
NF-Y coassociates with FOS at promoters, enhancers, repetitive elements, and inactive chromatin regions, and is stereo-positioned with growth-controlling transcription factors
- Joseph D. Fleming1,
- Giulio Pavesi2,
- Paolo Benatti3,
- Carol Imbriano3,
- Roberto Mantovani2 and
- Kevin Struhl1,4
- 1Department of Biological Chemistry and Molecular Pharmacology, Harvard Medical School, Boston, Massachusetts 02115, USA;
- 2Dipartimento di BioScienze, Università degli Studi di Milano, 20133 Milano, Italy;
- 3Dipartimento di Scienze della Vita, Università di Modena e Reggio Emilia, 41125 Modena, Italy
Abstract
NF-Y, a trimeric transcription factor (TF) composed of two histone-like subunits (NF-YB and NF-YC) and a sequence-specific subunit (NF-YA), binds to the CCAAT motif, a common promoter element. Genome-wide mapping reveals 5000–15,000 NF-Y binding sites depending on the cell type, with the NF-YA and NF-YB subunits binding asymmetrically with respect to the CCAAT motif. Despite being characterized as a proximal promoter TF, only 25% of NF-Y sites map to promoters. A comparable number of NF-Y sites are located at enhancers, many of which are tissue specific, and nearly half of the NF-Y sites are in select subclasses of HERV LTR repeats. Unlike most TFs, NF-Y can access its target DNA motif in inactive (nonmodified) or polycomb-repressed chromatin domains. Unexpectedly, NF-Y extensively colocalizes with FOS in all genomic contexts, and this often occurs in the absence of JUN and the AP-1 motif. NF-Y also coassociates with a select cluster of growth-controlling and oncogenic TFs, consistent with the abundance of CCAAT motifs in the promoters of genes overexpressed in cancer. Interestingly, NF-Y and several growth-controlling TFs bind in a stereo-specific manner, suggesting a mechanism for cooperative action at promoters and enhancers. Our results indicate that NF-Y is not merely a commonly used proximal promoter TF, but rather performs a more diverse set of biological functions, many of which are likely to involve coassociation with FOS.
Transcriptional regulatory proteins and the RNA polymerase II (Pol II) machinery recruit chromatin-modifying activities to their target loci, thereby determining the genomic pattern of histone modifications and nucleosome occupancy. Activator proteins, functioning combinatorially at distal enhancers and in proximity to core promoters, recruit nucleosome remodeling and histone acetylase complexes, thereby generating nucleosome-depleted regions that nevertheless have peaks of histone acetylation. The Pol II machinery recruits H3K4 histone methylases near the core promoter, and upon transcriptional elongation recruits H3K36 and H3K79 histone methylases to active coding regions. Although less well defined, other DNA-binding proteins and nascent RNA can recruit H3K27 or H3K9 methylases to other genomic regions, resulting in heterochromatic silencing by polycomb complexes (PcG) or HP1, respectively.
As a consequence of the above and other mechanistic relationships between TFs and chromatin-modifying activities, the genome-wide pattern of histone modifications and nucleosome occupancy can be used to classify promoters, enhancers, insulators, and distinct types of heterochromatic regions in a given cell type under a given physiological condition. Using chromatin immunoprecipitation (ChIP), formaldehyde-assisted isolation of regulatory elements (FAIRE), and DNase I hypersensitivity techniques coupled to massively parallel DNA sequencing, such classification of functional genomic regions has been done in several cell lines in the context of ENCODE (The ENCODE Project Consortium 2004, 2007, 2011, 2012). In addition, ENCODE has performed genome-wide mapping of binding sites for ∼80 TFs (at the time of writing), most notably in the leukemia cell line K562. These genome-wide maps provide an invaluable resource for uncovering new functional aspects of individual TFs.
NF-Y (also known as CBF, CP1) is a heterotrimeric, DNA-binding TF that is conserved in all eukaryotes (Romier et al. 2003). NF-Y binds specifically to the CCAAT motif (Sinha et al. 1995; Bi et al. 1997) that is frequently found in eukaryotic promoters (Suzuki et al. 2001; Marino-Ramirez et al. 2004). The NF-YB and NF-YC subunits (protein products of NFYB and NFYC) contain histone-fold domains (HFDs) structurally related to H2B and H2A, respectively (Baxevanis et al. 1995), which mediate formation of a stable histone-like heterodimer (Romier et al. 2003). NF-YA (protein product of NFYA) binds to this heterodimer, such that the resulting heterotrimeric complex can bind specifically to the CCAAT motif (Sinha et al. 1995). NF-YA contains the sequence-specific CCAAT recognition domain, and NF-YB and NF-YC also contact DNA through their HFDs (Kim et al. 1996; Sinha et al. 1996; Zemzoumi et al. 1999). All bases of the core pentanucleotide are critical for NF-Y binding, with immediate flanking sequences on both ends also being important for efficient DNA binding in vitro (Hooft van Huijsduijnen et al. 1987; Kim et al. 1990) and in vivo (Testa et al. 2005; Ceribelli et al. 2006, 2008).
At many promoters, the CCAAT motif is highly positioned ∼80 bp upstream of the transcriptional start site (TSS), in either orientation, suggesting that its location is important for gene expression. In essentially all promoters tested, mutation of the CCAAT motif reduces or eliminates transcriptional activity (Dolfini et al. 2009). In addition, functional inactivation of NF-Y subunits or the use of a dominant-negative NF-YA mutant indicates that NF-Y binding is important for the pattern of histone modifications at promoters (for review, see Dolfini et al. 2012). Interestingly, bioinformatic studies comparing gene expression patterns in tumors vs. normal tissues indicate that NF-Y sites are highly enriched in promoters of genes overexpressed in tumors (Rhodes et al. 2005; Sinha et al. 2008; Goodarzi et al. 2009), particularly in the most aggressive cohorts. The importance of NF-Y is further underscored by the early embryonic lethality of an NF-YA mouse knockout model due to defects in cell proliferation and extensive apoptosis (Bhattacharya et al. 2003).
Here we describe the genome-wide analysis of NF-Y binding in three tumor cell lines. Using data generated by ENCODE, we analyze the bound loci with respect to chromatin states and binding by 78 chromatin-associating factors. Our results uncover many new and unexpected aspects of NF-Y biology.
Results and Discussion
Unbiased genome-wide identification of NF-Y binding sites
We performed ChIP with anti-NF-YA and anti-NF-YB antibodies in three cell types (K562, GM12878, and HeLa S3) followed by deep DNA sequencing. Antibodies (Dolfini et al. 2009) were validated by Western blot and IP-WB, showing that NF-YA and NF-YB are specifically recognized (Supplemental Fig. 1A,B). Immunoprecipitated DNA was validated on known NF-Y targets (Supplemental Fig. 1C,D) and the reproducibility between biological replicates was high (Pearson correlations: >0.8).
Using a stringent cut-off (P-value < 10−9), we identify 12655, 7932, and 5457 NF-YB binding sites and 4726, 289, and 3726 NF-YA binding sites in K562, GM12878, and HeLa S3 cells, respectively (Fig. 1A). Applying the de novo motif discovery tool MEME to NF-YB peaks in K562 cells, we identify an NF-Y binding motif (Fig. 1B) that corresponds well to the motif derived from ChIP-chip experiments (Dolfini et al. 2009). These high-confidence binding sites, 83% of which have at least one CCAAT motif within each site (with a mean of 1.7 motifs per site), will be used for subsequent bioinformatic analyses. At lower stringency, we identify 14,772 (P < 10−7) and 18,523 (P < 10−5) NF-YB sites in K562, 81% and 77% of which, respectively, have CCAAT motifs. NF-YB sites with relatively high P-values in the range from 10−5 to 10−7 contain CCAAT motifs at a rate of ∼60%, whereas the genomic background is ∼5% for similarly sized regions (Supplemental Fig. 2A). Based on these observations and a peak saturation analysis (Supplemental Fig. 2B), we estimate that there are an additional ∼4000 low-affinity NF-Y binding sites in the genome of K562 cells.
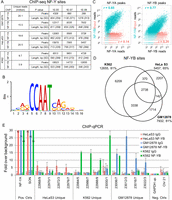
ChIP-seq of two components of the NF-Y complex in three cell types. (A) MACS peak analysis indicating peak numbers, mean peak lengths, and standard deviations at three different P-value thresholds for NF-YA and NF-YB ChIP-seq data sets in GM12878, HeLa S3, and K562. (B) Identification of the NF-Y DNA-binding site motif de novo from 12,655 K562 NF-YB peaks depicted as a sequence logo (Schneider and Stephens 1990). (C) Scatter plots of NF-YA, NF-YB, and input read counts at NF-YA or NF-YB sites in K562 showing correlation between data sets. (Blue shading) Correlation amongst NF-YA and NF-YB. (Orange shading) NF-YA or NF-YB correlation to input. (D) Venn diagrams depicting the overlap between NF-YB peak populations in GM12878, HeLa S3, and K562. Integers represent peak numbers called at the 10−9 P-value threshold. The percentages of peaks with CCAAT motifs are indicated (%). (E) ChIP-qPCR validation of NF-YB peaks unique to each cell type. (Error bars) Standard deviation of three biological replicates. “Pos. Ctrls” are loci known to be bound by NF-Y. “Neg Ctrls” are loci known to be devoid of NF-Y. Data represents a fold over background measurement compared with a non-NF-Y bound region (“GAPDH up”). (Solid and striped bars) ChIPs performed with NF-YB specific antibody and nonspecific rabbit IgG, respectively.
The apparently higher number of NF-YB sites with respect to NF-YA sites could be due to target loci bound only by NF-YB. In this regard, in nuclei, NF-YB is more abundant than NF-YA, and NF-YB is present in certain post-mitotic cells whereas NF-YA is not detected (Dolfini et al. 2012). However, the NF-YA and NF-YB data sets are highly correlated (Pearson correlation ∼0.7) (Fig. 1C), and quantitative PCR analysis of individual sites reveal threefold higher enrichments for NF-YB than for NF-YA. Furthermore, analysis of 21 NF-YB sites that appear to lack NF-YA show low occupancy of NF-YB, such that an NF-YA peak was below the detection limit (Supplemental Fig. 3A,B). These results indicate that the NF-YB antibody is more “immuno-efficient” than the NF-YA antibody, and that there are few, if any, genomic sites that are bound by NF-YB, but not NF-YA. For this reason, we will use the NF-YB data set to define NF-Y binding sites in subsequent analyses.
Approximately 39% of NF-Y sites are occupied in at least two cell types, whereas the remaining are cell-type specific (Fig. 1D). In accord with this observation, examination of 14 NF-Y target genes identified previously in different cell lines (Ceribelli et al. 2008) revealed that 13 are bound in K562 and eight are bound in Hela S3. We validated the cell-type specificity of a small number of these loci by ChIP-qPCR (Fig. 1E).
Asymmetric binding of NF-YA and NF-YB to the CCAAT motif
Linking the high-resolution positioning data of NF-Y subunits to the CCAAT motif, we confirm that NF-YA binds directly over the CCAAT sequence (Fig. 2A). Interestingly, the NF-Y complex is asymmetric, with NF-YB binding ∼15 bp downstream from the CCAAT motif, as defined by the CCAAT strand (Fig. 2A). This asymmetry fits extremely well with the available biochemical knowledge of NF-Y/DNA contacts (Dolfini et al. 2009) and with the crystal structure of trimer interactions with DNA (Romier et al. 2003; Nardini et al. 2013).
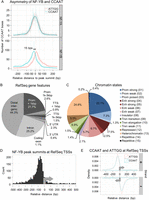
Annotation of NF-Y peaks to genomic features. (A) Kernel density estimate of the distribution of the 5′-CCAAT-3′ and 5′-ATTGG-3′ sequences under NF-YA and NF-YB peaks in relation to the peak summit centered at 0 bp. Only the position of the best matching CCAAT motif within 100 bp of the peak summit is considered and plotted. (Solid and dashed lines) Raw and Gaussian smoothed data, respectively. (B) Annotation of K562 NF-YB sites to RefSeq gene features. (C) As in B, except chromatin state maps are used. (Prom) promoter; (enh) enhancer; (trxn) transcription. Numbering is from the chromatin state maps of Ernst et al. (2011). (D) Frequency distribution of K562 NF-YB peak summits at RefSeq TSSs showing a preferential location between −50 and −100 bp upstream of the TSS. (E) Gaussian kernel density estimate of the distribution of positive and negative strand 5′-CCAAT-3′ and 5′-ATTGG-3′ sequences at K562 NF-YB-bound RefSeq TSSs. Only the best motif per region is considered. Bandwidth is equal to the standard deviation of the smoothing kernel. (Gray arrows) Direction of transcription.
NF-Y targets cell signaling, DNA repair, cell-cycle, metabolic, and gene expression genes
GREAT gene ontology analysis of NF-Y bound loci from K562, GM12878, and HeLa S3 reveals a strong enrichment of genes involved in cell-signaling pathways (ITGA2B, MAPK12, MAPK13), cell cycle (G2/M checkpoints, regulation of DNA replication), DNA repair (homologous recombination and base excision), and metabolism (cholesterol biosynthesis, polyamines) (Supplemental Fig. 4A,B). The connection to the cell cycle and metabolism are in line with previous findings, and further stress the central role of NF-Y in growth-controlling decisions.
In addition, just below our fold-enrichment cutoff, we found a preponderance of GO terms associated with gene expression, in multiple cell lines, that were highly significant. Upon further analysis, it was apparent that NF-Y significantly targeted genes involved in transcription, mRNA splicing, mRNA editing, mRNA 3′-end processing, and mRNA transport. Included is a large and diverse set of TFs, including the NF-Y genes themselves, members of the transcriptional machinery, and coactivators and corepressors (Supplemental Fig. 5A,B; Supplemental Data Tables 10,11,12). Thus, NF-Y appears to be a regulator of gene expression regulators.
In a separate analysis (IPA, Ingenuity) of signaling pathways, we found that NF-Y preferentially associates with genes involved in the interrelated TP53 (protein product of TP53, also known as p53) and TNF-related apoptosis-inducing ligand (TRAIL) pathways (Supplemental Fig. 5C,D). This observation reinforces the notion of a direct and indirect NF-Y/TP53 interplay, with opposing functional consequences depending on the TP53 status of the cell, i.e., proliferation or apoptosis (Imbriano et al. 2012). In addition, it is consistent with anecdotal evidence about the role of NF-Y in apoptosis (Morachis et al. 2010; Hughes et al. 2011), which helps explain the phenotypes of NF-YA overexpression and inactivation experiments (Gatta and Mantovani 2011).
NF-Y binds to a diverse set of genomic features including nongenic regions
We annotated the NF-Y bound regions in K562 to RefSeq genes, TSSs, maps of histone modifications and nucleosome-depleted regions, and RNA levels. Unexpectedly, ∼25% of the NF-Y binding sites are not situated near RefSeq promoters, or the following types of genic regions: lncRNAs (Khalil et al. 2009); miRBASE (Kozomara and Griffiths-Jones 2011); UCSC RNA genes (Fujita et al. 2011); NONCODE (He et al. 2008); loci bound by Pol II or Pol III (Moqtaderi et al. 2010). These sites are not false positives, because the vast majority (88%) have CCAAT motifs, and 46% of them are present in at least one other cell type. Based on the patterns of colocalized histone modifications and Pol II, NF-Y-bound regions in K562 and HeLa S3 reproducibly partition into 20 clusters that can be grouped into five major classes: promoter, enhancer, gene body, PcG repressed, and LTR/nonmodified-chromatin. As discussed below, these results indicate that NF-Y binding is prevalent in tissue-specific enhancers and specific types of repetitive sequences, in addition to proximal promoters, where NF-Y has traditionally been observed.
Only a minority of NF-Y binding sites are located at proximal promoter regions
Although NF-Y is typically described as a factor that binds to proximal promoter regions, only 22% of NF-Y sites are located within 1 kbp upstream of a RefSeq TSS (Fig. 2B; Supplemental Fig. 6). A similar analysis shows ∼30% of NF-Y sites are located within chromatin states marked by histone modifications characteristic of promoters (Fig. 2C). This is consistent with our previous analysis of 2% of the human genome (Ceribelli et al. 2008). For such proximal promoter binding sites, a frequency distribution plot of peak summits indicates that NF-Y is highly positioned upstream of the TSS at −40 to −100 bp (Fig. 2D), in line with the position of the CCAAT motif at TSSs (Fig. 2E), in agreement with previously published observations (Dolfini et al. 2009). Though NF-YA and NF-YB bind asymmetrically to the CCAAT motif, the orientation with respect to the TSS is largely irrelevant for transcription, as only a small difference in the frequency of CCAAT and its complement ATTGG are noticed on the same strand (Fig. 2E). More generally, only a third of NF-Y loci (clusters B, K, L, N, P, S, U, V; n = 4061) (Fig. 3A) are associated with active promoters, as defined by high levels of di- and trimethylated H3K4, acetylated H3K27 and H3K9, Pol II, and nucleosome depletion (defined by a “valley” of low enrichment of mono-methylated H3K4 at NF-Y summits and a FAIRE signal) (Fig. 3A,B). By comparison, essentially no sites are located within nonmodified chromatin regions, and only a few MYC sites are located within weak enhancer-like regions (low K4me1 signals; Supplemental Fig. 7).
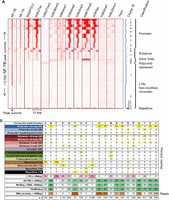
NF-YB bound loci reside within five epigenetic domains. (A) K-means clustering of K562 NF-YB loci based on the distribution of histone PTM, RNA Pol II, NF-YB, and NF-YA ChIP-seq reads within a region spanning ±5 kbp from the summit of NF-YB peaks (centered at 0 bp). Clustering was carried out on transformed, rank normalized read counts. Raw read count intensity is depicted in red. The interpretation and classification of clusters into functional categories are shown at right. (B) NF-YB summits from clusters derived from A are annotated to genomic features: chromatin states, LTRs, dbTSS, RefSeq promoters, and FAIRE-seq regions. The percentage of peak summits within each cluster overlapping a specific feature is indicated. Overlap with LTRs is assayed within a window of ±250 bp from the ends of the LTR feature. RefSeq promoters are considered within a window of −2500:+500 bp from the TSS. A direct overlap with FAIRE-seq regions and chromatin states is used. Long poly(A) purified RNA reads were counted within a window of ±500 bp about the NF-YB peak summit, and the median value of that cluster is shown (n = size of cluster in peaks).
A subset of NF-Y sites is located at tissue-specific enhancers
Although NF-Y is typically described as a proximal promoter factor, binding to enhancers has been described, e.g., the 5′ upstream regions of the MHC class II genes (Dorn et al. 1988) and the intronic enhancer of the HOXB4 gene (Gilthorpe et al. 2002). In this regard, all four enhancer chromatin states, as defined by Ernst et al. (2011), are bound by NF-YB (Fig. 2C), totaling 25% of NF-Y peaks in K562. From clustering analysis of histone modifications and Pol II, 12% of all NF-Y sites (clusters E, R, and T; n = 1525) have histone modification patterns typical of enhancers: high H3K4me1, low H3K4me2/me3, low Pol II, and only a modest overlap with RefSeq TSSs (Fig. 3A,B). This is also observed with NF-Y sites in HeLa S3 (Supplemental Fig. 8). The apparent discrepancy in the NF-Y sites in K562 designated as enhancer (25% vs. 12%) is likely due to our more conservative definition of enhancer and wider region used for interpretation. Clusters E and R (Fig. 3A) are exceptional in that they represent NF-Y sites located close to (∼2.5 kb), but not within regions of high enrichment for H3K27ac, H3K9ac, H3K4me1/me2/me3 (i.e., strong actively transcribing promoters), unlike all other clusters from the enhancer and promoter groups, where NF-Y is directly within the domains enriched for acetylation and methylation.
Interestingly, cell-type-specific NF-Y sites are enriched for enhancers and are, on average, located further away from TSSs as compared with NF-Y sites common to all cell types (Supplemental Fig. 9A,B). GO analysis of cell type-specific NF-Y loci reveals categories enriched in individual cell types. NFkappaB cascade and regulation of IL12 is enriched in GM12878, a cell type in which NFkappaB is constitutively active (Gubler et al. 1991; Wolf et al. 1991), and HeLa S3 shows enrichment for epidermis morphogenesis and establishment of tissue polarity, commonly associated with cells of epithelial origin (Supplemental Fig. 4B).
Functional inactivation of NF-YA supports a transcriptional role for NF-Y located distally to TSSs
The large number of NF-Y locations at distal enhancers that are functional as defined by the histone modification pattern (Fig. 2C) and previous analysis of individual genes (Dorn et al. 1988; Gilthorpe et al. 2002) strongly suggests that NF-Y binding to distal locations can have functional consequences. To provide additional evidence for this idea, we performed expression array analysis on HeLa S3 cells depleted for NF-YA by lentiviral small hairpin RNA (shRNA) (Supplemental Fig. 10A,B) and correlated these changes to the location of NF-Y (Supplemental Figs. 10C, 11A,B). At a P-value cutoff of 10−4, 84 genes are down-regulated and 252 genes are up-regulated (Supplemental Fig. 10C) upon NF-YA knockdown. Of these, only 11% (n = 9) and 39% (n = 98) have NF-Y bound to their proximal promoters, respectively. The topmost differentially down- and up-regulated genes both trend toward having a higher percentage of their promoters occupied by NF-Y than nondifferentially regulated genes (Supplemental Fig. 11A). Of the 1059 NF-YA peaks in HeLa S3 located within 250 bp of a RefSeq TSS, only 5.2% are differentially regulated at a P-value of 10−4 (n = 55). The low percentage of differentially regulated genes bound by NF-Y is similar to that found with other TFs (Yang et al. 2006; Strub et al. 2011; Martynova et al. 2012) and could be exacerbated by the incomplete functional inactivation of NF-Y.
We ranked NF-Y sites by the fold change in RNA expression of the nearest associated gene upon NF-YA inactivation. Importantly, for both promoters and distal regions, the topmost differentially down- and up-regulated genes both trend towards having a higher percentage of NF-Y occupancy than nondifferentially regulated genes (Supplemental Fig. 11A). In addition, the most strongly down-regulated genes have NF-Y sites that are much more distal to the TSS, with the median distance being >10 kb; this preference for distal sites is not true for MYC (Supplemental Fig. 11B). This observation suggests the possibility that distal NF-Y sites might be more important for differential regulation. Whatever the relative importance of NF-Y at promoters and enhancers, our results support the idea that NF-Y located at both promoters and enhancers can be important for transcription of neighboring genes.
LTRs are the most prevalent class of NF-Y sites in the human genome
Of all NF-Y binding sites in K562, 40% directly overlap an LTR, the promoter elements of endogenous retroviruses, making LTRs the most prevalent class of NF-Y loci in the human genome (Fig. 4A). NF-Y selectively associates with the MLT1 and LTR12 families of LTRs (Fig. 4B,C). NF-Y does not bind to all LTR families, irrespective of the presence of a CCAAT motif in the consensus sequence. The R66 tandem repeat (which is related to LTR12B) (Benachenhou et al. 2009a,b), MER51A and MER51E are also associated with NF-Y. In general, there is no significant cell-type specificity in LTR binding (Supplemental Fig. 12).

NF-YB binds extensively to long terminal repeats. (A) The percentage of all K562 NF-YB peak summits that occupy the indicated feature. Core and proximal promoters are defined as −250:+50 bp and −2500:+500 bp from the TSS of RefSeq promoters, respectively. (B) Mapping of ChIP-seq reads from K562, GM12878, and HeLa S3 to Repbase consensus sequences showing an abundance of NF-Y ChIP-seq reads mapping to repetitive elements. Ratios reflect the enrichment of reads in the NF-YB ChIP sample as compared with input. Only Repbase entries with a read ratio ≥5 are shown. Orange shading indicates enriched repeats present in all cell lines. Green and red shading indicate the presence and absence, respectively, of a CCAAT motif match at P-value < 10−4 in the consensus sequence. (C) Frequency of overlap between NF-YB peak summits and the genomic locations of LTR families. Only LTR elements that overlap at least one NF-YB summit in each cell line are shown. The two most highly overlapping repeat families are indicated, LTR12 and MLTJ1. (D) Distribution of NF-YB bound LTRs from K562 and GM12878 at chromatin states. No chromatin state map is available for HeLa S3.
Most NF-Y bound sites at LTRs lack any detectable histone modifications within 5 kbp of the NF-Y peak summit (Figs. 3A,B, 4D, clusters D and J) . These NF-Y loci appear to be inactive, yet maintain substantial NF-Y occupancy. In contrast, a sizeable minority of LTRs (27% K562; 20% GM12878) (Fig. 4D) are associated with high levels of H3 acetylation and/or H3K4 methylation and appear to be transcriptionally active. This minority class most likely represents NF-Y-bound functional regulatory elements derived from transposable repetitive elements and regulating endogenous genes.
LTRs function as promoter elements of endogenous retroviruses and they can act as regulatory elements for certain host genes (Bourque 2009). NF-Y sites abound in viral LTRs (Graves et al. 1986; Dutta et al. 1990; Faber and Sealy 1990; Greuel et al. 1990; Scheef et al. 2002). The selectivity for the gamma-retrovirus LTR family, and within it for certain members, likely reflects the presence of CCAAT in the original viral LTRs. Thus, our results suggest a strong genetic pressure on their genomic transduced copies to maintain NF-Y binding. This is not unprecedented, as evidenced by the preference of particular TFs for specific repetitive sequences (Bourque et al. 2008; Kunarso et al. 2010). Genetic analysis of the ERV-9/LTR12 element located 5′ of the globin locus-control region indicates a crucial role of the 14 CCAAT and GATA containing E3 repeats for expression of the β-globin locus (Yu et al. 2005; Pi et al. 2010). Despite this precedent, most NF-Y sites are associated with heterochromatin-like domains and are apparently devoid of any transcriptional signal. As the vast cohort of endogenous LTR proviral sites are under strong control by the host organism and, in most cases, actively repressed (Bourque 2009), we are tempted to speculate that NF-Y plays a role in the repression of these LTRs in somatic tissue and/or in their activation during embryogenesis, where many repetitive elements are demethylated and become expressed (Maksakova et al. 2008).
NF-Y binds CCAAT motifs in nonmodified chromatin domains in vivo, unlike most TFs
The majority of NF-Y sites (n = 6169; 49%) are in two similar clusters (D and J, i.e., LTR/nonmodified-chromatin class in Fig. 3A,B) that display no positive or repressive histone modifications tested for, negligible Pol II and polyA RNA, and overlap few open regulatory regions (11%, 25%) and RefSeq TSSs (7%, 11%). An analysis of MYC sites reveals the absence of binding at nonmodified chromatin sites (Supplemental Fig. 7). Interestingly, most of these loci overlap LTRs, 58% and 82%, respectively (Fig. 3B). These NF-Y sites are interesting, as most TFs are believed to not be able to bind to their DNA motifs within closed, transcriptionally inactive chromatin domains.
To further explore this issue, we calculated the percentage of motifs residing within NF-Y peaks within distinct chromatin states, over a range of motif quality scores. Interestingly, and unlike other TFs such as E2Fs and MYC, NF-Y is not excluded from any chromatin state assayed (Fig. 5A,B). At strong and weak promoters, >80% of CCAAT motifs (scores >16) are occupied by NF-Y (Fig. 5A). CCAAT motifs at enhancers and insulators are also well occupied by NF-Y (30%–75%, respectively) (Fig. 5A), although the percent occupancy is lower than at strong promoters, indicating that binding to these genomic regions is more selective. More generally, CCAAT motifs situated within open chromatin regions, as defined by FAIRE, are exceptionally well occupied to near-saturated levels by NF-Y, with 80% occupancy (Fig. 5A). Interestingly, many CCAAT motifs within the nonmodified chromatin, PcG repressed and transcription elongation states are occupied by NF-Y at a rate of ∼20% (Fig. 5A).
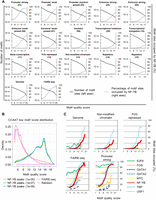
NF-YB can occupy its motif in closed chromatin. (A) The percentage of genome-wide computationally discovered CCAAT motifs within each chromatin state, FAIRE-seq regions or the entire genome, that directly overlap NF-YB K562 sites plotted as a function of CCAAT motif quality (right axes). Also shown are the numbers of discovered CCAAT motifs as a function of quality (left axes). Numbering is derived from Ernst et al. (2011) and kept for consistency. (B) Distribution of CCAAT motif quality scores under NF-YB K562 peaks, called at three different P-values, a random genomic background sample set of 400 k 500-bp regions and K562 FAIRE-seq regions. (C) Similar to A, except motifs of different TFs are plotted as a function of motif quality. Only a subset of TFs is shown; see Supplemental Figure 13 for all TFs analyzed.
To test whether the substantial occupation of CCAAT motifs within nonmodified chromatin and repressed genomic contexts is unique to NF-Y, we performed the same analysis on 22 additional TFs, whose binding sites in K562 cells have been determined by ENCODE. As expected, most TFs show high levels of motif occupancy at nucleosome-depleted regulatory regions at high levels, comparable to that of NF-Y (Fig. 5C; Supplemental Fig. 13). In contrast, GATA1 and GATA2, thought to be “pioneer” TFs (Magnani et al. 2011; Zaret and Carroll 2011), are highly selective and unable to saturate their motifs that reside within these nucleosome-depleted regulatory regions. However, most TFs lack the ability to occupy even their highest quality motifs within nonmodified and repressed chromatin states. For the 23 factors tested, only USF1, MAFK, and NF-Y can bind to motifs in the context of nucleosomes lacking some of the most common “positive” histone modifications or containing the repressive H3K27me3 mark.
By preventing accessibility to target sites, chromatin is a formidable barrier for binding by most TFs. This creates a dilemma as to how cis-regulatory motifs can provide transcriptional competency if they cannot be accessed by their corresponding TFs. There are a small number of “pioneer” factors that can efficiently bind to their DNA motif located within nonmodified, closed chromatin. Once bound, these pioneer TFs can recruit chromatin-modifying activities to generate open chromatin for the subsequent binding of partnering TFs (Magnani et al. 2011; Zaret and Carroll 2011). NF-Y can associate with a CCAAT motif after nucleosome assembly in vitro, and the NF-YB/NF-YC HFD dimer can physically interact with H3/H4 in solution and on DNA (Caretti et al. 1999). Indeed, NF-Y binding is not mutually exclusive with nucleosomes in vitro, giving NF-Y the theoretical functional ability to interact efficiently with chromatin-bound CCAAT motifs in vivo. NF-Y binds to a sizeable number of sites either in functionally “hostile” environments or sites lacking all of the common positive histone modifications. Perhaps the structural features of the HFD heterodimer are instrumental for this. We propose that NF-Y is a type of “pioneer” TF that retains histone-like features while possessing high-sequence specificity, with the ability to access its motif irrespective of the chromatin state.
NF-Y extensively coassociates with FOS, typically at loci lacking an AP-1 motif
Unexpectedly, we observe a remarkable coassociation of NF-Y and FOS (Pearson = 0.74) that is only marginally lower than that observed between the NF-Y subunits (Pearson = 0.77) (Fig. 6A). This coassociation is observed over all chromatin states, cluster classes, and genic features, with 45% of NF-Y sites directly overlapping a FOS site and 39% of FOS sites directly overlapping an NF-Y site (Fig. 6B). Similar results are observed in HeLa S3 cells, although the overlap is slightly lower (26% and 16%, respectively). Interestingly, NF-Y does not significantly coassociate with JUN (Pearson = 0.14), a TF that forms heterodimers with FOS (Fig. 6A). NF-Y and FOS sites are located just as close (<50 bp) as that observed between the NF-Y subunits or between FOS and JUN (Fig. 6C). Interestingly, most sites bound by NF-Y and FOS lack detectable AP-1 motifs (Fig. 6D), with the notable exception being sites at LTR loci (see below). In contrast, and as expected, most sites bound by FOS and JUN have an AP-1 motif (Fig. 6D). A representative example of the interplay is shown in Figure 6E.
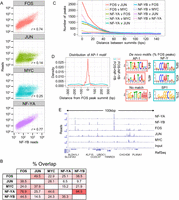
NF-Y and FOS are closely coassociated at loci that lack JUN and the AP-1 motif. (A) Correlation between ChIP-seq read counts at NF-YB peak summits, within a window of ±500 bp, between NF-YB and NF-YA, FOS, JUN, or MYC in K562 cells. (B) Values represent the percentage of peak populations (left row) directly overlapping the peak population of a second factor (top column). All binding sites are called at a P-value < 10−9. FOS (n = 14404); JUN (n = 18480); MYC (n = 13693); NF-YA (n = 4726); NF-YB (n = 12655). (C) The number of ChIP-seq peaks at the indicated distance between adjacent peak summits is plotted. All peaks were called at a 10−9 P-value threshold in K562. (D) The top 1000 K562 FOS ChIP-seq sites, as ranked by site P-value, that directly overlap an NF-YB site (“FOS+NF-YB”) and the top 1000 that do not overlap an NF-YB site (10−5 P-value site list, “FOS-NF-YB”) are assayed for the distribution of the AP-1 motif in relation to the FOS peak summit centered at 0 bp. Plotted is the Gaussian kernel density estimate of the AP-1 motif using a bandwidth of 0.5 of the standard deviation of the smoothing kernel. The top three motifs discovered de novo from each FOS peak set, as above, are depicted with the percentage of FOS peaks containing a match to that motif indicated. (E) Representative view of a locus on chromosome 3 of the K562 ChIP-seq read counts from NF-YA, NF-YB, FOS, JUN, and MYC ChIPs, with an input control.
NF-Y and FOS protein–protein interactions have not been described, but the striking colocalization of these factors suggests the possibility of a direct interaction in the context of chromatin. Alternatively, the coassociation at genomic sites in vivo may not be due to a direct interaction between these factors. In this regard, ChIP-seq analysis reveals that sites bound by JUN N-terminal kinases (JNKs) generally lack AP-1 motifs, but often contain CCAAT motifs (Tiwari et al. 2012). Furthermore, NF-Y is necessary for the JNK association with these genomic sites. We speculate that FOS, together with another protein(s) such as JNK, binds to NF-Y at CCATT motifs in the context of chromatin.
NF-Y coassociates with different TFs depending on genomic context
Given the availability of 78 ChIP-seq data sets in K562 for chromatin associating factors involved in diverse functions, we explored combinatorial genomic interactions of these factors with NF-Y on promoters and enhancers. On a pairwise basis, we observe coassociation between NF-Y and 44 factors at promoters and 50 factors at enhancers (P < 10−10, Supplemental Fig. 14A), consistent with the general requirement for multiple TFs to stimulate transcription in mammalian cells. Hierarchical clustering (Supplemental Fig. 14B), and analysis of higher order combinations of factors (Supplemental Fig. 14C), reveals distinct groups of factors at proximal promoters and enhancers. At promoters, the core group of NF-Y coassociating factors includes FOS, CHD2, TBP, Pol II, CCNT2, HMGB2, MYC, and E2F4/-6 (Supplemental Fig. 14B). A comparison of promoters that are or are not bound by NF-Y reveals that only FOS and (to a lesser extent) CHD2 are specifically associated with NF-Y. FOS associates with 59% of promoters bound by NF-Y, but only 8% of promoters not bound by NF-Y. The other factors were common promoter bound TFs (Supplemental Fig. 14C). At enhancers, NF-Y forms a well-defined cluster consisting of FOS, USF1/2, MAX, CHD2, and E2F4 (Supplemental Fig. 14B), a slightly different grouping compared with promoters. FOS and USF1 are highly prevalent, being present, respectively, at 39% and 27% of NF-Y enhancers, and were the most common 2-way overlap at 13% (Supplemental Fig. 14C). A summary of the interactions at promoters and enhancers is shown in Figure 7.
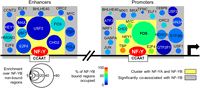
NF-Y coassociates with many factors at promoters and enhancers. Illustration of the factors that significantly associate with NF-YB-bound strong promoters and enhancers. Only those factors that satisfy the following criteria are shown: greater than the median fold enrichment with respect to NF-YB-nonbound regions (enrichment indicated by circle size); greater than the median value of percent occupancy of NF-YB-bound regions (percentage occupied indicated by color); significantly coassociate with NF-Y (gray box, see Supplemental Fig. 14A). Factors enclosed within a yellow box are, additionally, the subset of factors that cluster with NF-YA and NF-YB (see Supplemental Fig. 14B). A black arrow indicates the start of a transcribed region. Two vertical slashes are used to represent being distal to a promoter area.
The widespread partnership of NF-Y with a group of TFs (FOS, MYC, and E2Fs) that control cellular proliferation and play important roles in cancer are consistent with the importance of NF-Y for expression of growth-regulating genes. The close association of the E2Fs and NF-Y is consistent with the high enrichment of their motifs at promoters of genes overexpressed in tumors (Rhodes et al. 2005; Sinha et al. 2008; Goodarzi et al. 2009). In addition, apoptosis mediated by overexpression of NF-Y is abolished in cells lacking E2F1 (Gurtner et al. 2010). E2F4 is part of the DREAM complex (Litovchick et al. 2007; Schmit et al. 2007), which binds to the CDE motif, and cooperates with the CCAAT motif to negatively regulate expression of G2/M-specific genes during the cell cycle (Muller and Engeland 2010; Muller et al. 2012). CCAAT motif and CDE containing G2/M genes are significantly overexpressed in a model of stepwise transformation of primary fibroblasts (Tabach et al. 2005).
Essentially, all E box binding TFs present in ENCODE are statistically enriched at NF-Y locations (Supplemental Fig. 14A), suggesting a pervasive partnership between CCAAT and E boxes. Interestingly, the number of MYC/NF-Y bound promoters exceeds those with MAX/NF-Y, suggesting that either MYC heterodimerizes with another E box binding partner, or that it binds in an E box-independent manner, possibly directly to NF-Y (Izumi et al. 2001; Ravasi et al. 2010).
At the LTR/nonmodified chromatin class, NF-Y extensively colocalizes only with FOS, USF1, and to a lesser degree, USF2 and SP1, although specific groupings occur upon clustering (Supplemental Fig. 15A). The chromatin at colocalized NF-Y/USF sites is not acetylated, suggesting that USF1 and USF2, in the context of binding with NF-Y, do not behave as barrier elements of acetylated chromatin in intergenic regions that inhibit the spread of heterochromatin (West et al. 2004; Huang et al. 2007; Li et al. 2011). It should also be noted that cluster HL2 (n = 147; Supplemental Fig. 15A) is enriched for four members of the CTCF–cohesin insulator complex (CTCF, CTCFL, RAD21, and SMC3) in direct proximity with NF-Y, and a similar small cluster is also observed in the PcG repressed class (data not shown). Interestingly, the NF-Y-bound regions in the LTR/nonmodified chromatin class show a remarkable paucity of known DNA motifs, with the exception of the CCAAT motif and in K562, but not GM12878, the KLF4 motif (P = 1.6 × 10−10; data not shown). De novo motif analysis of the same NF-Y sites again reveals the CCAAT and KLF4 motifs as well as two unknown motifs (Supplemental Fig. 15B). As KLF4 can act as a transcriptional activator or repressor (Turner and Crossley 1998; van Vliet et al. 2000; Schuierer et al. 2001; Yoon and Yang 2004; Evans et al. 2007; Oishi et al. 2008) and is expressed in K562 cells (Kalra et al. 2011), it may cooperate with NF-Y to repress LTR elements.
NF-Y sites contain positionally biased TFs
To investigate whether there is a specific distance relationship between NF-Y and coassociating factors, we plotted the distribution of the relative position of the TATA element, E box, E2F, and AP-1 motifs (termed “predicted”) at NF-Y peaks in relation to the position of the best-scoring CCAAT motif, while maintaining strandedness. We then plotted the subset of motifs (termed “verified”) that were actually occupied in vivo by the TF of interest. Remarkably, there is an AP-1 motif 10- to 11-bp upstream of CCAAT, which corresponds to verified FOS target sites (Fig. 8A). However, this positioning was only found at NF-Y-bound LTRs, as sites with NF-Y and FOS generally do not contain an AP-1 motif (Fig. 6D). The TATA element (+50), E box (−12/−11) and E2F (+6/+7, +31, +55, and +72) motifs are also highly positioned in a CCAAT orientation-specific manner (Fig. 8B). The position of the TATA element is maintained in TBP-bound locations at NF-Y sites. The E2F motif is unusual in that multiple stereo alignments are present and only one, the closest to CCAAT, is maintained at E2F6, but not at E2F4 occupied sites (Fig. 8B; data not shown). The positioning of the E box is only maintained when MAX or USF1 but not MYC loci are considered, suggesting that MYC, when associating with NF-Y, is either not positioned or does not bind DNA directly.
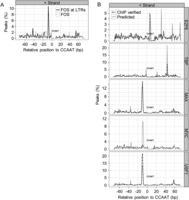
Motif pairings with the CCAAT motif are stereo positioned. (A) The percentage of NF-Y sites that have an AP-1 motif at the specified distance from the best scoring CCAAT motif centered at 0 bp. NF-YB peaks overlapping LTRs are categorized as “predicted,” while the subset of NF-YB sites overlapping the respective ChIP-seq sites of FOS are categorized as “verified.” The negative strand plots are near identical mirror images of the positive strand plots and are not shown. (B) Similar to A, except that all genomic regions are considered. The percentage of NF-YB peaks that have a TATA element (TBP), E box (MYC, MAX, USF1), and E2F motif (E2F6) are plotted. All NF-Y peaks are categorized as “predicted,” while those NF-Y peaks overlapping the respective ChIP-seq peaks of the other TF are categorized as “verified.” Only the top 500 peaks in each category are plotted.
The USF1 observation is interesting because it is one of the few factors that partners with NF-Y in the LTR/nonmodified chromatin class and can bind its motif within a repressive nucleosomal structure. Perhaps the precise positioning may facilitate the cooperation of NF-Y and USF1 to penetrate inactive, nonmodified chromatin domains.
Cooperativity mediated by precise spacing between NF-Y and other TFs has been observed at MHC class II promoters, NF-Y/ATF6 sites in ER-stress responsive promoters (Yoshida et al. 2000), and multiple CCAAT motifs in G2/M promoters (Salsi et al. 2003). Our results greatly extend these findings of precise spacing relationships with NF-Y with its most common TF partners, notably those that play crucial roles in the control of cell proliferation, cell cycle, and metabolism genes. In the vast majority of NF-Y-bound promoters, where NF-Y synergizes with neighboring TFs, it appears to be more of a promoter organizer and facilitator of transcription than a strong activator per se. Our results strongly suggest that cooperativity mediated by precise spacing is a general mechanism utilized by NF-Y to regulate transcription of its target genes.
Conclusions
Our comprehensive analysis of NF-Y confirms many functions including its prevalence at proximal promoters, particularly those of growth controlling genes, at a much higher degree of precision and completion. More interestingly, our analyses uncover several novel and unexpected aspects of NF-Y function. In particular, NF-Y binds asymmetrically at its target sites, plays an important role at many tissue-specific enhancers, is capable of binding “closed” chromatin including at LTRs, coassociates pervasively with FOS but not other AP-1 factors, and displays precise stereo positioning with a restricted group of TFs involved in cellular proliferation. Lastly, we note that comprehensive bioinformatic analyses of the type performed here have been done on relatively few TFs. Similar analyses on other TFs whose target sites have been or will be defined by ChIP-seq are likely to uncover new functional properties and relationships of biological relevance.
Methods
Cell culture
K562, GM12878, and HeLa S3 were grown as per standard ENCODE protocols (The ENCODE Project Consortium 2011) and a detailed protocol is available at http://genome.ucsc.edu/ENCODE/.
ENCODE data sets
ChIP-sequencing data sets for histone PTMs, TFs, and RNA-seq for K562 and/or HeLa S3 cell lines were provided by ENCODE via the UCSC Genome Browser and are described there and elsewhere (The ENCODE Project Consortium 2011; http://genome.ucsc.edu/ENCODE/). ChIP-seq data sets were mapped and peaks called as described in the Supplemental Methods. RNA-seq data was prepared by Helicos as long (>200 nt), poly(A)-enriched, cytosolic RNA, and mapped using rSeq (Jiang and Wong 2008, 2009). Chromatin state maps and the associated numbering are from ENCODE and are detailed at http://genome.ucsc.edu/ENCODE/ and in Ernst et al. (2011). The chromatin state “heterochromatin” was renamed to “non-modified-chromatin.”
Lentiviral knockdown and gene expression arrays
Scrambled control (shSCM) and NF-YA pLKO.1-shRNAs were designed by Sigma-Aldrich. The puromycin resistance cassette was replaced with an EGFP cassette. Viral production and transduction were carried out as previously described (Benatti et al. 2011). HeLa S3 cells were transduced with shSCM (scrambled control) or shNF-YA viral supernatants, in triplicate, and cells were collected after 48 h of incubation. The distribution of cells within the cell cycle was checked via FACS as previously described (Benatti et al. 2011). Knockdown efficiency was assayed by PCR on cDNA to known NF-YA target genes and by Western Blot on whole-cell protein extracts using anti-NF-YA and anti-actin antibodies. For arrays, total RNA was prepared by TRIzol extraction and Qiagen RNeasy kit purification, converted to biotinylated aRNA, and hybridized to U133 Plus 2.0 GeneChip expression arrays using the 3′ IVT Express Kit (Affymetrix) following the manufacturer's protocol. Arrays were RMA normalized (Irizarry et al. 2003), gene expression levels calculated, differential expression determined, and probes annotated using the following R packages from the Bioconductor project: affy (Gautier et al. 2004), limma (Smyth 2004), and annaffy (http://www.bioconductor.org/packages/devel/bioc/html/annaffy.html).
Annotation of peaks to gene features, GO analysis (GREAT/IPA)
Genomic locations of peak summits (where summit is the local maxima in read counts) were submitted to the annotation tool GREAT (McLean et al. 2010) using the following parameters: whole-genome background set, basal plus extension, proximal upstream, 5 kbp; proximal downstream, 1 kbp; distal, 1 mbp; or whole-genome background set, basal, proximal upstream, 5 kbp; proximal downstream, 1 kbp. Molecular signaling pathways were visualized using IPA (Ingenuity Systems: http://www.ingenuity.com) where a gray-shaded node represents a K562 NF-YB binding site located within the putative regulatory region, as defined by GREAT, of that molecule. Peak summits were annotated to genomic features using in-house scripts.
Motif stereo-positioning
NF-YB summit locations from K562 were scanned using Pscan (Zambelli et al. 2009) for matches to the NF-Y matrix in the JASPAR_CORE_2009 database (MA0060.1) (Portales-Casamar et al. 2010). For NF-Y loci with the best matrix match on the positive strand, the first C (of CCAAT) of the best match was set to 0 bp. Genomic sequences ±75 bp from the motifs were retrieved and scanned with Pscan using the collection of matrices in the JASPAR_CORE_2009 database (Portales-Casamar et al. 2010). For each JASPAR matrix, only regions containing a best matrix match >0.8 (computed as described in Zambelli et al. 2009) were considered for further analyses. This population was deemed “predicted.” For each “predicted” population, the subpopulation of regions that overlapped the relevant TF ChIP-seq peak data set were deemed “ChIP verified.” The frequency of the best motif occurrences for each motif matrix at each base pair from the CCAAT motif was determined for each population and plotted as the percentage of motifs.
Histone modifications and chromatin-associated factor clustering
Density arrays at NF-YB peak summits spanning either ±5 kbp or ±500 bp representing ChIP-seq read counts of histone PTMs (H3K79me2, H3K4me3, H3K27me3, H3K4me1, H4K20me1, H3K36me3, H3K4me2, H3K9ac, H3K9me1, H3K27ac), NF-YA, NF-YB, and RNA Pol II or NF-YA, NF-YB, and 78 chromatin-associated factors (see Supplemental Fig. 15A for the full list) with appropriate input samples, were computed using the ranked based correlation method of seqMINER v1.2 (Ye et al. 2011). Clustering was carried out using the following parameters: T = 10, K-means. Clusters from three to 50 were considered. Non-normalized raw read counts are depicted in Figure 3A and Supplemental Figures 7, 8, and 15A.
Mapping to repeats
Bowtie (Langmead et al. 2009) was used to map the NF-YB and input ChIP-seq data sets to a reference genome composed of Repbase v15.08 (Jurka et al. 2005) entries—simple.ref, humrep.ref, humsub.ref, and pseudo.ref—allowing ≤2 mismatches per read, and reads with >1 alignment had one alignment selected at random. Read counts for each Repbase entry were tallied and the ChIP:input ratio calculated. Individual consensus sequences of repeat elements were scored for the presence or absence of the CCAAT motif using the matrix derived from this study and FIMO (Grant et al. 2011), with matches called at a significance P-value threshold of 10−4.
Hierarchical clustering of binding events to promoters and enhancers
Regions considered promoters and enhancers were taken from the K562 chromatin state maps of Ernst et al. (2011). Regions were considered “bound” if an NF-YB peak summit directly overlapped the region. Regions were considered “nonbound” if no NF-YB peak overlapped the region of interest and the region had <1.5× the normalized fold-over-input ChIP-seq enrichment. At all NF-YB bound or NF-YB nonbound regions, chromatin associated factors were scored as present (1) or absent (0) based on directly overlapping peak summits. The R packages pvclust (Suzuki and Shimodaira 2006) and snow (http://cran.r-project.org/web/packages/snow/) were used to cluster the matrices and to calculate P-values using multiscale bootstrap resampling. Parameters were: method.dist=”binary”, method.hclust=”ward”, nboot=10000. Red and blue numbers in plots indicate the approximately unbiased (AU) P-values and the bootstrap probability (BP), respectively, as detailed in Suzuki and Shimodaira (2006).
Statistical test of TF coassociation with NF-YB
NF-YB-bound regions were as above. We assessed promoters or enhancers occupied by NF-YB for individual co-occupancy of 78 transcriptional regulators. The significance of the overlap was tested by a 2 × 2 contingency table using Fisher's exact test and calculated (Carlson et al. 2009), and P-values <10−9 were deemed significant.
Data access
Microarray gene expression data from this study have been submitted to the NCBI Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE40215.
Acknowledgments
The NF-Y ChIP-sequencing data was generated as part of ENCODE. We thank the members of the Snyder and Gerstein labs, and the ENCODE Project Consortium for support and access to pre-release data sets; Hannah Monahan for preparing sequencing libraries; WQCG and RITG for technical support and access to computing facilities; Koon Ho Wong, Rajani Gudipatti, Nathan Lamarre-Vincent, and Joseph Geisberg for advice and help with figures; Benoit Miotto for performing Orc2 ChIP-seq. This work was supported by grants to K.S. from the National Institutes of Health (GM30186; HG4558), to R.M. from Lombardy Region (NEPENTE) and AIRC, and to C.I. from AIRC (MFAG 6192).
Author contributions: J.D.F. and K.S. conceived the project. J.D.F., K.S., R.M., G.P., P.B., and C.I. participated in experimental design. J.D.F. performed biological experiments and analyzed the data; G.P. analyzed the motif stereo-positioning data; P.B., C.I., and J.D.F. performed shRNA experiments. J.D.F., R.M., and K.S. wrote the paper. All authors have read and accepted the manuscript.
Footnotes
-
↵4 Corresponding author
E-mail kevin{at}hms.harvard.edu
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.148080.112.
- Received August 20, 2012.
- Accepted April 11, 2013.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see http://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 3.0 Unported), as described at http://creativecommons.org/licenses/by-nc/3.0/.








