 and 1.2.1), BEDTools (version 2.23.0), and RseQC (version 2.6) packages for basic quality control, duplicate read removal, filtering, extending, and counting reads (Li et al. 2009; Quinlan and Hall 2010). The reads mapping within gene bodies were counted and normalized to the total number of reads, and the statistical analysis using the DESeq2 Bioconductor package was performed (Love et al. 2014).
Quantitative PCR (qPCR) validation, Northern blotting, and other standard methods
Standard methods were used for RNA isolation, cDNA preparation, qPCR, and Northern blotting. The vectors and cell lines were produced in similar way as described previously (Tomecki et al. 2014). Refer to Supplemental Data for details.
Paraspeckles analysis
Paraspeckles were visualized by transient transfection with the construct coding for NONO fused with mCherry. Fixed cells were imaged with a FluoView 1000 confocal system (Olympus) using a PLANAPO 60.0 × 1.40 oil objective. Images were 3D rendered and analyzed using Imaris 7.2.3 software (Bitplane). For a more detailed procedure, see Supplemental Data.
Analysis of nontemplated oligoA
In PAR-CLIP data, we counted the number of partially mapped second reads with untemplated A (from two to nine in a row). We calculated the percentage of these reads in all mapped second reads.
Genome Browser visualization tracks
The UCSC Genome Browser was used for visualization of sequencing data (Kent et al. 2002). Numbers in visualization tracks represent the normalized mean read count values from the three repeats of a given track for RNA-seq samples and the values from one repeat for PAR-CLIP samples (the library with ∼8 × 106 read pairs). When only WT and PIN RNB double mutant tracks of RNA-seq are presented, the data is from deeply sequenced libraries. For the Pol II ChIP experiment, the summarized data from the three replicates was visualized after extending the reads to the expected fragment length and normalizing the signal to total library size of 10 million reads. The mock control signal is overlaid in scale on the same track as the Pol II ChIP signal but in gray color.
Distribution of reads over different classes of transcripts
To calculate this distribution, we uniquely assigned reads to classes using a hierarchical procedure. Reads that mapped to more than one class of transcript annotated in the genome were assigned to a class that was higher in the hierarchy. Reads were assigned ignoring strand specificity. We did not want to overestimate the number of reads mapped to unannotated parts of the genome because of inaccurate strand specificity of the sequencing technique. As defined by us, the hierarchy of transcript classes is included in Supplemental Data. Calculation of the distribution using uniquely versus nonuniquely mapped reads yielded nearly identical results.
Differential expression analysis
We used the DESeq2 R package (Love et al. 2014) for differential expression analysis of annotated transcripts, filtered out transcripts with very low expression, and required at least one count per million mapped reads in at least two probes.
Assembly of new transcripts
We used reads from deeply sequenced libraries from PIN RNB double mutants to assemble new transcripts. We used Cufflinks v2.1.1 (Trapnell et al. 2010) to assemble reads in each from three replicates separately. We applied all default parameters, with the exception of increasing “–max-bundle-length” to 107 and “–max-bundle-frags” to 107. We used cuffmerge from the Cufflinks package to merge assemblies from the replicates. New transcripts were only counted as those transcripts that did not overlap with any other known transcript from the annotation. The genome coverage by the new transcripts was calculated without introns predicted in those transcripts. Including intronic positions, new transcripts covered 27.1% of the genome. Transcript length was measured from genomic start till genomic end of a transcript.
Genome coverage
Each deeply sequenced library was normalized to 100 × 106 read pairs per sample. BEDTools “genomecov” output tracks were scaled based on this normalization. The percentage of nucleotides covered by at least 100, 10, or 1 read was calculated.
eRNA analysis
We defined enhancers as 500 nt from the middle point on the forward strand and 500 nt from the middle point on the reverse strand from the list of 35,265 probable enhancers (Andersson et al. 2014). To exclude from our analysis those enhancers that did not show bidirectional expression, we applied a “directionality” parameter defined by Andersson et al. (2014) and a “strandedness” parameter that was defined by us. Enhancer directionality was calculated as (F − R)/(F + R), where F and R is the sum of the RNA-seq fragments aligned on the forward and reverse strands. Directionality close to −1 or 1 indicates a unidirectional behavior, whereas 0 indicates perfectly balanced bidirectional transcription (Andersson et al. 2014). Enhancer strandedness was defined as (S − A)/(S + A), where S is the sum of fragments mapped to the proper forward and reverse strand of an enhancer (i.e., the region from the middle point to the middle point plus 500 nt on the forward strand, and the region from the middle point to the middle point minus 500 nt on the reverse strand), and A is the sum of fragments mapped to the antisense strand of the enhancer proper forward and reverse (i.e., the region from the middle point to the middle point minus 500 nt on forward strand, and the region from the middle point to the middle point plus 500 nt on the reverse strand). We counted as proper, expressed enhancers only those regions with an absolute value of directionality less than 0.5 and an absolute value of strandedness greater than 0.5.
We defined genes located in close proximity to the enhancers as (1) neighboring genes—one gene for each enhancer that has closest transcription start; or (2) genes located within chromatin domain marked by CTCF sites. We took CTCF site positions for HEK293 cells from Euskirchen et al. (2007).
Data access
PAR-CLIP, RNA-seq, and Pol II ChIP-seq data from this study have been submitted to the NCBI Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/) (Edgar et al. 2002) under accession number GSE64332.
Acknowledgments
We acknowledge Joanna Kufel and Roman Szczesny for critical reading of this manuscript. The work was supported by the Polish–Swiss Research Programme (PSPB-183/2010), the National Science Centre (NCN Maestro: UMO-2011/02/A/NZ1/00001), The National Centre for Research and Development (NCBR LIDER/35/46/L-3/11/NCBR/2012), and The European Social Fund (373/ES/ZS-III/W-POKL/14). Experiments were carried out with the use of CePT infrastructure that was financed by the European Union via the European Regional Development Fund (Innovative economy 2007–13, Agreement POIG.02.02.00-14-024/08-00).
Author contributions: A.D. conceived and directed the studies. T.S. performed bioinformatics analyses. K.K. performed PAR-CLIP experiments and participated in validation experiments. R.T. generated the model cell lines, analyzed snoRNA processing, and participated in the design of experiments. A.L. established model cell lines for EXOSC10 analysis and performed validation experiments. D.A. prepared RNA-seq libraries. T.M.K. participated in bioinformatics analyses. J.K. provided access to the sequencing facility. L.S.B. performed paraspeckle analysis and ChIP-seq experiments. A.D. wrote the paper with contributions from R.T. and T.S.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.189597.115.
- Received January 16, 2015.
- Accepted July 16, 2015.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see http://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








