
Maps of open chromatin highlight cell type–restricted patterns of regulatory sequence variation at hematological trait loci
- Dirk S. Paul1,2,13,16,
- Cornelis A. Albers1,3,4,13,
- Augusto Rendon3,5,6,13,
- Katrin Voss3,
- Jonathan Stephens3,
- HaemGen Consortium15,
- Pim van der Harst7,8,
- John C. Chambers9,10,11,12,
- Nicole Soranzo1,
- Willem H. Ouwehand1,3,14 and
- Panos Deloukas1,14,16
- 1Wellcome Trust Sanger Institute, Hinxton, Cambridge CB10 1SA, United Kingdom;
- 2UCL Cancer Institute, University College London, London WC1E 6BT, United Kingdom;
- 3Department of Haematology, University of Cambridge and National Health Service (NHS) Blood and Transplant, Cambridge CB2 0PT, United Kingdom;
- 4Department of Human Genetics, Radboud University Nijmegen Medical Center, 6500 HB Nijmegen, The Netherlands;
- 5MRC Biostatistics Unit, Institute of Public Health, Cambridge CB2 0SR, United Kingdom;
- 6NIHR Biomedical Research Centre, Cambridge CB2 0PT, United Kingdom;
- 7Department of Cardiology,
- 8Department of Genetics, University of Groningen, University Medical Center Groningen, 9713 GZ Groningen, The Netherlands;
- 9Department of Epidemiology and Biostatistics, Imperial College London, London W2 1NY, United Kingdom;
- 10Imperial College Healthcare NHS Trust, Hammersmith Hospital, London W12 0HS, United Kingdom;
- 11Royal Brompton and Harefield Hospitals NHS Trust, London SW3 6NP, United Kingdom;
- 12Ealing Hospital NHS Trust, Southall, Middlesex UB1 3HW, United Kingdom
-
↵13 These authors contributed equally to this work.
Abstract
Nearly three-quarters of the 143 genetic signals associated with platelet and erythrocyte phenotypes identified by meta-analyses of genome-wide association (GWA) studies are located at non-protein-coding regions. Here, we assessed the role of candidate regulatory variants associated with cell type–restricted, closely related hematological quantitative traits in biologically relevant hematopoietic cell types. We used formaldehyde-assisted isolation of regulatory elements followed by next-generation sequencing (FAIRE-seq) to map regions of open chromatin in three primary human blood cells of the myeloid lineage. In the precursors of platelets and erythrocytes, as well as in monocytes, we found that open chromatin signatures reflect the corresponding hematopoietic lineages of the studied cell types and associate with the cell type–specific gene expression patterns. Dependent on their signal strength, open chromatin regions showed correlation with promoter and enhancer histone marks, distance to the transcription start site, and ontology classes of nearby genes. Cell type–restricted regions of open chromatin were enriched in sequence variants associated with hematological indices. The majority (63.6%) of such candidate functional variants at platelet quantitative trait loci (QTLs) coincided with binding sites of five transcription factors key in regulating megakaryopoiesis. We experimentally tested 13 candidate regulatory variants at 10 platelet QTLs and found that 10 (76.9%) affected protein binding, suggesting that this is a frequent mechanism by which regulatory variants influence quantitative trait levels. Our findings demonstrate that combining large-scale GWA data with open chromatin profiles of relevant cell types can be a powerful means of dissecting the genetic architecture of closely related quantitative traits.
Genome-wide association (GWA) studies have discovered many non-protein-coding loci associated with complex traits. The precise localization of the causative sequence variant(s) at GWA loci is often impeded due to the extent of high linkage disequilibrium (LD), even when fine-mapping data are available. In addition, the functional impact of noncoding sequence variants at the molecular level is difficult to evaluate (Donnelly 2008; McCarthy et al. 2008; Cooper and Shendure 2011). Recent studies have shown that a large proportion of GWA signals are located within active gene regulatory elements in selected cell lines and primary tissues (The ENCODE Project Consortium 2012; Maurano et al. 2012). The ENCODE Project Consortium (2012) mapped deoxyribonuclease I (DNase I) hypersensitive and transcription factor binding sites in 147 cell types, and found that 34% and 12%, respectively, of GWA lead SNPs overlapped with these regulatory regions. Maurano et al. (2012) expanded the catalog of DNase I hypersensitive sites to 349 cell types (including 85 ENCODE cell types), and showed that 57% of GWA lead SNPs were located within these regulatory sites. Additional candidate functional variants were retrieved by considering proxy SNPs that are in high LD with the lead SNP. Despite the ambitious scale of ENCODE and related efforts, biologically relevant effector (primary) cell types have not yet been assayed for many traits.
We recently demonstrated that the formaldehyde-assisted isolation of regulatory elements (FAIRE) technique is a valuable tool in mapping nucleosome-depleted regions (NDRs) at selected genetic loci associated with hematological traits, and in prioritizing candidate variants for experimental validation (Paul et al. 2011). Hematological traits, such as the count and volume of cells in peripheral blood and the hemoglobin content of erythrocytes, are under genetic control and vary extensively between individuals (Evans et al. 1999; Garner et al. 2000). Such traits offer an excellent means of investigating the genetic architecture of closely related complex traits, because the cellular components of the hematopoietic system are well understood and primary precursor cells can be relatively easily accessed for experimental assays.
In this work, we used FAIRE-seq to map NDRs genome-wide in primary human megakaryocytes (MKs) and erythroblasts (EBs), the precursor cells of platelets and erythrocytes, respectively, as well as in monocytes (MOs). We also mapped NDRs in two immortalized cell lines commonly used as models for MKs and EBs, i.e., CHRF-288-11 and K562, respectively. First, we characterize the open chromatin profiles with respect to hematopoietic cell type and lineage, as well as FAIRE signal strength. Second, we assess the cell type–dependent enrichment patterns of sequence variants associated with two platelet and six erythrocyte indices at NDRs, using the results from the largest GWA meta-analyses conducted so far for these traits (Gieger et al. 2011; van der Harst et al. 2012). For these analyses, we also consider unrelated quantitative traits, i.e., fasting glucose (FG) and insulin (FI) levels, body mass index (BMI), and height (Dupuis et al. 2010; Lango Allen et al. 2010; Speliotes et al. 2010), as well as an open chromatin data set in a nonhematopoietic cell type, i.e., pancreatic islets (Gaulton et al. 2010). Finally, we experimentally validate a set of candidate regulatory variants identified within NDRs at platelet quantitative trait loci (QTLs).
Results
Preparation of open chromatin profiles of human myeloid cells
Cord blood–derived CD34+ hematopoietic progenitor cells (HPCs) from two unrelated individuals were differentiated in vitro into either MKs in the presence of thrombopoietin and interleukin-1β, or into EBs in the presence of erythropoietin, interleukin-3, and KIT ligand (also known as stem cell factor). MOs were purified from peripheral blood from another two individuals (Supplemental Fig. 1A–C). In addition, we prepared FAIRE samples from CHRF-288-11 megakaryocytic cells and retrieved publicly available FAIRE-seq data for K562 erythroblastoid cells and pancreatic islets (Gaulton et al. 2010; The ENCODE Project Consortium 2012). Figure 1 gives an overview of the study design. All FAIRE-seq data sets were processed in a standardized manner, as described in the Methods section.
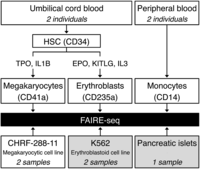
Overview of the study design. Cord blood–derived CD34+ hematopoietic progenitor cells from two unrelated individuals were differentiated in vitro into either megakaryocytes (MKs) or erythroblasts (EBs). Monocytes (MOs) were purified from peripheral blood from another two individuals. We also prepared FAIRE samples from CHRF-288-11 megakaryocytic cells. In addition, we retrieved publicly available FAIRE-seq data sets for K562 erythroblastoid cells and pancreatic islets from The ENCODE Project Consortium (2012) and Gaulton et al. (2010), respectively, and reanalyzed the data sets in concordance with all other FAIRE data sets. (HSC) Hematopoietic stem cell; (TPO) thrombopoietin; (IL1B) interleukin 1, beta; (EPO) erythropoietin; (KITLG) KIT ligand (also known as SCF, or stem cell factor); (IL3) interleukin-3.
We determined FAIRE-derived NDRs (peaks) using a Gaussian kernel density estimator implemented in the software F-Seq (Supplemental Tables 1, 2; Boyle et al. 2008). As the Illumina DNA sequencing platform provides a large dynamic range and high sensitivity using discrete, digital sequencing read counts, we hypothesized that a subclassification of FAIRE peaks based on signal strength may allow more precise downstream functional analyses. Therefore, we stratified the peaks according to their signal strength (F-Seq peak score) into four equally spaced intensity bins, termed “Bins 1–4” (Supplemental Table 3).
The reproducibility of the peak calls across biological replicates in CHRF-288-11 and K562 cell lines increased with signal strength and was consistently >80% for peaks in the top three intensity bins, i.e., Bins 2–4. In contrast, we observed limited reproducibility (<50%) of the peaks in the lowest intensity bin, i.e., Bin 1 (Supplemental Fig. 2A–K; Supplemental Table 4A). Furthermore, the fraction of overlap between peaks in Bin 1 and other regulatory marks (H3K4me1/3 histone modifications and transcription factor binding sites) was small compared with that of peaks in Bins 2–4 (Supplemental Table 4B); i.e., the overlap of FAIRE peaks with transcription factor binding sites in MKs was 4% in Bin 1 and on average 50% across Bins 2–4. Thus, we excluded peaks in Bin 1 from subsequent analyses, as a large fraction of its peaks were neither reproducible nor appeared to bear hallmarks of regulatory chromatin.
Hematopoietic cell type–restricted and lineage-restricted open chromatin signatures
We first investigated to what extent individual myeloid cell types have distinct open chromatin signatures. We constructed distance matrices based on the overlap of NDRs across all sampled cell types (Supplemental Fig. 2A–K) and assessed the uncertainty of the clustering using bootstrap resampling (Suzuki and Shimodaira 2006).
We found that the hierarchical clustering is dominated by cell type identity rather than interindividual variation (Fig. 2A). The observed hierarchical tree branches reflected the established relation of the myeloid hematopoietic lineages. For example, MKs and EBs were found to cluster together, reflecting that the two cell types share a common progenitor, termed the MK-erythroid progenitor. MOs did not co-cluster with MKs/EBs and formed an out-group, corresponding to the split of the common myeloid progenitor into the MK-erythroid and granulocyte-macrophage lineages. As expected, pancreatic islets formed another out-group (Supplemental Fig. 2K) due to the limited overlap between NDRs from endoderm-derived pancreatic islets and from mesoderm-derived hematopoietic cells. The difference in clustering between MOs and pancreatic islets as out-groups was marginal, based on their small number of shared NDRs across cell types (Supplemental Fig. 3). These data suggest that globally, the open chromatin signature of MOs is as unrelated to MKs/EBs as pancreatic islets, and indicate clear differences in chromatin profiles even within related hematopoietic cells of the same myeloid lineage.
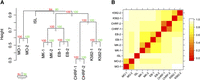
Hierarchical clustering of the overlap of FAIRE-derived nucleosome-depleted regions (NDRs). (A) The hierarchical clustering is based on the overlap of NDRs across different cell types, as shown in Supplemental Figure 2. The dendrogram shows that the clustering is dominated by cell type identity rather than individual preparation. The observed hierarchical tree mirrors the hematopoietic tree, where MKs and EBs share a common progenitor. MKs and EBs do not co-cluster with their representative cell lines, i.e., CHRF-288-11 and K562, respectively, indicating that the open chromatin structure of immortalized lines does not fully reflect that of primary cells. Both MOs and pancreatic islets form out-groups, due to the limited overlap of NDRs with the other cell types tested. This suggests that MOs, despite being one of the myeloid types of cells akin to MKs and EBs, have a marked different open chromatin profile. The hierarchical cluster analysis was performed using the R package Pvclust (distance: binary; cluster method: complete) (Suzuki and Shimodaira 2006). The uncertainty of the clustering was assessed using bootstrap resampling. (B) The heatmap of the binary distances complements the cluster plot. Relationships between NDRs across all samples are observable. The binary distances were plotted using the levelplot function of the R package lattice (http://cran.r-project.org/web/packages/lattice/). (MO) Monocyte; (MK) megakaryocyte; (EB) erythroblast; (ISL) pancreatic islet; (CHRF) CHRF-228-11 megakaryocytic cell; (K562) K562 erythroblastoid cell; (au) approximately unbiased P-value; (bp) bootstrap probability value.
We then compared the open chromatin profiles of MKs and EBs with that of CHRF-288-11 and K562 cells, respectively. The two investigated immortalized lines clustered closer to each other than to their respective primary cell type (CHRF-288-11/MKs and K562/EBs), suggesting differences in open chromatin structure between immortalized lines and that of primary cells (Fig. 2A,B). For example, although there was extensive overlap between NDRs found in MKs and CHRF-288-11 cells (Supplemental Fig. 2A,B), the latter cell type possessed a large number of additional NDRs that overlapped with K562 cells but not with MKs (Supplemental Fig. 2G,H).
Hematopoietic lineage-restricted gene expression patterns
Next, we examined if the identified NDRs mark lineage-specific elements involved in regulation of expression of genes relevant to blood cell lineage commitment. For each cell type, we pooled the sequence fragments of the two individual FAIRE preparations and processed the data as for the individual preparations described above. We then assessed the expression levels of the single closest gene (defined as in terms of the distance to its transcription start site [TSS]) to each cell type–restricted NDR, interrogated over several time points during in vitro differentiation of HPCs into MKs (Supplemental Table 5A) and EBs (Supplemental Table 5B).
Transcripts close to MK-restricted NDRs (i.e., NDRs found only in MKs but not in the other cell types assayed) were more likely to be up-regulated during MK differentiation relative to all expressed transcripts (1.41-fold enrichment; P = 1.00 × 10−29, two-tailed χ2 test). We observed the same effect directionality for transcripts close to EB-restricted NDRs during EB differentiation (1.50; P = 1.23 × 10−59). Transcripts close to MO-restricted NDRs were down-regulated during both MK and EB differentiation with a fold change of 0.91 (P = 3.62 × 10−9) and 0.85 (P = 1.23 × 10−19), respectively.
Interestingly, transcripts in proximity to NDRs shared between MKs and MOs were also up-regulated during MK differentiation (1.19; P = 1.13 × 10−5). The corresponding genes (n = 265) mostly encode signaling proteins downstream from integrins, cytokine receptors, and G protein–coupled receptors that are expressed in both MKs and MOs. We annotated the gene set using the Ingenuity Knowledge Base and found an enrichment of genes in the canonical pathways “Fcγ receptor-mediated phagocytosis in macrophages and monocytes” (P = 4.53 × 10−4, Benjamini-Hochberg corrected for multiple testing; n = 10 genes) and “integrin signaling” (P = 4.53 × 10−4, n = 14). We suggest that the 265 genes may also include a subset of coexpressed genes in MKs and MOs that play a role in the molecular events that link cell surface growth factor receptors to platelet integrin activation.
Functional classification of NDRs based on signal strength
We then investigated whether NDRs of different signal strength have different functional properties: in particular, their distance to the TSS, correlation with promoter and enhancer histone marks, and ontology classes of nearby genes.
First, we examined the location of NDRs of different signal strength relative to the TSS. We observed that for both MKs and EBs but not for MOs, NDRs in the lowest retained intensity bin (Bin 2; Supplemental Fig. 4A) were less often located close to the TSS than NDRs in the highest intensity bin (Bin 4; Supplemental Fig. 4B). We further investigated these observations by performing chromatin immunoprecipitation (ChIP) combined with high-throughput next-generation sequencing (ChIP-seq) of the histone modifications H3K4me3 and H3K4me1, which mark active promoters and enhancers, respectively. In MKs and EBs, NDRs in the highest intensity bin showed stronger overlap with promoters proximal to TSSs compared with NDRs at the other extreme, which showed stronger overlap with enhancers distal to TSSs (Fig. 3A,B). NDRs that did not overlap with either histone mark were more likely to be in the lowest intensity bin and located far from promoters. In contrast, we found that NDRs in the highest intensity bin in MOs were depleted close to the TSS compared with MKs and EBs (Fig. 3C).
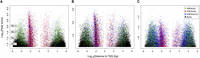
Overlap of H3K4me3 (promoter) and H3K4me1 (enhancer) histone marks with NDRs. In (A) MKs and (B) EBs, NDRs in the highest intensity bin (Bin 4) showed stronger overlap with gene promoters close to TSSs compared with NDRs in the lowest retained intensity bin (Bin 2), which showed stronger overlap with enhancer elements distal to the closest TSS. NDRs that did not overlap with histone marks were more likely to be in the lowest intensity bin and far from promoters. (C) In MOs, however, we found that NDRs in the highest intensity bin were depleted close to the TSS compared with MKs and EBs. The peak bins are indicated with a dashed gray line. These results suggest that NDRs of different signal strength may have different functional properties.
Second, we applied the Genomic Regions Enrichment of Annotations Tool (GREAT) (McLean et al. 2010) to aid the functional interpretation of NDRs of different signal strength by analyzing the annotations of the single closest flanking gene (Supplemental Table 6). In MKs and EBs, NDRs in the lowest intensity bin were enriched in cell type–specific genes, while NDRs in the highest intensity bin were enriched in housekeeping genes. However, in MOs we observed an enrichment of cell type–specific gene sets close to NDRs irrespective of their signal strength. One possible explanation for the lack of enrichment in housekeeping genes could be that mature MOs, as studied here, do not proliferate but rather differentiate into various macrophage classes upon stimulation (Geissmann et al. 2010).
Cell type–dependent enrichment of genome-wide significant SNPs associated with hematological traits at NDRs
We assessed the enrichment of genome-wide significant SNPs associated with platelet and erythrocyte phenotypes at NDRs in a cell type–dependent context. We retrieved proxy SNPs (r2 > 0.8) (The 1000 Genomes Project Consortium 2010) of all GWA lead SNPs (P < 5 × 10−8) at 68 platelet and 75 erythrocyte QTLs (Gieger et al. 2011; van der Harst et al. 2012). By use of these criteria, we obtained 1680 and 4632 SNPs at platelet and erythrocyte QTLs, respectively. Then, we intersected the SNP positions with the composite map of open chromatin in myeloid cell types.
At 18 (26.5%) and 25 (33.3%) of the platelet and erythrocyte QTLs, respectively, we found at least one trait-associated SNP located within an NDR across MKs, EBs, and MOs (Fig. 4A,B; Supplemental Table 7A–D). Next, we compared the extent of overlap with 100,000 random sets of 68 and 75 SNPs that were matched for possible confounding factors such as minor allele frequency (MAF), distance to a TSS, and number of proxy SNPs per locus (Methods). At platelet QTLs, significant overlap with NDRs in MKs (P = 2.0 × 10−5) and MOs (P = 1.7 × 10−3) was observed. The extent of overlap with NDRs in EBs was not more than expected by chance when compared to random sets of SNPs (Fig. 4C). At erythrocyte QTLs, we found significant (P < 1 × 10−5) overlap with NDRs in EBs, but not with NDRs in MKs or MOs (Fig. 4D). At both platelet and erythrocyte QTLs, there was no significant enrichment of GWA signals at NDRs in pancreatic islets.
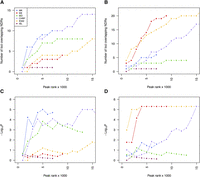
Cell type–dependent enrichment of GWA signals associated with hematological quantitative traits at NDRs. (A,B) Cumulative number of GWA loci harboring platelet (A) and erythrocyte (B) trait-associated SNPs at NDRs across different cell types as a function of rank tranches for decreasing NDR signal strength (F-Seq peak score). (C,D) To determine whether such overlap was expected by chance, we compared the number of overlapping SNPs with 100,000 random samples of 68 and 75 SNPs at the platelet (C) and erythrocyte (D) QTLs, respectively. These random sets of SNPs were matched for possible confounding factors such as minor allele frequency, distance to a TSS, and number of proxy SNPs per locus. The achieved significance level is displayed across the cumulative rank tranches to better appreciate the effect of increasing the number of NDRs in the analysis. The strongest enrichment of genome-wide significant sequence variants at platelet and erythrocyte QTLs was found at NDRs in MKs and EBs, respectively. However, the enrichment was equally clear at NDRs in the respective immortalized lines, i.e., CHRF-288-11 megakaryocytic cells and K562 erythroblastoid cells, respectively. NDRs identified in CHRF-288-11 cells but not MKs were enriched for SNPs associated with erythrocyte indices, indicative of the less differentiated state of cell lines of leukemic origin relative to the primary cells.
Compared with immortalized cell lines representative of MKs and EBs, the same trends of enrichment as for the primary cell types were observed in platelet and erythrocyte traits, respectively. However, NDRs identified in CHRF-288-11 cells were also enriched for SNPs associated with erythrocyte indices (Fig. 4D). This is consistent with the notion that CHRF-288-11 cells are immature and therefore more closely related to MK-erythroid progenitor cells (Nürnberg et al. 2012), and is in agreement with the extensive overlap we found between NDRs in CHRF-288-11 and K562 cells (Supplemental Fig. 2G–J).
The NDRs overlapping platelet trait-associated SNPs were more likely to be restricted to MKs than expected by chance (P = 2.83 × 10−4, Bonferroni-adjusted binomial test) (Fig. 5A; Supplemental Table 8A). We observed the same cell type–dependent effect for erythrocyte trait-associated SNPs at EB-restricted NDRs (P = 4.62 × 10−7) (Fig. 5B; Supplemental Table 8B). These results suggest that regulatory variation may underlie the association signals observed at several of the genetic loci identified in hematological trait GWA studies. Importantly, the cell type corresponding to the hematological trait may play a pivotal role in the genetic architecture of the complex trait.
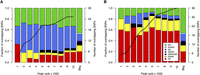
Cell type distribution of NDRs containing candidate functional variants. We considered GWA index SNPs associated with platelet (A) and erythrocyte (B) parameters, as well as their proxy SNPs in high LD (r2 > 0.8; located within 1 Mb of index SNPs). NDRs were ranked by signal strength (F-Seq peak score). Then, these rankings were used to divide the NDRs into cumulative tranches (x-axis) to investigate the impact of peak calling thresholds on results. For example, the first bar represents the tranche containing the 1000 top-ranked NDRs, whereas the penultimate bar represents the tranche containing the 10,000 top-ranked NDRs of each cell type. The bars summarize the cell type distribution of candidate functional SNPs at NDRs as a percentage of the tranche-specific total. The last bar, labeled “Bkg,” represents the expected cell type distribution for the SNPs under the null hypothesis. The solid line indicates the number of SNPs overlapping the tranche-specific NDRs. The results showed that for both platelet and erythrocyte QTLs, the candidate functional variants were most commonly found at MK- and EB-restricted NDRs, respectively. This was true across the spectrum of peak calling thresholds.
We further validated the statistical enrichment (Fig. 4C,D) by estimating the fold change of the number of GWA signals overlapping NDRs, relative to random sets of SNPs (Supplemental Fig. 5A,B). Despite the statistical enrichment and strong fold enrichment in relevant cell types, it is important to note that about half of the observed overlaps between candidate SNPs and NDRs are expected by chance (Supplemental Fig. 5A,B). Taken together, our findings suggest that the intersection of trait-associated SNPs with NDRs, and particularly for NDRs identified in relevant tissues, is likely to provide an informative ranking for selection of candidate causative variants for experimental validation.
Identification of candidate functional variants at platelet QTLs
To provide evidence that the SNPs we identified using the above approach are indeed valid functional candidates, we performed electrophoretic mobility shift assays (EMSAs). Here, we define “candidate functional variant” as a variant that affects DNA–protein binding, a possible mechanism for causality at non-protein-coding regions. We tested 13 functional candidates at 10 of the 18 identified platelet QTLs (Supplemental Table 9). These 13 selected SNPs were located within 11 NDRs that were present in both MKs and CHRF-288-11 megakaryocytic cells. Specifically, we identified two NDRs at the PTGES3-BAZ2A GWA locus, while the NDRs at the FAR2 and DNM3 loci each contained two candidate SNPs (Table 1). Importantly, seven of these 11 NDRs also coincided with binding sites of transcription factors key in regulating megakaryopoiesis (Tijssen et al. 2011), i.e., FLI1, GATA1, GATA2, RUNX1, and TAL1 (also known as SCL), suggesting identification of physiologically relevant regulatory elements.
Summary of the functional evidence obtained for platelet candidate functional SNPs through FAIRE, ChIP, and EMSA experiments, as well as annotation from RegulomeDB
For 10 of the 13 tested SNPs, we observed by visual inspection of the EMSA blot, differential binding of nuclear proteins between alleles in CHRF-288-11 cells (Supplemental Figs. 6A,B,E–I,N,O, 7B). For the three remaining SNPs, we observed comparable protein binding between allelic probes (Supplemental Fig. 6K–M).
We then annotated the candidate SNPs using RegulomeDB, a database containing known and predicted regulatory elements in the human genome (Boyle et al. 2012), and found all but one SNP to coincide with at least one RegulomeDB feature. Table 1 summarizes the obtained functional evidence for the platelet candidate variants.
As an example, the platelet count-associated SNP rs4148450 was located at an MK-restricted intronic NDR of ABCC4. The open chromatin region coincided with a RUNX1 transcription factor binding site in MKs (Supplemental Fig. 7A). ABCC4 encodes the ATP-binding cassette protein ABCC4, also known as multidrug resistance protein 4 (MRP4). Several studies indicated that ABCC4 is involved in the accumulation of the platelet-activating signaling molecule adenosine diphosphate (ADP) in platelet-dense granules (Jedlitschky et al. 2004, 2010). Our data suggest the noncoding SNP rs4148450 to be the functional variant at the 13q32.1 platelet count locus. As a further example, we recently described the molecular mechanism underlying the DNM3 platelet count and volume locus (Nürnberg et al. 2012). In brief, the SNP rs2038479 was located at intron 2 of DNM3, which encodes Dynamin 3, a mechanochemical enzyme involved in MK progenitor proliferation and maturation (Reems et al. 2008; Wang et al. 2011). The corresponding MK-restricted NDR (Table 1) was found to be bound by the MK-specific transcription factor MEIS1 and to mark an alternative promoter of a truncated DNM3 transcript, which is uniquely expressed in MKs and whose level depends on the rs2038479 genotype (Nürnberg et al. 2012).
Enrichment patterns of SNPs associated with closely related quantitative traits at cell type–restricted NDRs
Next, we investigated whether the various hematological parameters showed different patterns of enrichment at NDRs in the primary cells (see Fig. 6). In contrast to the analyses presented in Figure 4, where the different platelet and erythrocyte parameters were combined, here, we investigated the cellular parameters individually, and did not focus only on genome-wide significant signals. We considered sequence variants associated with two platelet indices (PLT, platelet count; MPV, mean platelet volume) and six erythrocyte indices (Hb, total concentration of hemoglobin; PCV, packed red cell volume; RBC, red blood cell count; MCHC, mean red cell hemoglobin concentration; MCH, mean red cell hemoglobin; MCV, mean red cell volume). In addition, we examined sequence variants associated with four nonhematological quantitative traits, i.e., FG and FI levels, BMI, as well as height (Dupuis et al. 2010; Lango Allen et al. 2010; Speliotes et al. 2010). As cell type–restricted NDRs showed stronger enrichment for sequence variants associated with the relevant trait compared with NDRs shared across cell types (Fig. 5A,B), we focused this in-depth analysis on NDRs restricted to MKs, EBs, MOs, or pancreatic islets.

Enrichment patterns of quantitative trait-associated variants with small effect sizes at cell type–restricted NDRs. The data points shown as circles and rectangles represent the deviation of the P-value distribution of SNPs at NDRs restricted to MKs, EBs, MOs, or pancreatic islets (ISLs) from the P-value distribution of matched randomly sampled SNPs at the 0.005 quantile (Supplemental Fig. 8). Thus, this deviation measures the level of enrichment of associated sequence variants at NDRs, where the circle and rectangle surface areas represent level of enrichment (mean ratios > 1) and depletion (mean ratios < 1), respectively. Gray symbols represent ratios that are not significantly different from 1; i.e., the mean ratio across replicates was within 2 SDs of 1. The level of enrichment is indicated for sequence variants associated with two platelet traits ([PLT] platelet count; [MPV] mean platelet volume), six erythrocyte indices ([Hb] total hemoglobin concentration; [PCV] packed red cell volume; [RBC] red blood cell count; [MCHC] mean red cell hemoglobin concentration; [MCH] mean red cell hemoglobin; [MCV] mean red cell volume), as well as four nonhematological quantitative traits ([FG] fasting glucose; [FI] fasting insulin; [BMI] body mass index; height). The circle area labeled “Power” gives a quantification of the amount of signal present in each GWA data set. Specifically, it represents the deviation of the P-value distribution of all tested SNPs from the expectation under the null at the 0.005 quantile.
To improve the statistical power of this analysis, we compared the distribution of P-values for all SNPs located at NDRs to the distribution of randomly selected SNP sets from the genome, matched for possible confounding factors (Methods). Specifically, we calculated the ratio at the 0.005 quantile between the P-value for a random, matched set of SNPs and the P-value for the SNPs within NDRs. Then, we estimated the ratio in 5000 bootstrap samples (Supplemental Fig. 8). In the absence of enrichment at NDRs, this ratio is expected to be one. Thus, this analysis quantifies to what extent SNPs at NDRs tend to have lower P-values than SNPs located outside NDRs, providing an indication of the extent to which SNPs at NDRs in a given cell type are enriched for potential causative variants compared with randomly selected SNPs. However, this analysis does not quantify the overall contribution from these variants to the phenotypic variation.
We observed diverse cell type–dependent enrichment patterns at NDRs for the various cellular parameters. For example, we found strong enrichment of SNPs associated with erythrocyte indices at EB-restricted NDRs. MCH and MCV, which are highly correlated quantitative traits (r = 0.91) (Supplemental Table 10), showed substantially stronger enrichment (ratios > 106.2 ± 1.5) compared with the other four erythrocyte traits investigated (ratios < 101.0 ± 0.3). This suggests that MCH and MCV may be governed by molecular processes that are regulated at an intracellular level within the erythroid lineage. Conversely, variants associated with Hb were not significantly enriched in EB-restricted NDRs or any other cell type investigated. Indeed, hemoglobin concentration is tightly regulated by the level of bioavailable iron and is therefore dependent on several iron hemostasis processes involving the absorption, storage, and mobilization of iron. These processes comprise several organs in addition to erythroid cells, including the gut, the liver, and macrophages.
Both PLT and MPV association signals were enriched at MK-restricted NDRs. Interestingly, PLT-associated SNPs were also enriched at NDRs restricted to MOs and pancreatic islets. To shed light on the properties of the genes closest to these NDRs that contained a PLT-associated SNP (P < 10−4; n = 75 genes), we performed canonical pathway analyses using the Ingenuity Knowledge Base. We detected a modest enrichment of genes involved in “cell-to-cell signaling and interaction” (range, P = 4.52 × 10−2 to 1.17 × 10−1, Benjamini-Hochberg corrected for multiple testing; n = 10). Notably, these genes included THBS1 (encoding thrombospondin 1), WASL (Wiskott-Aldrich syndrome-like), and EDN1 (endothelin 1), which have a marked role in the activation of blood platelets (Dorahy et al. 1997; Falet et al. 2002; Jagroop et al. 2005). This suggests the involvement of non-cell-autonomous mechanisms for the regulation of platelet count, whereby extrinsic factors expressed by MOs or their progeny regulate the differentiation and proliferation of the hematopoietic stem cells toward MKs and the removal of senescent platelets from the circulation by liver-residing MO-derived macrophages.
The variation in enrichment patterns for cellular parameters of the same cell type was further illustrated by a trend of depletion of MCHC- and PCV-associated SNPs at MK-restricted NDRs. MCH- and MCV-associated SNPs showed the opposite trend and were enriched at MK-restricted NDRs.
Some quantitative traits showed enrichment at NDRs in several different cell types. For example, PLT-associated SNPs were enriched at NDRs restricted to all four assayed cell types. A subset of platelet (i.e., PLT) and erythrocyte traits (i.e., MCH, MCV, RBC, and PCV) was enriched at pancreatic islet-restricted NDRs.
SNPs associated with the four nonhematological quantitative traits tested were either not enriched or only weakly enriched at NDRs restricted to hematopoietic cells. Notably, height-associated SNPs were not significantly enriched at NDRs, even though this GWA study was very well powered. In contrast, FG-associated SNPs showed evidence for enrichment in three cell types. We note that the NDRs considered here may in fact be shared across other cell types not tested in this study, and thus, the corresponding gene expression pattern may be more global.
Taken together, these findings demonstrate that distinct cell type–restricted patterns can be identified for closely related quantitative traits, and for different hematological parameters of the same cell type, which we suggest reflect aspects of the different underlying molecular mechanisms. Thus, candidate variants may be subdivided based on their overlap with NDRs restricted to certain cell types, allowing for more informative downstream functional analyses.
Discussion
We generated genome-wide maps of open chromatin in human myeloid cells and used these to define cell type–dependent enrichment patterns of sequence variants associated with hematological quantitative traits at NDRs. These patterns allowed us to dissect platelet and erythrocyte quantitative trait associations in effector cell types within the myeloid arm of hematopoiesis.
Although immortalized cell lines are valuable tools for the discovery of NDRs, there were clear differences in patterns of chromatin accessibility compared with primary cells (Fig. 2A,B; Supplemental Table 2; Supplemental Fig. 2A–K). Among many factors, these differences may arise through serial subculturing of immortalized cell lines, resulting in a more homogenous cell population. Furthermore, the primary cells obtained by culture, i.e., MKs and EBs, will be more heterogeneous populations of cells at different stages of lineage commitment and maturation. Of the 68 and 75 GWA loci associated with platelet and erythrocyte traits, respectively, only five overlapped. This suggests that the effect of common variants on the formation of platelets and erythrocytes occurs after the MK-erythroid progenitor has committed to the megakaryocytic and erythroid lineages, making our findings with primary lineage-committed MKs and EBs particularly valuable for biological interpretation of NDRs.
We stratified NDRs based on their signal strength (peak score) into four intensity bins and excluded the lowest intensity bin (Bin 1) from further analyses due to the lack of reproducibility of FAIRE peaks between replicates, and limited overlap with other regulatory marks (Supplemental Table 4A,B). We recognize that division of peaks into four bins is arbitrary, but it represented the simplest approach to yield a sufficient number of peaks per bin to carry out the statistical analyses. With this caveat in mind, we provided evidence that NDRs of different signal strength have different functional features. For example, NDRs in the lowest retained intensity bin (Bin 2) were found to be located distal from the TSS and to overlap with H3K4me1 sites, similar to features of enhancer elements (Ernst and Kellis 2010; Ernst et al. 2011). Therefore, we suggest that stratification of peaks with respect to signal strength can be used to tailor downstream functional analyses.
We showed that there is cell type–dependent enrichment of hematological trait-associated variants at NDRs. For the genome-wide significant SNPs, we found that ∼50% of the overlaps are not due to chance (estimated as the asymptotic enrichment of about two for unrelated cells in Supplemental Fig. 5A,B). Given our observation that ∼25% of the SNPs overlap a FAIRE peak (Fig. 4A,B), we suggest that ∼50% × 25% = 12.5% could be used as an estimate for the contribution from SNPs at NDRs to phenotypic variance explained by additive genetic effects using the cell types investigated. Since we did not find an overlap for every genome-wide significant SNP with a FAIRE peak, we do not claim that NDRs in the three primary cell types can explain all association signals.
We tested 13 candidate regulatory variants at platelet QTLs in EMSA studies, and provided evidence that all but three (76.9%) of the tested SNPs exerted their effect through disruption or introduction of nuclear protein binding sites. This suggests that the impact of trait-associated sequence variants on protein binding sites may prove to be a key molecular mechanism at non-protein-coding regions. Indeed, results from our tightly focused analysis of hematopoietic QTLs confirm recent observations made by ENCODE and others that GWA variants frequently affect transcription factor occupancy as well as alter allelic chromatin states (The ENCODE Project Consortium 2012; Maurano et al. 2012). We tested by EMSA three additional candidate SNPs that were located within FAIRE peaks in the lowest intensity bin (Bin 1), which was excluded from analyses (see above). All three tested SNPs, i.e., rs11731274 and rs11734099 at the KIAA0232 gene locus and rs2735816 at the BRF1 locus (Supplemental Table 7A), did not show differential binding of nuclear proteins (Supplemental Fig. 6C,D,J). Although these findings further justify our decision to exclude FAIRE peaks in Bin 1 from analyses, we also expect that a fraction of these peaks is likely to correspond to functional elements. A more refined approach, for example by incorporating overlaps with additional histone marks, will be needed to dissect more accurately this set of peaks.
As shown in Table 1, the functional evidence we obtained for the platelet candidate variants through EMSAs did not consistently correlate with publicly available regulatory annotation data sets. It is important to note that mere overlap of regulatory features (such as NDRs, transcription factor binding sites, and others) with a candidate SNP is not proof of a functional role of that SNP. For example, the SNP rs3214051 was located at an NDR shared across MKs, EBs, and MOs, and coincided with transcription factor binding sites, including FLI1, at the PTGES3-BAZ2A locus. However, EMSA experiments did not reveal differential protein binding between the allelic probes of the candidate SNP. Further experiments have to be carried out to investigate whether such SNPs affect the platelet phenotype through alternative molecular mechanisms. Several examples of such suitable experimental strategies have recently been described in the literature (Pomerantz et al. 2009; Tuupanen et al. 2009; Gaulton et al. 2010; Musunuru et al. 2010; Harismendy et al. 2011; Paul et al. 2011).
Sequence variants that are strongly associated with a quantitative trait but without necessarily surpassing the genome-wide threshold of significance (P = 5 × 10−8) are very likely to include additional true positive signals in well-powered GWA studies, such as the two large GWA meta-analyses we examined here (Gieger et al. 2011; van der Harst et al. 2012). There is an increasing body of evidence that functional data, e.g., gene expression QTLs, can be successfully correlated with GWA studies to reduce the false-discovery rate and identify novel association signals (Nicolae et al. 2010). In that context, we calculated the enrichment at the 0.005 quantile (Fig. 6; Supplemental Fig. 8), which places more emphasis on weaker regulatory associations that did not reach the threshold of genome-wide significance. This may explain the different results for the pancreatic islets, for which no enrichment was found using the genome-wide–associated SNPs (Fig. 4C,D; Supplemental Fig. 5A–D), while enrichment was found in the analysis that used all SNPs (Fig. 6). The observed enrichment of hematological trait-associated SNPs (P > 5 × 10−8) at cell type–restricted NDRs suggests that maps of open chromatin have the potential to pinpoint candidate sequence variants below the genome-wide significance threshold, effectively reducing the number of false-positive associations. Integration of such variants in network analyses and subsequent functional studies may provide valuable biological insights.
A more complete catalog of chromatin profiles will be needed to address whether the candidate functional SNPs have truly cell type–specific effects (i.e., out of all possible cell types). This can be addressed by large collaborative efforts such as ENCODE (The ENCODE Project Consortium 2012), BLUEPRINT (Adams et al. 2012), and the Roadmap Epigenomics Project (Bernstein et al. 2010). Incorporation of these genome- and epigenome-wide data sets in a multitude of different primary cell types will greatly facilitate the systematic functional interpretation of noncoding trait-associated sequence variants in terms of effector cell type and underlying molecular mechanism.
Methods
MO isolation
We isolated MOs from residual leukocytes obtained following apheresis platelet collections from Cambridge BioResource volunteers at NHS Blood and Transplant, Cambridge. Each sample (7.5 mL) was diluted 1:2 with PBE buffer (PBS [Sigma-Aldrich] at pH 7.2, 2 mM EDTA [Sigma-Aldrich], and 0.5% BSA [Sigma-Aldrich]) and gently layered onto the membrane of a 50-mL Leucosep tube (Greiner Bio-One). Samples were centrifuged for 15 min at 800g at room temperature (RT). The peripheral blood mononuclear cell (PBMC) layer was transferred into a fresh 50-mL tube. PBMCs from different Leucosep tubes were pooled, washed three times with 25 mL PBE buffer, and centrifuged for 5 min at 500g at RT. PBMCs were counted, diluted to 1 × 108 cells/mL with PBE buffer, and transferred into 5-mL polystyrene round-bottom tubes (BD Biosciences). MO isolation was performed using the EasySep Human CD14 Positive Selection Kit (StemCell Technologies) according to the manufacturer's instructions.
MK and EB culture
Umbilical cord blood was obtained after informed consent under a protocol approved by the NHS Cambridgeshire Research Ethics Committee (07/MRE05/44). Cord blood was collected into cord blood collection bags (MacoPharma). CD34+ HPCs were purified using the CD34 MicroBead Kit (Miltenyi Biotec) following the manufacturer's instructions. We tested purity (92%–98%) and viability of HPCs by flow cytometry. For in vitro differentiation of HPCs into MKs, 150,000 cells/mL/well were seeded in serum-free medium (CellGro SCGM, CellGenix) supplemented with 50 ng/mL human recombinant thrombopoietin (rhTPO; CellGenix) and 10 ng/mL interleukin 1, beta (rhIL-1β; Miltenyi Biotech). To differentiate HPCs into EBs, we seeded 5000 cells/mL/well in serum-free medium supplemented with 6 units/mL erythropoietin (rhEPO; R&D Systems), 10 ng/mL interleukin-3 (rhIL-3; Miltenyi Biotech), and 100 ng/mL stem cell factor (rhSCF; R&D Systems). Cells were cultured for 10 d at 37°C and 5% CO2. On the day of harvest, a cell aliquot was stained with 0.2% Trypan blue, and live cells were counted using a hemocytometer (InCyto C-Chip, VWR International).
Cell morphology and flow cytometric analysis
For cell morphological analysis, aliquots of 50,000 cells were centrifuged onto a glass slide for 5 min at 400g at RT and stained with modified Wright's stain using an automated slide stainer (HemaTek 1000, Miles Laboratories). Stained cytospins were microscopically analyzed (Axiovert 40 CFL, AxioCam HSc, and AxioVision v4.5; Carl Zeiss MicroImaging). Aliquots of 300,000 cells were used for flow cytometry. We stained MOs with human anti-CD14-PE clone TUK4 and anti-CD45-FITC clone c29/33 (Alere), as well as FITC and PE mouse monoclonal IgG1 isotype control (BD Biosciences). After antibody incubation, 500 μL washing buffer PBE buffer (PBS [Sigma-Aldrich] at pH 7.2, 2 mM EDTA [Sigma-Aldrich], and 0.5% BSA [Sigma-Aldrich]), and 5 μg/mL 7-amino actinomycin D (7-AAD; Invitrogen) were added. Flow cytometric analysis of MKs and EBs was performed according to the method previously described (Macaulay et al. 2007; Tijssen et al. 2011) using the following antibodies: human anti-CD41a-APC clone HIP8, anti-CD42a-FITC clone ALMA.16, anti-CD235a-FITC clone GA-R2 (HIR2), and anti-CD34-PE clone 581 (BD Biosciences). All samples were analyzed on the CyAn ADP 9-Color flow cytometer using the software Summit v4.3.02 (Beckman Coulter).
Ploidy stain of MKs
An aliquot of 1 × 106 MKs was fixed with 70% (w/v) ethanol (Sigma-Aldrich) for 30 min at RT, washed once with PBE buffer (PBS [Sigma-Aldrich] at pH 7.2, 2 mM EDTA [Sigma-Aldrich] and 0.5% BSA [Sigma-Aldrich]) and stained human anti-CD41a-APC clone HIP8 (BD Biosciences) or matched isotype control, as described above. After centrifugation, cells were resuspended in 500 μL staining buffer (465 μL PBE buffer, 5 μL 10% Tween-20 [Sigma-Aldrich], 5 μL of 10 mg/mL RNase A [Sigma-Aldrich], and 25 μL propidium iodide [Sigma-Aldrich]). After incubation for 30 min at 37°C, DNA content was determined using a flow cytometer.
Formaldehyde-assisted isolation of regulatory elements
CHRF-288-11 cells were cultured according to the method previously described (Paul et al. 2011). We used approximately 10 × 106 CHRF-288-11 cells for each FAIRE experiment. Each FAIRE assay in primary human MKs, EBs, and MOs was performed with approximately 15 × 106 cells from two independent extractions. FAIRE was performed according to the method previously described (Paul et al. 2011), except that cross-linked washed cell pellets were resuspended in 2 mL of lysis buffer (10 mM Tris [Thermo Fisher Scientific] at pH 8.0, 10 mM NaCl [VWR BDH Prolabo], 1× EDTA-free Protease Inhibitor [Complete Mini, Roche] and 0.2% Tergitol solution [Type NP-40, Sigma-Aldrich]). The sample was incubated for 10 min on ice. FAIRE DNA was processed following the Illumina paired-end library generation protocol. Genomic libraries derived from MO extractions and CHRF-288-11 cells were sequenced on Illumina HiSeq 2000 with 50-bp and 75-bp paired-end reads, respectively. Libraries derived from EB and MK cultures were sequenced on Illumina GAIIx with 54-bp paired-end reads.
Sequence data processing
Raw sequence reads were aligned to the human reference sequence (NCBI build 37) using the algorithm Stampy (Lunter and Goodson 2011). Reads were realigned around known insertions and deletions (The 1000 Genomes Project Consortium 2010), followed by base quality recalibration using the Genome Analysis Toolkit (GATK) (McKenna et al. 2010). Duplicates were flagged using the software Picard (http://picard.sourceforge.net/) and excluded from subsequent analyses. We retrieved FAIRE raw sequencing data for K562 erythroblastoid cells (GEO accession no. GSM864361) (The ENCODE Project Consortium 2012) and pancreatic islets (GEO accession no. GSM491290) (Gaulton et al. 2010), and remapped the data as described above. An overview of the sequencing statistics is provided in Supplemental Table 1.
Peak calling and binning strategy
Regions of enrichment (peaks) were determined using the software F-Seq v1.84 (Boyle et al. 2008). We applied a feature length of L = 600 bp and two different SD thresholds of T = 6.0 (“moderate”) and T = 8.0 (“stringent”) over the mean across a local background. In order to reduce false-positive peak calls, we removed regions of collapsed repeats according to the method recently described (Pickrell et al. 2011), applying a threshold of 0.1% (http://eqtl.uchicago.edu/Masking). For comparison of open chromatin profiles, all read fragments were merged into one data set for each cell type. Then, peaks were called as described. For the K562 and pancreatic islets single-end sequencing data sets, we adjusted the mode of the peak width distribution to the mean of the modes across all non-K562 cells/pancreatic islets. We defined four equally spaced intensity bins between the first and 99th percentile of the log10-transformed F-Seq peak score distribution (termed “Bins 1–4”). Then, we added the peaks below the first percentile and above the 99th percentile of the peak score distribution to Bin 1 and Bin 4, respectively. Supplemental Table 2 and Supplemental Table 3 give an overview of the peak data sets. ChIP-seq data sets in primary MKs were obtained from Tijssen et al. (2011) (GEO accession no. GSE24674). The peak coordinates were remapped to hg19 (minimum ratio of bases that must remap: 0.95) using the Lift-Over tool v1.0.3 of the web-based analysis platform Galaxy (http://main.g2.bx.psu.edu/).
Hierarchical cluster analysis
First, we created the union set of all peaks across all samples. Next, we defined a vector of binary values for each sample s, where the length of this vector is given by the total number of peaks in the union set and is therefore the same for all samples. Position i in the vector for sample s was set to a value of one, if the peak i in the union peak set overlaps with a peak in sample s. If there was no overlap with a peak in sample s, position i was set to zero. From these vectors, we constructed bin-specific vectors based on the binning scheme described above. For each bin and sample, we defined a vector where all entries with a peak score not between the lower and upper peak scores defined for that bin were set to zero. We then used the R package Pvclust (Suzuki and Shimodaira 2006) to perform a bootstrapped hierarchical cluster analysis of the samples based on these binary vectors, using the “binary” distance measure and the “complete” method for defining the clusters (Suzuki and Shimodaira 2006). Here, 1000 bootstrap samples were applied. All analyses were carried out in the R/Bioconductor environment.
Gene expression analysis during in vitro differentiation of cord blood–derived HPCs
Experiments and statistical analyses were performed according to the method previously described (Gieger et al. 2011). Briefly, MKs and EBs were differentiated from cord blood–derived HPCs as described above. Time points were taken at days 3, 5, 7, 9, 10, and 12. Whole-genome gene expression levels were measured using Illumina HumanWG-6 v3 Expression BeadChips. Expressed probes were selected based on stringent thresholds, and the slope of expression was determined using standard linear regression. To every FAIRE peak, we assigned the single closest Ensembl transcript (release 69) with a HGNC symbol.
H3K4me1 and H3K4me3 ChIP
MKs and EBs were differentiated from cord blood–derived HPCs as described above. ChIP assays were performed according to the method previously described (Forsberg et al. 2000), using rabbit polyclonal antibodies against H3K4me1 (ab8895, Abcam) and H3K4me3 (07-473, Millipore). Chromatin-immunoprecipitated DNA was sequenced on Illumina GAII with 54-bp single-end reads. Sequence reads were aligned using the algorithm BWA (Li and Durbin 2009). Areas of enrichment were determined using the slice function of the R package IRanges (http://bioconductor.org/packages/2.10/bioc/html/IRanges.html). Histone modification raw sequencing data for MOs were retrieved from ENCODE (GEO accession nos. GSM1003535 and GSM1003536) (The ENCODE Project Consortium 2012) and reanalyzed as described above. For H3K4me1, we identified 79,049 regions of enrichment in MKs, 66,410 in EBs, and 77,051 in MOs. For H3K4me3, 17,402 regions were found in MKs, 16,871 in EBs, and 33,842 in MOs.
Annotation of NDRs using GREAT
We analyzed the ontology of genes flanking FAIRE peaks using GREAT v1.8.2 (McLean et al. 2010) with the following parameters: association rule: single nearest gene; 1 Mb maximal extension; curated regulatory domains excluded. The genomic distances between FAIRE peaks and TSSs were exported from the genomic region–gene association table in GREAT.
Overlap of NDRs with genome-wide significant SNPs associated with hematological traits
For each GWA locus, candidate functional SNPs were selected by identifying all biallelic SNPs with an r2 > 0.8 and within 1 Mb of the lead SNP in the European samples of the 1000 Genomes Project data set (interim phase I release of June 2011). We determined if at least one of these candidate SNPs overlapped with a FAIRE peak. Since this analysis is sensitive to the number of peaks, the overlap was carried out for a successively increasing number of peaks (cumulative tranches) by considering peaks with decreasing peak rank (F-Seq peak score). As more peaks are considered, the chance of finding an overlap increases. Therefore, we estimated the significance of our findings by resampling. Samples were drawn from the Phase II HapMap panel of approximately 2.6 × 106 SNPs (The International HapMap Consortium 2007), such that the MAF, the distance to a TSS, and the number of proxy SNPs (r2 > 0.8) had the same distribution as the genome-wide significant lead SNPs. This was achieved by estimating the joint distribution of the signed distance to a TSS and the number of proxy SNPs using a two-dimensional Gaussian kernel density estimate as implemented in the R package KernSmooth (http://cran.r-project.org/web/packages/KernSmooth/). The MAF was treated as an independent variable and was also estimated using a one-dimensional Gaussian kernel density estimate. We sampled with replacement 100,000 sets of lead SNPs (loci) of equal size as in the GWA study and with similar distribution of MAF, distance to TSS, and number of proxy SNPs per locus, representing loci drawn from the null distribution. Fold enrichment and Z-scores were calculated relative to the mean and SD of the observed number of GWA loci overlapping with an NDR for the null lead SNP sets. All analyses were carried out in the R/Bioconductor environment.
Canonical pathway analysis using the Ingenuity Knowledge Base
Genes were subjected to the core analysis module of the software Ingenuity IPA v14197757 (http://www.ingenuity.com) and analyzed using the following parameters: reference set: Ingenuity Knowledge Base (genes only); relationship to include: direct and indirect; filter: only molecules and/or relationships where species=human and confidence=experimentally observed. We report Benjamini-Hochberg multiple test corrected P-values.
Electrophoretic mobility shift assays (EMSAs)
EMSAs using nuclear extracts from CHRF-288-11 cells were performed according to the method previously described (Paul et al. 2011). Oligonucleotide probes were designed based on the genomic sequence surrounding the candidate SNPs. A list of the oligonucleotides is provided in Supplemental Table 9. Competitor probes were prepared without biotin labels. All oligonucleotides were synthesized by Sigma-Aldrich. For competition assays, we used 100- or 200-fold molar excess (as indicated in Supplemental Figs. 6A–O, 7B) of the unlabeled probes.
Enrichment analysis using bootstrapped quantile distributions
The association analysis for the hematological quantitative traits was performed by imputation to the Phase II HapMap panel (The International HapMap Consortium 2007). To improve the coverage, we determined for each HapMap SNP which 1000 Genomes SNPs (interim phase I release of June 2011) within a distance of 50 kb had an r2 > 0.95 with the imputed HapMap SNPs. For each trait, we assigned the P-value of the HapMap SNP from the meta-analysis to the 1000 Genomes SNP. To prevent chance inflation from LD and to obtain confidence estimates, SNPs were randomly removed until the genomic distance between remaining SNPs was at least 50 kb. For each combination of trait and cell type, we created 5000 bootstrap samples of 1000 Genomes SNPs located at a cell type–restricted NDR. Then, for each bootstrap sample of SNPs located at an NDR, we created a matched null set of SNPs by sampling from the full set of 1000 Genomes SNPs that had an r2 > 0.95 with a HapMap SNP. Each SNP was annotated for (1) MAF in the 1000 Genomes data set (bin boundaries: 0.0, 0.05, 0.1, 0.2, 0.3, 0.4, 0.5, 1.0); (2) genomic annotation (categories: exon, intron/3′ UTR, promoter/TSS/5′ UTR, intergenic/noncoding); (3) 36-bp mappability (bin boundaries 0.0, 0.95, 1.0) (http://hgdownload.cse.ucsc.edu/goldenPath/hg19/encodeDCC/wgEncodeMapability/ wgEncodeCrgMapabilityAlign36mer.bigWig); and (4) absolute distance from the TSS of the nearest gene (bin boundaries: 0 kb, 1 kb, 10 kb, 100 kb, 1 Gb). We note that many of these categories are correlated, e.g., genomic annotation and distance to TSS. The distribution of annotation categories of SNPs in the null set was matched to that of the SNPs located at NDRs by sampling 10 random SNPs with the same annotation in the four categories for each SNP located at an NDR. The enrichment was quantified as the mean difference between the −log10(P-value) at the 0.005 quantile in the sample of SNPs located at NDRs and the −log10(P-value) at the 0.005 quantile in the matched null set. The enrichment may be interpreted as the relative genomic inflation factor at the 0.005 quantile. The 0.005 quantile provides a trade-off between highlighting differences in enrichment between different cell types and reducing uncertainty in the estimates of the relative genomic inflation factors. All analyses were carried out in the R/Bioconductor environment.
Data access
The sequence data from this study have been submitted to the NCBI Gene Expression Omnibus (GEO) (http://www.ncbi.nlm.nih.gov/geo/) under accession no. GSE37916.
Acknowledgments
We thank S.F. Garner (Department of Haematology, University of Cambridge & NHS Blood and Transplant, Cambridge, UK) and D.M. Bloxham (Department of Haematology, University of Cambridge & Addenbrooke's Hospital, Cambridge) for support. We thank the core informatics, library-making, and sequencing teams at the Wellcome Trust Sanger Institute. D.S.P. and K.V. are supported by the Marie-Curie Initial Training Network NETSIM (EC-215820). D.S.P. is further supported by the EU-FP7 Project BLUEPRINT (282510). C.A.A. and A.R. are funded by the British Heart Foundation Program Grant RG/09/12/28096. J.S. and W.H.O. are supported by a grant from the National Institutes for Health Research (RP-PG-0310-1002). N.S. and P.D. are supported by the Wellcome Trust (098051).
Author contributions: D.S.P., K.V., and P.D. conceived and designed the experiments. D.S.P., K.V., and J.S. performed the experiments. C.A.A., A.R., and D.S.P. performed statistical analysis. C.A.A., A.R., D.S.P., and K.V. analyzed the data. P.vdH., J.C.C., and N.S. contributed GWA meta-analysis data sets. P.D. and W.H.O. jointly supervised the research. D.S.P. and P.D. prepared the manuscript, with major contribution from C.A.A., A.R., and W.H.O.
Footnotes
-
↵14 These authors jointly directed this work.
-
↵15 A full list of members is provided in the Supplemental Material.
-
↵16 Corresponding authors
E-mail d.paul{at}ucl.ac.uk
E-mail panos{at}sanger.ac.uk
-
[Supplemental Material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.155127.113.
Freely available online through the Genome Research Open Access option.
- Received January 18, 2013.
- Accepted April 2, 2013.
This article, published in Genome Research, is available under a Creative Commons License (Attribution-NonCommercial 3.0 Unported), as described at http://creativecommons.org/licenses/by-nc/3.0/.








