
A genomic portrait of the genetic architecture and regulatory impact of microRNA expression in response to infection
- Katherine J. Siddle1,2,11,
- Matthieu Deschamps1,2,3,11,
- Ludovic Tailleux4,
- Yohann Nédélec5,6,
- Julien Pothlichet1,2,
- Geanncarlo Lugo-Villarino7,8,
- Valentina Libri9,
- Brigitte Gicquel4,
- Olivier Neyrolles7,8,
- Guillaume Laval1,2,
- Etienne Patin1,2,
- Luis B. Barreiro6,10 and
- Lluís Quintana-Murci1,2,12
- 1Institut Pasteur, Unit of Human Evolutionary Genetics, 75015 Paris, France;
- 2Centre National de la Recherche Scientifique, CNRS URA3012, 75015 Paris, France;
- 3Université Pierre et Marie Curie, Cellule Pasteur UPMC, 75015 Paris, France;
- 4Institut Pasteur, Unit of Mycobacterial Genetics, 75015 Paris, France;
- 5Department of Biochemistry, Faculty of Medicine, University of Montréal, Montréal H3T 1C5, Canada;
- 6Ste-Justine Hospital Research Centre, Montréal H3T 1C5, Canada;
- 7Centre National de la Recherche Scientifique, Institut de Pharmacologie et de Biologie Structurale, 31077 Toulouse, France;
- 8Université de Toulouse, Université Paul Sabatier, Institut de Pharmacologie et de Biologie Structurale, 31077 Toulouse, France;
- 9Institut Pasteur, Centre d’Immunologie Humaine, 75015 Paris, France;
- 10Department of Paediatrics, Faculty of Medicine, University of Montréal, Montréal H3T 1C5, Canada
-
↵11 These authors contributed equally to this work.
Abstract
MicroRNAs (miRNAs) are critical regulators of gene expression, and their role in a wide variety of biological processes, including host antimicrobial defense, is increasingly well described. Consistent with their diverse functional effects, miRNA expression is highly context dependent and shows marked changes upon cellular activation. However, the genetic control of miRNA expression in response to external stimuli and the impact of such perturbations on miRNA-mediated regulatory networks at the population level remain to be determined. Here we assessed changes in miRNA expression upon Mycobacterium tuberculosis infection and mapped expression quantitative trait loci (eQTL) in dendritic cells from a panel of healthy individuals. Genome-wide expression profiling revealed that ∼40% of miRNAs are differentially expressed upon infection. We find that the expression of 3% of miRNAs is controlled by proximate genetic factors, which are enriched in a promoter-specific histone modification associated with active transcription. Notably, we identify two infection-specific response eQTLs, for miR-326 and miR-1260, providing an initial assessment of the impact of genotype-environment interactions on miRNA molecular phenotypes. Furthermore, we show that infection coincides with a marked remodeling of the genome-wide relationships between miRNA and mRNA expression levels. This observation, supplemented by experimental data using the model of miR-29a, sheds light on the role of a set of miRNAs in cellular responses to infection. Collectively, this study increases our understanding of the genetic architecture of miRNA expression in response to infection, and highlights the wide-reaching impact of altering miRNA expression on the transcriptional landscape of a cell.
The responses of host immune cells to microbial stimuli are accompanied by marked changes in gene expression, with transcriptional programs that can be shared among different microbes or be specific to each (Huang et al. 2001; Amit et al. 2009; Chevrier et al. 2011; Gat-Viks et al. 2013). The regulatory networks that control the initiation, peak magnitude, and resolution of host responses must all be properly tuned to achieve optimal immunity. In this context, microRNAs (miRNAs), a group of endogenous small RNAs (∼22 nt), play a critical role in the epigenetic regulation of gene expression in eukaryotes (Ambros 2004; Bartel 2004). MiRNAs bind complementary sequences of target mRNAs to promote translational repression and/or mRNA degradation (Guo et al. 2010; Huntzinger and Izaurralde 2011). For an individual target, miRNAs have only subtle regulatory effects (Hornstein and Shomron 2006; Baek et al. 2008; Selbach et al. 2008), though a single miRNA may target over 100 genes. Over 60% of human genes are expected to be directly regulated by miRNAs (Friedman et al. 2009), with many predicted to be cotargeted by multiple miRNAs (Krek et al. 2005; Stark et al. 2005; Tsang et al. 2010). The importance of such complex and tightly regulated miRNA–mRNA networks is highlighted by the strong evolutionary constraints acting on both miRNAs and mRNA target sites, across species and within humans (Chen and Rajewsky 2006; Saunders et al. 2007; Friedman et al. 2009; Quach et al. 2009; Christodoulou et al. 2010; Berezikov 2011).
In addition to their involvement in a wide range of biological processes, including development, cell differentiation, and apoptosis, the role of miRNAs in regulating mammalian immune systems is increasingly well established (Lodish et al. 2008; O’Connell et al. 2012). MiRNAs regulate the development and function of cells of innate and adaptive immunity (Chen et al. 2004; Johnnidis et al. 2008; Lodish et al. 2008; O’Connell et al. 2012), and can have either pro-inflammatory or anti-inflammatory effects, indicating that the immune system utilizes multiple miRNAs to balance its response (O’Connell et al. 2012). Furthermore, experimental data indicate that viral, parasitic, and bacterial pathogens induce marked changes in miRNA expression in host cells (Cullen 2011; Eulalio et al. 2012). For example, activation of the innate immunity Toll-like receptor (TLR) pathway influences the expression of a well-defined group of miRNAs, while miRNAs can also regulate TLR expression as well as that of TLR signaling molecules, transcription factors, and cytokines (O’Neill et al. 2011).
There is growing evidence indicating that there is strong variation in miRNA expression in human populations, within a given cellular state, cell type, or tissue (Wang et al. 2009; Huang et al. 2011; Lu and Clark 2012; Lappalainen et al. 2013). The extent to which this variation is under genetic control (i.e., miRNA expression quantitative trait loci, miR-eQTLs) has recently begun to be investigated (Borel et al. 2011; Rantalainen et al. 2011; Gamazon et al. 2012; Parts et al. 2012; Civelek et al. 2013; Gamazon et al. 2013; Lappalainen et al. 2013). However, as has been shown for mRNAs in yeast and mammals, genetic variants can differentially affect gene expression after perturbation by various treatments or environmental variables (i.e., response eQTLs) (Gargalovic et al. 2006; Smith and Kruglyak 2008; Smirnov et al. 2009; Yang et al. 2009; Dombroski et al. 2010; Romanoski et al. 2010; Maranville et al. 2011; Barreiro et al. 2012). In humans, recent studies of protein-coding gene expression have identified response eQTLs related to oxidative stress (Romanoski et al. 2010), ionizing radiation (Smirnov et al. 2009), drug treatment (Maranville et al. 2011), and infection (Barreiro et al. 2012; Idaghdour et al. 2012). Conversely, as the few miR-eQTL studies to date have all used steady-state expression measurements, the degree of population variation in miRNA expression upon external stimulation, and the extent to which gene-environment interactions may affect the regulation of miRNA responses, remain to be determined.
Here, we aimed to dissect the genetic architecture and regulatory impact of miRNA expression in response to an external, infectious stimulus. To do so, we first characterized the population variation of miRNA transcriptional responses to infection using, as a model, Mycobacterium tuberculosis (MTB) infection of human dendritic cells (DCs). We then investigated the extent to which miRNA expression variation upon infection is under genetic control, providing the first attempt to map response miR-eQTLs. We next explored the relationship between miRNA and mRNA expression levels to understand how infection not only affects miRNA responses but also impacts upon broader miRNA-mediated regulatory networks. Finally, we performed miRNA gain- and loss-of-function experiments to assess the impact of altered miRNA expression on downstream transcriptional and protein responses to infection.
Results
Genomic characterization of miRNA transcriptional responses to infection
We profiled genome-wide patterns of miRNA expression in monocyte-derived DCs, untreated and after infection with a virulent strain of MTB, from a panel of 65 healthy individuals of European descent. The presence of an infection-related response in these samples has been previously confirmed at the mRNA level, by the altered expression of genes involved in immune responses, and at the protein level, by the strong induction of cytokines known to play a critical role in protective immunity against tuberculosis (TB) (Barreiro et al. 2012). After quality checks and normalization of the data, we assessed differences in miRNA expression levels upon infection using a final set of 346 miRNAs from 63 individuals (Supplemental Fig. S1; Supplemental Methods). We found that 155 miRNAs were differentially expressed upon infection (P < 1 × 10−5; Bonferroni-corrected P < 0.01), of which 64 were up-regulated and 91 down-regulated (Fig. 1; Supplemental Table S1). Among these, down-regulated miRNAs exhibited lower fold changes than those that were up-regulated, with only three (3%), compared with 20 (31%), showing at least a twofold difference in expression levels upon infection (Fig. 1). These maximal fold changes are markedly smaller than those observed for protein-coding genes in the same system (Barreiro et al. 2012), consistent with previous observations (e.g., Sharbati et al. 2011).
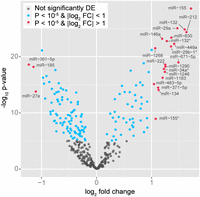
Changes in miRNA expression levels upon infection. Volcano plot showing the differential expression of miRNAs in DCs upon infection with MTB. Red dots denote significantly differentially expressed miRNAs whose expression changed by more than twofold following infection, whereas blue dots represent significantly differentially expressed miRNAs whose fold change (FC) was less than two. (DE) Differentially expressed.
The most differentially expressed miRNAs upon infection include, among others, the up-regulated miR-155, miR-132, and miR-29a, and the down-regulated miR-361-5p, miR-185, and miR-27a. These miRNAs are involved in the modulation of immune functions, such as the activation of core signaling pathways or the response to bacterial infections (O’Neill et al. 2011; Eulalio et al. 2012; Qi et al. 2012). More generally, although we found a substantial overlap between the 40% of differentially expressed miRNAs in our study and previous analyses using similar settings (Ceppi et al. 2009; Liu et al. 2009; Fu et al. 2011; Sharbati et al. 2011; Maertzdorf et al. 2012; Yi et al. 2012), we also identified distinctive signatures for some miRNAs in our model. These include the down-regulation of miR-125b and members of the miR-148 family, which have been reported to be up-regulated upon MTB infection of macrophages (Rajaram et al. 2011) and activation of DCs with lipopolysaccharide (Liu et al. 2010), respectively. Additionally, we identified previously unreported miRNAs, such as miR-630 and miR-339-3p, as being differentially expressed upon infection. Collectively, our results are consistent with a general pro-inflammatory response (Turner et al. 2011), together with putatively cell or stimulus-dependent responses that could impact the ways in which infection is established and maintained.
Genetic regulation of miRNA expression upon infection
To identify genetic variants that affect the response to MTB infection, we mapped miR-eQTLs by testing the association between miRNA expression profiles and genome-wide genotyping data from the same 63 individuals (Methods). This sample size affords sufficient power to detect eQTLs, even in some cases where genetic variation has a moderate impact on expression levels (Supplemental Fig. S2; Supplemental Methods). To map cis-acting variants, we focused on SNPs located within a 200-kb window centered on the mature miRNA, and analyzed the data separately for non-infected and infected samples to discern miR-eQTLs that are shared between conditions from those that are unique to a particular state. In total, we identified miR-eQTLs for six miRNAs (miR-326, miR-338-3p, miR-451, miR-1260, miR-769-5p, and miR-130b) in infected samples, of which one (miR-338-3p) was also significant in non-infected samples, at a false discovery rate (FDR) of 0.2 (Table 1; Supplemental Table S2). These associations accounted for 20%–50% of the variance in the expression of these miRNAs.
miR-eQTLs identified in cis for miRNA expression variation in non-infected and/or MTB-infected samples
Despite the fact that most of these miR-eQTLs displayed a significant association only in infected samples, we observed similar tendencies in the effect of the genotype on miRNA expression before and after infection (Supplemental Fig. S3). To identify response eQTLs, where a genetic variant has a stimulus-specific impact on transcript abundance, we focused on miR-eQTLs detected in one condition that had no observed effect in the other condition (P > 0.05). Using this threshold, we detected two putative response miR-eQTLs for miR-326 and miR-1260 (Fig. 2A). Using BRIdGE, a Bayesian approach for identifying genetic associations under different models of gene-environment interactions (Maranville et al. 2011), we confirmed five of the six associations detected, including the two response miR-eQTLs, at a posterior probability >0.7 (FDR = 0.15; Supplemental Table S3). This approach further enabled us to identify a general interaction effect for miR-338-3p, where the miR-eQTL had a different effect in each condition (Supplemental Fig. S3).
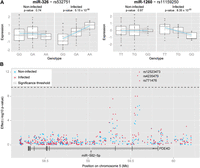
MiR-eQTLs upon infection. (A) Boxplots showing the detected response miR-eQTLs in cis for miR-326 and miR-1260, in non-infected and infected samples. (B) Regional association plot for miR-582-5p and genotyped SNPs in the region of the gene PDE4D showing the location of a cluster of significantly associated SNPs ∼500 kb upstream of the miRNA in infected samples. An additional region, in between the detected eQTL and the miRNA, also showed a strong tendency of association; however, this did not reach genome-wide significance. All annotations are based on UCSC hg19.
We next searched for trans-eQTLs by associating miRNA expression levels with genome-wide SNPs. We identified one miRNA, miR-582-5p, whose expression was associated with a cluster of SNPs in infected samples (Fig. 2B), of which the most strongly associated was rs12523473 (Bonferroni-corrected P = 1.49 × 10−4). While this association lies outside the 200-kb region considered for cis-acting variants, its physical proximity to miR-582-5p suggests that this miR-eQTL is likely to be a long-distance cis-eQTL. The detection of trans-eQTLs suffers, however, from a high burden of multiple testing while effects may be only of modest size (Gilad et al. 2008; Majewski and Pastinen 2011; Montgomery and Dermitzakis 2011). Given that regulatory variants have been observed to overlap with SNPs associated with complex phenotypes (Nica et al. 2010; Nicolae et al. 2010), we restricted our analysis to SNPs that have been suggestively associated (P < 1 × 10−5) with TB susceptibility by genome-wide association studies (GWAs) (Hindorff et al. 2009; Thye et al. 2010; Thye et al. 2012). In doing so, we identified a putative trans-eQTL for let-7i, a strongly induced, pro-inflammatory miRNA, the expression of which was associated with rs9373523 upon MTB infection (Bonferroni-corrected P = 7.78 × 10−3). The mechanism underlying this association remains unclear, however, that this SNP lies in an intron of the gene STXBP5, which is a target of let-7i, may point to a complex regulatory interaction between these transcripts.
Genomic and functional context of miR-eQTLs
To understand how eQTLs may influence miRNA expression, we studied the genomic context of the detected miR-eQTLs. We first investigated the overlap between miR-eQTLs and signatures of regulatory regions, ChIP-seq peaks and DNase I signals, reported by the ENCODE Project for human monocytes, the closest available cell-type to our model (The ENCODE Project Consortium 2012). We observed a number of overlaps with histone modifications (Supplemental Table S4). In particular, we found that the detected miR-eQTLs are significantly enriched in regions associated with the histone modification H3K4me3 (P = 1.6 × 10−3). That this is a mark of regulatory elements primarily associated with promoters/transcription start sites (The ENCODE Project Consortium 2012), and that three of the four SNPs overlapping such regions are located 5′ of the miRNA with which they are associated (miR-1260, miR-582-5p, and miR-769-5p), further supports the functional relevance of the miR-eQTL regions in modulating miRNA expression.
To gain further insight into the putative regulatory mechanisms underlying the detected miR-eQTLs, we refined our association signals by genotype imputation (Supplemental Fig. S4; Supplemental Methods). We then searched for our miR-eQTL SNPs, and those in strong linkage disequilibrium with them, among (1) DNase I sensitivity QTLs (dsQTLs) (Degner et al. 2012), (2) mRNA-eQTLs identified in the same cellular infection model (Barreiro et al. 2012), and more generally, (3) mRNA-eQTLs identified from the International HapMap project (Xia et al. 2012). We found that two miR-eQTL SNPs for miR-451 were associated with variation in the chromatin accessibility of a nearby region (rs9279, P = 3.77 × 10−10, and rs2320588, P = 9.55 × 10−8). This DNase I-sensitive region, which lies 4 kb upstream of the precursor of miR-451, is further associated with the binding of a number of transcription factors, especially POLR2A and GATA1 (The ENCODE Project Consortium 2012), suggesting that this region may directly regulate the transcription of miR-451. With respect to mRNAs, miR-eQTL SNPs for miR-582-5p were associated with the expression of DEPDC1B in cis (FDR = 0.05–0.11) and PNMAL1 in trans (FDR < 0.2), the latter gene being a predicted target of miR-582-5p, in HapMap samples. Interestingly, four of the reported miR-eQTLs (miR-326, miR-338-3p, miR-130b, and miR-582-5p) are noncanonical intronic miRNAs (mirtrons) (Okamura et al. 2007; Ruby et al. 2007). However, no cis-eQTLs were identified for the corresponding host genes (ARRB1, AATK, PPIL2, and PDE4D, respectively) in the same samples. This, together with the observation that only miR-582-5p expression was moderately positively correlated with that of its host gene (r = 0.311; P = 0.013), supports the notion that mirtrons are regulated and/or processed independently of their host genes (Parts et al. 2012; Civelek et al. 2013).
Extensive remodeling of genome-wide miRNA–mRNA interactions upon infection
To understand the impact of infection on broader miRNA-mediated regulatory networks, we next investigated the genome-wide relationships between variation in miRNA expression and that of protein-coding genes. To do so, we calculated Pearson correlation coefficients between the expression levels of mature miRNAs and mRNAs, obtained from the same 63 individuals, before and after infection (see Methods). We detected an overwhelming 35-fold increase in the number of significant correlations between miRNAs and mRNAs in infected samples with respect to non-infected samples (FDR < 0.005; Supplemental Table S5; Supplemental Fig. S5). Furthermore, the patterns of genome-wide correlations strongly differed between the two conditions: Correlations before infection were enriched in positive correlations (P = 6 × 10−4) whereas those after infection were skewed toward negative relationships (P < 1 × 10−20), compared with the null distribution. Specifically, only 23% of significant miRNA–mRNA correlations were negative in non-infected samples, compared with 52% in infected samples (Supplemental Fig. S6), a difference that became more pronounced when considering only the strongest correlations (|r|>0.7, 9% vs. 72% in non-infected and infected samples, respectively). These trends remained similar after accounting for variation in the percentage of infected cells among individuals (Supplemental Fig. S7). Notably, we observed an enrichment in predicted miRNA targets not only among negatively (P = 2.85 × 10−5) but also among positively (P = 6.45 × 10−7) correlated genes in infected cells. This observation is consistent with the fact that miRNAs are often involved in incoherent feed-forward loops and other complex network relationships with their targets (Hornstein and Shomron 2006; Tsang et al. 2007; Vasudevan et al. 2007; Martinez et al. 2008; Ebert and Sharp 2012; Lu and Clark 2012).
The majority of significant miRNA–mRNA correlations in both conditions were accounted for by a small number of miRNAs, as previously observed (Nunez-Iglesias et al. 2010; Su et al. 2011; Gamazon et al. 2012). Upon infection, 46 miRNAs were each correlated with at least 10 mRNAs, of which 15 were found to be associated with over 100 mRNAs and cumulatively accounted for 75% of all significant correlations (Fig. 3A; Supplemental Table S5). Furthermore, these 46 miRNAs were enriched in differentially expressed miRNAs upon infection (P = 0.03; N = 30), and included many of known importance in the regulation of the immune response (e.g., miR-155, miR-132, and miR-146a). These differentially expressed miRNAs and their significantly correlated gene sets, which were also enriched in differentially expressed genes (P < 1 × 10−20), formed a tightly connected regulatory network (Fig. 3B). This highlights the highly interrelated nature of miRNAs in gene regulation, consistent with their frequent cotargeting of mRNA transcripts (Krek et al. 2005; Stark et al. 2005; Tsang et al. 2010). Furthermore, among these correlated gene sets, 70% of those presenting an over/under-representation of at least one KEGG pathway and/or GO category (FDR<0.05) were associated with immune-related functions. These included innate immunity signaling pathways (e.g., TLR and JAK-STAT pathways) and activation and differentiation of lymphocytes (Supplemental Table S6), consistent with the expected functions of activated DCs. Taken together, these findings suggest that a subset of differentially expressed miRNAs may account for most of the functional associations between miRNAs and mRNAs upon MTB infection.
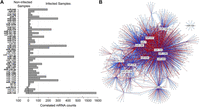
Relationship between the levels of expression of miRNAs and protein-coding genes. (A) Barplot showing the number of significantly correlated mRNAs per miRNA in non-infected and infected samples. Only miRNAs whose expression levels were significantly correlated with those of at least 10 mRNAs are shown. A total of 47 miRNAs were correlated with at least 10 mRNAs in non-infected and/or infected conditions, with three miRNAs satisfying this criterion in both conditions. A total of 31 of these 47 miRNAs were significantly differentially expressed upon MTB infection (marked in bold). (B) Regulatory network of significantly correlated mRNAs and differentially expressed miRNAs in MTB-infected samples. Nodes represent miRNAs and mRNAs. MiRNAs are labeled when correlated with more than 100 mRNAs, with the exception of miR-150, which is independent of the main network. Edge thickness reflects the strength of the correlation between one miRNA and one mRNA transcript. Edge color represents the direction of the correlation (red, negative; blue, positive).
Characterization of the impact of miR-29a on the response to infection
To experimentally assess the impact of altered miRNA expression on both miRNA–mRNA interactions and cellular responses to MTB infection, we studied one miRNA—miR-29a—using gain- and loss-of-function approaches. We chose this miRNA because (1) it is strongly induced upon infection (Fig. 1), (2) it is among those presenting the largest number of correlated mRNAs in infected cells (Fig. 3), and (3) it has been explicitly implicated in the response to various mycobacterial infections (Fu et al. 2011; Ma et al. 2011; Sharbati et al. 2011; Eulalio et al. 2012; Yi et al. 2012; Brain et al. 2013). We thus transfected DCs with a miR-29a mimic or a miR-29 family inhibitor, as all miR-29 family members (miR-29a,b,c) share the same seed sequence, and confirmed the perturbation of miR-29 expression before and after infection with MTB (Supplemental Fig. S8; Supplemental Methods). Using genome-wide expression arrays, we identified 193 and 539 differentially expressed genes, at the steady-state, and 59 and 307, after infection, in miR-29 overexpressing and inhibited cells, respectively, compared with control-transfected cells (FDR-corrected P < 0.01, Supplemental Table S7). These differentially expressed genes, in particular upon miR-29a overexpression, were enriched in predicted targets of miR-29a (P = 0.01–1 × 10−20).
Importantly, we found that the genes whose expression was significantly correlated with that of miR-29a after infection in our previous computational analysis (Fig. 3; Supplemental Table S5) showed significantly greater changes in their expression levels with respect to noncorrelated genes in infected samples (P = 2.31 × 10−5 and P = 9.34 × 10−3 in overexpressing and inhibited cells, respectively) (Fig. 4A,B). Moreover, miR-29a predicted targets whose expression was correlated with that of this miRNA showed a significant decrease and increase in their expression levels in overexpression and inhibition experiments, respectively (Fig. 4C,D). These results support that a significant proportion of miRNA-correlated transcripts, in particular those that are predicted to be direct targets, are indeed causally regulated by miR-29.
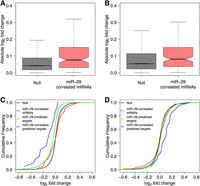
Functional validation of miRNA–mRNA relationships using miR-29a gain- and loss-of-function experiments. (A,B) Boxplots showing absolute fold changes in genome-wide mRNA levels of cells transfected with either an miR-29a mimic (A) or an miR-29 inhibitor (B) and subsequently infected with MTB. (C,D) Cumulative distributions of changes in genome-wide mRNA levels after transfection with the mimic (C) or the inhibitor (D) in MTB-infected cells.
Lastly, we assessed the impact of miR-29a on cellular responses to infection at the transcript and protein levels. Consistent with the profound effect of infection on DC maturation and function, the transcriptional response to MTB was highly concordant between control- and miR-29-transfected cells (82% and 91% overlap in mimic and inhibitor-transfected cells, respectively; FDR-corrected P < 0.01; Supplemental Table S7). However, a subset of differentially expressed genes upon infection responded differently following perturbation of miR-29 (64 and 235 in mimic and inhibitor-transfected cells, respectively; P < 0.01; Supplemental Table S7). Among these differentially responding genes, we observed an enrichment of a number of KEGG pathways and GO categories (Supplemental Table S8). In particular, genes that were differentially up-regulated following miR-29 inhibition were significantly enriched in genes involved in cytokine–cytokine receptor interaction and TLR signaling pathways. At the protein level, we similarly observed a major effect of infection, with a strong induction of many cytokines that play a major role in the DC response to infection, including TNF, IL12B, and IL10 (Giacomini et al. 2001; Hickman et al. 2002; Supplemental Table S9). However, the inhibition of miR-29 promoted an enhanced cytokine response, as attested to by the significantly higher induction of 12 cytokines and chemokines, with respect to control-transfected samples, after infection (Fig. 5A). Notably, three of these cytokines are miR-29a predicted targets (IL12B, IL2RA, and CXCL10), consistent with a direct effect of miR-29 on cytokine responses. Furthermore, despite the more modest effect of miR-29a overexpression on cytokine levels (Supplemental Table S9), one of these proteins, CXCL10, was significantly down-regulated compared with control-transfected samples (Fig. 5B), providing strong support for a direct regulation of this chemokine by miR-29 in MTB-infected DCs.
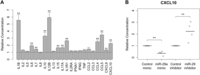
Inhibition of miR-29 up-regulates the secretion of multiple cytokines in MTB-infected DCs. Cytokine and chemokine concentrations in culture supernatants from transfected, infected cells were analyzed by multiplex bead immunoassay. (A) Relative cytokine/chemokine concentrations in miR-29 inhibitor-transfected cells compared with control-transfected samples ±SEM for duplicates of infection performed on four different donors. MiR-29 inhibition significantly increased the expression of 12 out of 22 cytokines. (**) P < 0.01. (B) Relative concentrations of the chemokine CXCL10 for mimic- and inhibitor-transfected cells, with respect to control-transfected samples, for duplicates of infection performed on four different donors.
Discussion
Identifying genetic variants that affect miRNA expression in the presence or absence of specific environmental variables can provide insights into the mechanisms underlying variation in transcript abundance. We first showed that the expression of 3% of miRNAs is explained by proximate genetic factors, consistent with several previous estimates (Rantalainen et al. 2011; Parts et al. 2012; Civelek et al. 2013). That 9% of protein-coding genes harbor cis-regulatory variants in the same samples, at an FDR of only 0.01 (Barreiro et al. 2012), supports the notion that miRNA expression is under less genetic control than that of mRNAs (Su et al. 2011; Civelek et al. 2013; Lappalainen et al. 2013). The fewer eQTLs detected for miRNAs may reflect a lower permissibility of large changes in their expression, because these would have extensive consequences on the multiple genes and pathways with which miRNAs are associated. Moreover, given the strong conservation observed in miRNA sequences and expression patterns (Quach et al. 2009; Christodoulou et al. 2010; Berezikov 2011), one may also anticipate greater sequence conservation in their regulatory regions, and hence a lower dependence of miRNA expression on proximate genetic variants.
Although several studies have mapped gene-environment interactions for expression phenotypes of protein-coding genes (Smirnov et al. 2009; Romanoski et al. 2010; Maranville et al. 2011; Barreiro et al. 2012; Idaghdour et al. 2012), there has been no effort to characterize the genetic architecture of miRNA expression upon perturbation by external stimuli. Here, we provide evidence of genotype-by-infection interactions that affect miRNA expression variation. The strongest signal among response miR-eQTLs is that observed for miR-326—associated with an eQTL exclusively upon infection (Fig. 2A). The increased expression of miR-326 in T cells promotes the generation of Th17 cells and is associated with the severity of autoimmune disease (Du et al. 2009). In MTB infection, a shift toward a stronger induction of the Th17 pathway has been associated with excessive neutrophil recruitment, tissue damage, and increased disease severity (Torrado and Cooper 2010; Jurado et al. 2012). The down-regulation of miR-326 that we observed upon infection points to a direct link between this miRNA and DC responses to MTB infection. Furthermore, that a subset of individuals characterized by the AA genotype show higher miR-326 expression after infection (∼10% of Europeans) (Fig. 2A), which may decrease protective immunity, suggests that regulatory variation at this locus might ultimately impact inter-individual differences in the host response to MTB. This example, together with the infection-dependent eQTL for miR-1260 expression and the interaction effect associated with miR-338-3p, provides experimentally testable hypotheses concerning the role of these miRNAs in immune responses and TB pathogenesis.
The integration of genome-wide expression data from miRNAs and mRNAs highlights the complexity of the miRNA-mediated regulatory system. Although differences in miRNA–mRNA relationships have been reported in diseased and healthy individuals or distinct cell subsets (Cheng et al. 2009; Nunez-Iglesias et al. 2010; Allantaz et al. 2012; Zhang et al. 2012), a systematic study of the impact of an external stimulus on these relationships has been lacking. Our findings showed that infection is accompanied by a rapid and strong remodeling of miRNA-mediated regulatory networks, with a shift toward negative miRNA–mRNA correlations. Such a marked shift, largely accounted for by a small number of differentially expressed miRNAs, emphasizes the wide-reaching impact of a subset of miRNAs in the transcriptional response of a cell to infection.
Through our gain- and loss-of-function experiments, using the model of miR-29a, we have confirmed that the miR-29a-correlated mRNAs detected by our computational analysis are associated with significantly greater changes in expression levels upon perturbation of miR-29 expression in infected cells. This points to a general causal regulation, direct or indirect, of correlated genes by miR-29. That not all genes showed such a change, however, is consistent with the expectation that miRNA–mRNA interactions reflect the coregulation of miRNA–mRNA pairs and/or miRNA sensing as well as signaling (Su et al. 2011). In addition, positively correlated miR-29a predicted targets displayed changes in their expression levels that are consistent with canonical miRNA-mediated repression, lending experimental support to the importance of regulatory loops in miRNA–mRNA interactions (Tsang et al. 2007; Martinez et al. 2008; Ebert and Sharp 2012). Given the large number of positive correlations that we and others report (Martinez et al. 2008; Nunez-Iglesias et al. 2010; Su et al. 2011; Lappalainen et al. 2013), and the enrichment in miRNA predicted targets observed among them, it thus seems likely that feedforward and feedback loops are widespread mechanisms in miRNA-mediated regulatory responses in the context of infection.
Our functional study of miR-29a has also provided new insight into the role of this miRNA in DC functions and responses to MTB infection. First, we observed more differentially expressed genes between miR-29- and control-transfected cells in non-infected conditions, with respect to infected samples. This suggests that, upon infection, miR-29 may drive more focused changes in a smaller set of genes. Second, not only was miR-29a strongly up-regulated in our setting but, most importantly, it had a substantial impact on cytokine secretion in response to infection. In particular, the secretion of CXCL10 is consistent with a direct, repressive effect of miR-29 on this chemokine, which may impact the recruitment of Th1 cells upon MTB infection (Giacomini et al. 2006).
In conclusion, our study has provided an initial assessment of the impact of genotype–environment interactions on miRNA molecular phenotypes by identifying response miR-eQTLs related to infection. This, together with the observed infection-dependent shift of miRNA–mRNA relationships driven by a few miRNAs, such as miR-29a, paves the way for additional studies to evaluate the biological contribution of these miRNAs to immunity to infection and disease outcome.
Methods
Samples and miRNA expression analyses
Blood samples were obtained from 65 healthy donors from Research Blood Components. Signed, written consent was obtained from all individuals, in accordance with the company’s independent ethics committee approval. Isolation and infection of DCs with Mycobacterium tuberculosis (H37Rv) for 18 h, RNA extraction and quality verification, DNA extraction, and genome-wide genotyping have been previously described (Barreiro et al. 2012). Genome-wide miRNA expression was profiled using the Agilent Human miRNA microarray (Release 16.0). After a series of quality checks, preprocessing, and normalization of the data (Supplemental Methods), we identified differentially expressed miRNAs upon MTB infection by applying a linear model with a fixed effect for MTB treatment, implemented in the Bioconductor package limma (Smyth 2004).
Mapping of expression quantitative trait loci (eQTLs)
Associations between SNP genotypes (GEO accession number GSE34588) (Barreiro et al. 2012) and miRNA expression levels were calculated using a linear regression model, assuming an additive effect of alleles on expression, in infected and non-infected samples. We improved the power to detect eQTLs by quantile normalization and regression of a number of principal components, to account for unknown confounders (Supplemental Fig. S9). FDRs were estimated by comparing the observed to a null distribution, generated using the lowest P-values observed for each miRNA in 100 permutations of expression values (Pickrell et al. 2010; Barreiro et al. 2012). Genotype-treatment interaction effects were detected by Bayesian regression with the software BRIdGE (Maranville et al. 2011). For trans-eQTLs, associations were calculated with both genome-wide genotyping data from the same individuals (Barreiro et al. 2012) and a subset of SNPs previously identified as susceptibility loci for TB by GWAs (http://www.genome.gov/26525384/) (Hindorff et al. 2009). Multiple testing corrections were performed using a Bonferroni correction at the 95% significance level. The overlap of miR-eQTLs with active genomic regions was assessed using data from the ENCODE Project (http://encodeproject.org/ENCODE/) (The ENCODE Project Consortium 2012). Enrichments were calculated using a Fisher’s exact test. For the fine-mapping of miR-eQTL regions, we imputed genotypes for SNPs not present on our genotyping array with IMPUTE2 (Howie et al. 2009), using integrated haplotype data from Phase 1 of the 1000 Genomes Project (The 1000 Genomes Project Consortium 2012). For details, see the Supplemental Methods.
Correlation of miRNA and mRNA expression levels
We calculated Pearson correlation coefficients between quantile normalized expression levels of miRNAs and mRNAs detected in at least 50% of samples in at least one condition. As clustered miRNAs show correlated expression profiles (Supplemental Fig. S10), we considered only the most abundant member of each pre-miRNA (N = 221). Expression levels of 12,958 protein-coding genes were previously obtained from the same 63 individuals (Barreiro et al. 2012). Significant miRNA–mRNA correlations were determined at an FDR <0.005 based on a null distribution of 1000 permutations and differences between the means of real and null distributions were determined using a t-test. Predicted targets of miRNAs were obtained from TargetScan (v6.2) (Friedman et al. 2009). The enrichment of predicted targets of all miRNAs in the network among all miRNA-correlated genes was calculated using a Fisher’s exact test. Enrichments of functional Gene Ontology categories and KEGG pathways among the same gene sets were computed using GeneTrail (Backes et al. 2007). We used all 12,958 expressed genes as a background set for over/under-representation analyses of correlated gene sets. Enrichment P-values were calculated using a hypergeometric test and we used the Benjamini and Hochberg (1995) approach to correct for multiple testing. Networks were visualized using Cytoscape (Cline et al. 2007).
Functional analyses of miR-29a using gain- and loss-of-function approaches
Immature DCs from four unrelated individuals were transfected on day 5 using HiPerFect transfection reagent. MiRCURY LNA Power Inhibitors were purchased from Exiqon (miR-29 family 460039, control 199020-00) and miRIDIAN microRNA mimics from Thermo Fisher (miR-29a C-300504-07, control CN-001000-01). Transfection efficiency was assessed by flow cytometry using a fluorescently labeled control oligonucleotide (Exiqon, 199020-04), and found to be on average 77% (Supplemental Fig. S11). Transfected cells were then infected for 24 h with MTB (H37Rv) (Supplemental Fig. S12). MiR-29 expression upon MTB infection and the extent of miR-29 perturbation in transfected cells were quantified by quantitative real-time PCR (qPCR). For gene expression analysis, genome-wide profiling of non-infected and MTB-infected samples was obtained by hybridizing RNA to Illumina HumanHT-12 v4 Expression BeadChip arrays. After a series of quality checks and preprocessing steps, differential expression analysis was performed using the Bioconductor package limma (Smyth 2004). For quantification of cytokine and chemokine levels, we measured supernatant levels of 25 cytokines/chemokines using the Human Cytokine Magnetic 25-Plex Panel (Invitrogen). Differences in secretion levels between conditions were calculated using a Wilcoxon paired rank sum test. For details, see the Supplemental Methods.
Data access
The miRNA and mRNA expression data reported in this manuscript have been submitted to the NCBI Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/) under accession numbers GSE49951 and GSE53143, respectively.
Acknowledgments
We thank Robin Friedman for critical reading of the manuscript and useful suggestions; Yoav Gilad for facilitating access to mRNA expression data; and Maud Fagny, Eddie Loh, and John Marioni for helpful discussions. The laboratory of L.Q.-M. has received funding from the Institut Pasteur, the Centre Nationale de la Recherche Scientifique (CNRS), the French government’s Investissement d’Avenir program, Laboratoire d’Excellence “Integrative Biology of Emerging Infectious Diseases” (grant no. ANR-10-LABX-62-IBEID), and the European Research Council under the European Union’s Seventh Framework Programme (FP/2007–2013)/ERC grant agreement no. 281297. The laboratory of L.B.B. has received funding from the Canadian Institute of Health Research (grant nos. 232519 and 285969) and the Human Frontiers Science Program (grant no. CDA00025-2012). L.B.B. is a scholar of the Fonds de la Recherche en Santé du Québec. The laboratory of L.T. and B.G. has received funding from the National Institutes of Health (grant no. NIH AI087658) and the European Commission within the 7th Framework Programme (grant no. HEALTH-F3-2009-241745). K.J.S. is a scholar of the Pasteur–Paris University (PPU) International PhD program and was supported by a stipend from the Direction Générale de l’Armement (DGA). Y.N. was supported by a fellowship from the Réseau de médecine génétique appliquée, Fonds de recherche du Québec–Santé (FRQS), and the Instituts de recherche en santé du Canada (IRSC, subvention no. TGF-96109).
Author contributions: The study was designed by K.J.S., L.B.B., and L.Q.-M. Computational analyses were performed by K.J.S. with contributions and input from Y.N., E.P., G.L., L.B.B., and L.Q.-M. Functional inhibition and overexpression experiments were performed by M.D. and differentiation and infection of dendritic cells by L.T., with contributions and guidance from J.P., G.L.-V., V.L., and O.N. B.G. contributed with materials and tools. The manuscript was written by K.J.S. and L.Q.-M. with input from all authors.
Footnotes
-
↵12 Corresponding author
E-mail quintana{at}pasteur.fr
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.161471.113.
- Received June 3, 2013.
- Accepted January 22, 2014.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see http://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








