
A systematic study of gene expression variation at single-nucleotide resolution reveals widespread regulatory roles for uAUGs
- Department of Genetics, Center for Genome Sciences and Systems Biology, Washington University School of Medicine, St. Louis, Missouri 63108, USA
Abstract
Regulatory single-nucleotide polymorphisms (rSNPs) alter gene expression. Common approaches for identifying rSNPs focus on sequence variants in conserved regions; however, it is unknown what fraction of rSNPs is undetectable using this approach. We present a systematic analysis of gene expression variation at the single-nucleotide level in the Saccharomyces cerevisiae GAL1-10 regulatory region. We exhaustively mutated nearly every base and measured the expression of each variant with a sensitive dual reporter assay. We observed an expression change for 7% (43/582) of the bases in this region, most of which (35/43, 81%) reside in conserved positions. The most dramatic changes were caused by variants that produced AUGs upstream of the translation start (uAUGs), and we sought to understand the consequences and molecular mechanisms underlying this class of mutations. A genome-wide analysis showed that genes with uAUGs display significantly lower mRNA and protein levels than genes without uAUGs. To determine the generality of this mechanism, we introduced uAUGs into S. cerevisiae genes and observed significantly reduced expression in 17/21 instances (p < 0.01), suggesting that uAUGs are functional in a wide variety of sequence contexts. Quantification of mRNA and protein levels for uAUG mutants showed that uAUGs affect both transcription and translation. Expression of uAUG mutants under the upf1Δ strain demonstrated that uAUGs stimulate the nonsense-mediated decay pathway. Our results suggest that uAUGs are potent and widespread regulators of gene expression that act by attenuating both protein and RNA levels.
Regulatory single-nucleotide polymorphisms (rSNPs) have garnered much attention in recent biomedical studies. Evidence has revealed that rSNPs contribute to human phenotypic variation and can affect disease susceptibility. Furthermore, many disease-associated SNPs identified in genome-wide association studies (GWAS) are noncoding and are most likely regulatory in nature (Hindorff et al. 2009). However, the identification of rSNPs remains challenging. Many researchers have applied computational methods to distinguish functional rSNPs from a large number of neutral noncoding variations, mostly focusing on SNPs in conserved regions. While such approaches have identified many functional regulatory regions, it is not clear whether they can identify the majority of regulatory elements. For example, a recent study analyzed transcription factor binding sites in five different vertebrates and found that most binding events were species-specific. In fact, for one of their transcription factors, CEPBA, only 0.3% of binding sites were conserved across all five species (Schmidt et al. 2010). Transcription factor binding site (TFBS) “turnover” and sequence mutation of binding sites are two mechanisms that may explain this high degree of species-specific binding (Odom et al. 2007; Schmidt et al. 2010). These results raise an important question: How sensitive are alignment-based conservation approaches in predicting regulatory elements? More specifically, what fraction of nucleotides regulating transcription or translation reside in conserved noncoding sequences?
In this study, we used the GAL1-10 regulatory region of yeast Saccharomyces cerevisiae as a model system and identified single nucleotides that affect gene expression in the region of 630 bases upstream of the GAL1 translation start site. We created a library of single point mutations covering 582 unique nucleotide positions in this region. Using a dual-color (CFP/YFP) reporter system (Elowitz et al. 2002; Raser and O'Shea 2004), we detected in vivo gene expression changes as small as 10%. This nearly exhaustive and uniformly distributed mutation library coupled with our sensitive detection assay allowed us to quantitatively study the effect of rSNPs in relationship with sequence conservation, TFBSs, and other functional sequence features. We identified 43 positions in the GAL1-10 regulatory region that reduced reporter gene expression by >10% upon mutation. The observed changes in expression ranged from small perturbations to complete abolishment of reporter gene expression. The majority of mutations affecting gene expression occurred in bases that are conserved, supporting the canonical view that conservation is a powerful predictor of function. For mutations within a binding site, we demonstrated that the in vivo expression change correlated with binding energy change predicted by the PWM of the Gal4 transcription factor.
We identified several mutations in our library that caused much larger expression changes than those in known TFBSs. These mutations produced frameshift uAUGs and completely silenced expression. It has previously been shown that uAUGs have strong effects on gene expression in both yeast and humans (Calvo et al. 2009; Hood et al. 2009), so we sought to further understand the mechanism by which this widespread and potent class of mutation affects gene expression. By performing a genome-wide analysis of uAUG sites in S. cerevisiae, we found a strong correlation between the reduction of gene expression and the existence of uAUG sites. We observed that ∼21% of genes in S. cerevisiae have preserved uAUG sites and are highly conserved among yeast species. To investigate the scope and the strength of uAUG cis-regulation in the yeast genome, we introduced the uAUG mutation into 21 randomly selected genes without native uAUGs. In 80% of the examined genes, the introduction of a uAUG significantly reduced gene expression. This effect was independent of the trinucleotide sequence context and other gene-specific features. Furthermore, we quantified the reduction of mRNA and protein in uAUG mutants for five genes and found that uAUGs exert both transcriptional and translational control. Finally, by analyzing these uAUG mutants in a nonsense-mediated decay (NMD) deficient yeast strain, we demonstrated that the NMD pathway plays a pivotal role in the degradation of mRNA transcripts caused by uAUG defects. Collectively, these results suggest that the creation or destruction of a uAUG site by a single nucleotide substitution is an rSNP of strong impact and might be a widespread feature in shaping the functional regulatory network in yeast.
Results
Creation of a nearly exhaustive single-nucleotide mutation library
We applied two mutagenesis methods to create a single-nucleotide mutation library for the GAL1-10 regulatory region: error-prone PCR and site-directed mutagenesis. To avoid over-representation of mutations at certain positions that were introduced by early PCR cycles, we modified the standard PCR mutagenesis protocol by performing linear template amplification, rather than exponential amplification (see Methods). We cloned the library into a pY10TY plasmid, transformed the plasmid into Escherichia coli, and identified mutations by Sanger sequencing. In total, 2764 constructs were found to contain one to seven nucleotide substitutions, and 65% of these constructs contained only a single nucleotide change. The average mutation rate of the library was 0.76 mutations per construct, a value that was close to the target mutation rate at one nucleotide per construct (Supplemental Fig. S1). The library covered 615 of the 630 nucleotide positions (98%) in the GAL1 regulatory region, and 533 nucleotide positions were covered by constructs containing only one mutation, indicating that our method was sufficient to create a highly enriched single-nucleotide mutation library (Supplemental Fig. S2). To increase the coverage of the mutation library and to validate the constructs generated by the above method, we also created constructs with an additional 140 single-nucleotide mutations by site-directed mutagenesis; some of the mutations overlapped with the previous set.
We selected one construct per nucleotide position and transformed them individually into yeast cells. In total, 582 constructs were chosen for expression analysis. Each construct contained a single-nucleotide mutation at a unique position in the GAL1-10 regulatory region. Among 582 mutated bases, 191 (33%) are transitions, and 391 (67%) are transversions (for a summary of the mutation spectrum, see Supplemental Tables S1, S2).
Detection of gene expression changes in GAL1-10 regulatory variants
Since small changes in gene expression can have important functional consequences, it is important to develop a highly sensitive assay to detect expression change caused by a single-nucleotide mutant. We implemented a dual-color reporter assay in which a mutant GAL1 construct drives a YFP reporter gene, and a wild-type GAL1 construct drives a CFP reporter gene (Fig. 1A). The CFP reporter acts as an internal control to eliminate the extrinsic noise from experimental measurements, estimated at 97% of the total noise (Raser and O'Shea 2004).
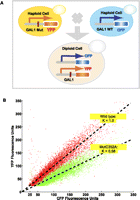
Overview of detecting expression variation for the mutation library. (A) Design of dual-color reporter system; (red bar) single nucleotide mutation; (WT) wild type; (MUT) mutant. (B) An example of determining the expression level (mutant strain Mut C352A, position 352, C → A mutation). Each dot represents the CFP and YFP fluorescence intensities from one cell. (Red dots) Cells from a wild-type diploid strain carrying a gal+-YFP and gal+-CFP fusion. The slope represents the mean of the YFP-versus-CFP ratio for a population of cells (normalized, k = 1). (Green dots) Cells from the Mut C352A strain carrying a gal--YFP and gal+-CFP fusion. The slope represents the mean for the mutant population (k = 0.58). The relative ratio between two slopes indicates the expression variation.
We individually transformed 582 constructs into yeast cells to create haploid strains that expressed YFP. Each strain was mated to a haploid cell expressing CFP under the control of a wild-type GAL1 regulatory construct. The resultant diploid strains expressed both CFP and YFP proteins at the homologous loci on sister chromosomes. We then used flow cytometry to measure the ratio of YFP to CFP in about 15,000 cells for each strain under the galactose-induction condition. The YFP-to-CFP ratio reports the mutation's effect on gene expression relative to the wild-type construct (Fig. 1B). We measured reporter gene expression in six independent transformants for each member of the mutation library, resulting in a total of 3492 measurements.
By combining this dual-color system and individual cell measurements, we achieved highly sensitive gene expression detection. Our analysis showed that we can reliably detect a 10% reduction in gene expression at a false discovery rate (FDR) of <0.005. Due to transformational variation, there was less power to detect increases in expression (see Supplemental Fig. S3; Supplemental Material I, II). In total, we identified 43 mutants that caused a significant reduction in gene expression (Fig. 2; Supplemental Fig. S4; Supplemental Table S3; Supplemental Material III, IV). We found that the efficacy of the mutation diminished as a function of the distance from the GAL1 translation start site, with the strongest effects observed for mutations in the 5′ UTR region, followed by those in the TATA-box and in the four Gal4 binding sites.

Multiple sequence alignments among four yeast species in the GAL1-10 regulatory region and gene expression of single-nucleotide mutated strains. The sequence alignment is shown according to Kellis et al. (2003). (Axis) The position from −1 to −630 before the GAL1 start codon. (*) Conserved positions among four species. (Sequences in blue) The mutated nucleotides. Validated Gal4 and TATA binding sites are boxed in red. Gene expression variations for a single nucleotide mutation are indicated with different colors along the position axis in the expression bar. The color bar indicates different expression levels (see color map). (Scer) S. cerevisiae; (Spar) S. paradoxus; (Smik) S. mikatae; (Sbay) S. bayanus; (Mut) mutated nucleotide.
The majority of single-nucleotide mutations that change gene expression reside within conserved regions
We first asked if the mutations that reduce gene expression reside predominantly in conserved regions. We defined conserved bases in two ways: first, as the invariant bases in the sequence alignment of four yeast species (Kellis et al. 2003); and second, as the bases with significant PhastCon conservation scores. PhastCon (Siepel and Haussler 2004) is a program for identifying evolutionarily conserved elements from a multiple alignment, and it correctly accounts for the phylogenetic relationships between sequences. Of the 582 nucleotide positions analyzed, 245 nucleotides were identical across four yeast species (Fig. 2), 160 nucleotides were defined as conserved with a PhastCon score >0.1 (Fig. 3), and 107 nucleotides were concordant by both methods.
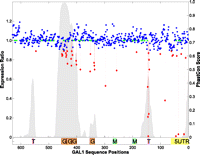
Expression variation versus phastCons scores. phastCons scores are plotted in sliding windows along the GAL1-10 regulatory sequences, with y-axis labeling on the right. (Gray peaks) The regions with pronounced signals of sequence conservation. The expression ratio (ER) for each nucleotide between YFP and CFP is plotted by colored dots, with y-axis on the left [(red) ER ≤ 0.9; (blue) 0.9 < ER < 1.2; (green) nucleotides with no mutation or expression data]. Below the graphic is a panel of known functional elements: (5UTR) 5′ UTR; (T) TATA; (M) MIG1 TFBS; (G) Gal4 TFBS.
Of our 43 bases whose mutation causes significant changes in gene expression, 35 (81%) were located in the conserved regions as defined by the alignment method (hypergeometric P < 5.3 × 10 −8), and 27 (63%) were defined as conserved by the PhastCon method (hypergeometric P < 1.3 × 10 −11). These 27 positions also happen to be invariant in the alignment. Both comparisons showed that the majority of single-nucleotide mutations that change gene expression reside within conserved regions, indicating that searching for rSNPs by focusing on conserved regions will likely capture a large fraction of, but not all, functional rSNPs.
Many single-nucleotide mutations that change gene expression reside within TFBS
We next sought to determine whether single-nucleotide mutations that change gene expression cluster solely within TFBSs or whether some fraction of mutations are located outside of these sequences. For example, regulatory variants that act by disrupting nucleosome positioning and therefore affect gene expression may be distributed throughout the regulatory region. The TATA-box and four Gal4 binding sites are known to up-regulate GAL1 under galactose induction (Giniger et al. 1985; Kellis et al. 2003). Of the 43 nucleotides whose mutation causes significant expression variation, 27 (63%) were located in or adjacent to these sites, indicating that the majority of single-nucleotide mutations that change gene expression reside within TFBS (Fig. 2). Also, nearly all changes in high information content positions of the PWM in these TFBSs significantly affected expression, suggesting that TFBSs are enriched with rSNPs. In the TATA-box, we observed significant changes in all six examined positions, and three of these reduced gene expression by >75%.
Three of the four Gal4 binding sites match the 17-bp Gal4 consensus sequence CGGN(11)CCG (Fig. 2). The three nucleotides at either end of the consensus sequence have the highest information content. We found 20 positions within the four Gal4 binding sites that produced a significant change in gene expression, 19 of which were at high information content positions. As expected, substitutions at the high information content positions in general caused larger changes in gene expression than substitutions at other positions. Compared with the TATA-box, the expression changes caused by substitutions in Gal4 sites were small: The largest variation was a decrease of 32%, and the majority of them showed a decrease of ∼15%. These relatively small effects may be explained by functional redundancy of the four coexisting Gal4 sites in one GAL1-10 regulatory region (Fig. 4A).
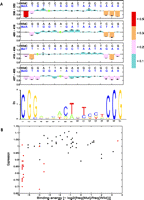
Expression variations among four Gal4 binding sites in the GAL1-10 regulatory region. (A) The motif pattern of the Gal4 binding site is created based on the position weight matrix reported in TRANSFAC. Expression levels of each position in the Gal4 sites are shown with different colors to discriminate the degree of changes (see color map). (Green circles) Nonmutated positions. Error bars are 1 standard deviation among different yeast transformants from the same mutant construction. (B) The correlation between gene expression and binding energy for the four Gal4 binding sites. The binding energy is proportional to the log [freq(Mut)/freq(Wild)] (Stormo 1998). (Red spots) The high information content sites of the Gal4 PWM; (black spots) the low information content sites.
We next examined how well the Gal4 PWM predicts changes in gene expression. We found a significant correlation (Pearson correlation, r = 0.58, P < 1.5 × 10 −8) between the changes in the binding energy predicted by the PWM (Matys et al. 2003) and the changes in gene expression among Gal4 binding sites (Fig. 4B). We also found that the changes in expression varied significantly between the different Gal4 binding sites, even for the mutations that occurred at the same position in the consensus site. Furthermore, within the six high information content bases, the change in binding energy does not explain all the variance of expression change. This suggests that in addition to binding energy, other factors such as the location of the TFBS play a significant role in gene regulation (Fig. 4A).
Creation of uAUG sites in the 5′ UTR of the GAL1-10 regulatory region abolishes gene expression
Sixteen of the positions that showed significant changes in gene expression upon mutation reside outside TFBSs and conserved regions. Surprisingly, mutations in these positions generated some of the strongest gene expression changes (Fig. 2; Supplemental Fig. S5). Nine of 16 positions were located in the 5′ UTR of the GAL1-10 regulatory region (between the major transcriptional start site at −62 and the AUG start codon) (Johnston and Davis 1984). These substitutions may affect gene expression by perturbing either transcriptional or translational regulation. Most strikingly, three mutations—at positions 16, 36, and 45—virtually abolished gene expression. Further analysis found that each of these mutations created a frameshift uAUG start codon in the 5′ UTR region. Each of these uAUGs created an upstream open reading frame (uORF), and all of these uORFs share the same termination codon, which overlaps with the first nucleotide of the canonical ATG start codon. Although all three of these mutations have large effects on gene expression, the nucleotides at these positions are variant across four yeast species; however, these sequences are probably still under some evolutionary constraint because no species contained a uAUG site at these positions. This example presents a nonconventional scenario of conservation, in that certain mutations can have major effects (strong functional constraint), but the bases at these positions show relaxed evolutionary constraint.
In the wild-type GAL1-10 regulatory region, there are no uAUGs. Nine positions in the 5′ UTR could potentially mutate to a frameshift uAUG by a point mutation with a correct substitution type. The three positions listed above are the only frameshift uAUGs created in the library, and all strongly impact gene expression. The remaining six positions (positions at 15, 21, 38, 46, 56, and 61) were mutated to trinucleotides other than AUG, and no gene expression change was observed (Supplemental Fig. S5). These results led us to further investigate the genome-wide regulatory roles of uAUGs.
uAUGs down-regulate both mRNA and protein expression levels genome-wide
We sought to determine if the uAUGs distributed throughout the genome caused expression changes of the same magnitude as those we observed at the GAL1 locus. We compared the mRNA (Nagalakshmi et al. 2008) and protein levels (Ghaemmaghami et al. 2003) between genes containing uAUGs and those without. Of 3042 genes with available mRNA and protein expression data, the median of mRNA levels for single uAUG genes was ∼15% lower (Wilcoxon, P < 7.8 × 10−17) than that of uAUG-free genes; the median of the protein levels for uAUG genes was ∼2.2-fold lower (Wilcoxon, P < 4.8 × 10−11) (Fig. 5A; Supplemental Material V). This suggests that uAUGs are involved in both transcriptional and translational regulation with a large impact at the translational level, an observation that agrees with a similar analysis of the human genome (Calvo et al. 2009).
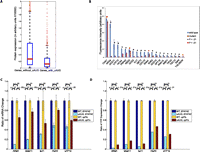
mRNA and protein expression of wild type and uAUG mutant. (A) A box-plot of protein expression between genes without uAUG and genes with one uAUG. (B) Comparison of the reporter protein expression driven by regulatory sequence between wild type and uAUG mutant in 21 genes. (C) mRNA expression quantified by qRT-PCR. (D) Protein expression quantified by YFP reporter gene. In C and D, the expression of wild type is normalized to 1, and the expression of the uAUG mutant is compared with its correspondent wild type. (Dark blue) Wild type in BY4742 strain; (light blue) uAUG mutant in BY4742 strain; (yellow) wild type in upf1-Δ strain; (brown) uAUG mutant in upf1-Δ strain.
Selection against uAUGs partially explains the large number of conserved bases in yeast 5′ UTR regions
Previous studies (Iacono et al. 2005; Hood et al. 2009) have shown that uAUGs in 5′ UTRs are under-represented. To confirm that purifying selection has indeed acted on the AUG trinucleotide and that the observed under-representation was not the result of selection against “AU” or “UG” di-nucleotides, we used a first-order Markov model to compute the expected uAUG frequency given the observed dinucleotide frequencies. We calculated the expected frequency and compared it with the observed uAUG frequency in yeast 5′ UTR sequences defined by RNA-seq (Nagalakshmi et al. 2008). We found that the AUG trinucleotide was the least common of the 64 possible trinucleotides, with the observed occurrence being 58% less than expected (simulation, P < 1 × 10 −16) (Supplemental Fig. S6), confirming that the uAUG trinucleotide is indeed under strong purifying selection.
We next asked if polymorphic uAUGs were less likely to be observed relative to the other trinucleotides in the 37 S. cerevisiae strains that have been sequenced. In the 365,753 bases of 5′ UTR sequences (Nagalakshmi et al. 2008), we observed 10,073 single-nucleotide changes relative to the reference strain. Only 1387 of these created an uAUG site in at least one of the other strains, a number significantly less than the expected 1813 SNPs from simulation (P < 0.0001). Thus, this orthogonal analysis provides additional support to the hypothesis that the presence of uAUGs in 5′ UTRs is under strong purifying selection.
It is known that 5′ UTRs in yeast are highly conserved, despite the fact that very few functional sequence elements have been found in this region. We hypothesized that selection against the formation of uAUGs might place evolutionary constraints on the 5′ UTR and explain the high level of conservation that is observed. We estimated that ∼14% of the nucleotides in the 5′ UTR of the 3499 non-uAUG genes of S. cerevisiae could be converted into AUGs by a single substitution. This may explain, in part, the high degree of conservation estimated in 5′ UTRs.
Some uAUGs are conserved and may play regulatory roles
Although uAUGs down-regulate gene expression and, in general, are under strong purifying selection, nearly 21% of the genes in yeast have maintained this trinucleotide in their 5′ UTRs, suggesting that this trinucleotide may play a regulatory role. To examine whether existing uAUGs were evolutionarily conserved, we analyzed the sequence conservation of the 5′ UTRs of uAUG-containing genes using alignments of four yeast genomes (Saccharomyces cerevisiae, Saccharomyces paradoxus, Saccharomyces mikatae, and Saccharomyces kudriavzevii) (http://genome.ucsc.edu). We used the phyloP score (Siepel and Haussler 2006) to evaluate the sequence conservation of each uAUG. Our data showed that in 5′ UTRs, AUG was indeed the most conserved among 64 trinucleotides, with an average score of 0.72 versus 0.48 for all 64 trinucleotides in this region (Wilcoxon rank sum test, P < 7.2 × 10−12).
We next asked if existing uAUGs in S. cerevisiae 5′ UTRs were more likely to be conserved in S. paradoxus than other trinucleotide sequences. Of the 4333 genes analyzed, we found 990 and 1045 genes with uAUGs in S. cerevisiae and S. paradoxus, respectively; 866 of these overlapped. Among all 64 trinucleotides, uAUG was the most highly conserved triplet with respect to the overlapping gene set (p < 0.05) (Supplemental Fig. S7). Taken together, our results suggest that while there is strong selection against the creation of new uAUGs, existing uAUGs play important regulatory roles and thus are conserved across yeast species.
Systematic introduction of uAUG mutations in yeast genes suggests their widespread regulatory role in gene expression
Since uAUGs are strongly conserved in many genes and appear to have a large effect on gene expression, we hypothesized that the presence of a uAUG in the 5′ UTR of a gene may be a general mechanism that a cell uses to tune down gene expression. However, it is possible that the effect of uAUG is dependent on certain sequence contexts. To examine if the creation of a uAUG at a random location universally impacts gene expression, we selected 21 genes whose 5′ UTRs do not contain an uAUG but have the potential to create a frameshift uAUG with a single-nucleotide substitution. To maintain consistency with our observation in the GAL1 mutation system, we also required that each uAUG site initiate a frameshift uORF. For each gene, we constructed a yeast strain with a YFP reporter gene cassette integrated at the TRP1 locus. The reporter gene is under the control of either a wild type or a mutant construct that contains a single base substitution creating a uAUG. We compared YFP reporter levels between both wild-type and mutant strains using six independent yeast transformants for each gene. In all cases, the average expression levels of the mutant were lower than in wild type. In 81% (17/21) of cases, the expression reduction was statistically significant (Student's t-test, P < 0.01), and in 14 cases, the expression level of the mutant was reduced by at least twofold. These results suggest that the presence of a uAUG in the 5′ UTR of a gene is a general mechanism by which the cell can modulate gene expression. The distribution of the distance for the first uAUG sites relative to the canonical ATG start site is shown in Supplemental Figure S8. All of the mutation sequences in this study can be found in Supplemental Material VII.
uAUGs regulate gene expression through both transcriptional and translational control
Previous studies suggest that uORFs can regulate gene expression through different mechanisms (Hood et al. 2009). To investigate whether uAUG regulates gene expression transcriptionally or translationally, we directly measured the reporter gene's mRNA levels for five of the 21 genes described above. We chose genes that displayed the most dramatic changes in the expression of the reporter gene upon the introduction of a uAUG.
All five genes showed a statistically significant reduction of mRNA levels between mutant and wild type (Student's t-test, P < 0.01) as measured by qRT-PCR. The average mRNA reduction was 4.5-fold (range: 2.2-fold to 10.3-fold) (Fig. 5C). Protein levels were even more dramatically reduced than the corresponding mRNA levels for each gene (Student's t-test, P < 0.01), with an average reduction of 15.7-fold (range: 2.8-fold to 43.7-fold) (Fig. 5D). The observed impact of uAUGs on mRNA and protein levels was consistent with our genome-wide analysis of transcriptomic and proteomic data and suggests that uAUGs govern gene expression at both the transcriptional and translational level.
mRNA molecules containing frameshift uAUGs are degraded by the NMD pathway
To test if the NMD pathway degrades uAUG-containing mRNAs, we introduced the uAUG mutants described above into a yeast strain, upf1-Δ, that is deficient for NMD. As a control, we also made NMD-deficient reporter strains with the wild-type 5′ UTRs. In the NMD-suppressive background, all five uAUG mutants showed mRNA reduction by qRT-PCR quantification (Student's t-test, p < 0.05 for four genes, and p < 0.1 for one gene), with an average reduction of 1.5-fold (range: 1.2-fold to 1.9-fold) (Fig. 5C). The degree of the mRNA reduction in the yeast upf1-Δ strain is much less than in the control BY4742 strain, and four genes showed statistically significant differences in mRNA reduction between the two backgrounds (Student's t-test, P < 0.05). On average, we observed a 36% increase in normalized mRNA levels of uAUG-containing genes in the upf1-Δ strain relative to the wild-type strain. This result demonstrates that the NMD pathway plays an important role in degrading transcripts with frameshift uAUGs.
Discussion
Using the well-studied yeast GAL1-10 regulatory region as a model system (Supplemental Material VI), our study comprehensively examined the relationship between sequence conservation and function at a single-nucleotide resolution. We found that the majority of mutations (81%) that cause gene expression changes are located in conserved regions or known regulatory regions. This supports the canonical view that regulatory elements can be identified by sequence conservation (Birney et al. 2007) and stands in contrast to a recent study in eutherian mammals suggesting that transcription factor binding sites can only rarely be identified by conserved bases in a multiple alignment, due in large part to binding site turnover (Schmidt et al. 2010). This discordance may be explained by the fact that, while yeast is an excellent model system for the study of gene regulation (Cliften et al. 2003; Kellis et al. 2003; Beer and Tavazoie 2004), its genome has smaller intergenic regions than are found in mammals, and thus binding site turnover occurs less frequently. Thirty-nine percent of the bases that have no effect on gene expression upon galactose induction are conserved (see Supplemental Table S2). Thus sequence conservation appears to have modest specificity (61%) as a predictor of rSNPs.
Although the GAL1-10 regulatory region has been extensively mutagenized (West et al. 1984), our approach identified six mutations within previously uncharacterized regulatory sites, located outside of known transcription factor binding sites and the 5′UTR region. Two out of the six mutations with unknown function are conserved among four yeast species. By searching with PWMs (Matys et al. 2003; Zhu et al. 2009) using cutoffs of reduced stringency, we could not find any convincing evidence that suggests either the creation or disruption of a TFBS, but three of these (−84, −101, and −547) are located near the region protected by GAL80 in footprinting experiments (Lohr et al. 1987). Also, although we could not find direct evidence that the novel sites themselves are involved in the creation or disruption of sequence motifs that alter nucleosome positioning signals (Kaplan et al. 2009), position −387 was located in a DNase I hypersensitive region and was close to a putative RSC/nucleosome complex binding site (Reagan and Majors 1998).
The biggest changes in gene expression were not due to mutations in TFBSs, but instead due to the creation of uAUGs. Although the effects of existing uAUGs present in the yeast genome have been noted previously (Vilela and McCarthy 2003; Hinnebusch 2005; Hood et al. 2009), it was not clear whether randomly created uAUGs would have consistently strong effects on gene expression. We showed that the efficacy of uAUGs is largely independent of sequence context. Because 14% of the bases in yeast 5′ UTRs can be converted to a uAUG by a single mutation, this represents a potent evolutionary mechanism for modulating the expression of virtually any gene.
We found evidence for strong purifying selection against uAUGs in yeast 5′ UTRs, an observation that is consistent with previous studies (Churbanov et al. 2005; Iacono et al. 2005). In agreement with Churbanov et al., we also found that, for the 20% of genes in yeast that do contain uAUGs in their 5′ UTRs, these trinucleotides tend to be conserved, suggesting that while in general uAUGs are deleterious, for a subset of yeast genes, they play an important regulatory role that confers a selective advantage to the organism. We further showed that selection against uAUGs may, in fact, partially explain the observation that 5′ UTRs are highly conserved, despite the fact that few regulatory elements have been found in these sequences. This observation suggests that some nucleotides may be under evolutionary constraint, not because they are functional so that a substitution would destroy that function, but instead, because certain substitutions may create an element with a new function that could be deleterious to the organism. We propose that selection against neomorphic mutations may explain a portion of the surprisingly high degree of conservation observed in regulatory regions.
Upstream AUGs can attenuate gene expression by at least two mechanisms: (1) translation from the uAUG causes a reduction in the translation rate at the canonical AUG (Hinnebusch 2005; Medenbach et al. 2011); and (2) the premature termination of polypeptides initiated at the uAUG can stimulate the degradation of mRNA transcripts via NMD (He et al. 2003). We measured the contribution of each mechanism in five yeast genes and found that both play significant roles in determining the final protein levels. Interestingly, NMD accounted for only part of the reduction in mRNA levels because we still observed a significant reduction in mRNA levels in NMD deficient yeast strains. This may suggest another, as yet unknown, mechanism by which these transcripts are reduced. Alternatively, it may be that the NMD pathway is not completely abolished in our upf1Δ deficient strain.
Because uAUGs have large effects on gene expression and occur in the 5′ UTRs of many yeast genes, they collectively have a substantial impact on protein expression in yeast. A similar purifying selection against uAUGs has also been observed in mammals (Iacono et al. 2005), indicating their functional roles would extend to multicellular eukaryotes (Medenbach et al. 2011). SNPs that create or destroy a uAUG may represent an important source of functional noncoding variation, and disease-associated SNPs in these regions would be strong candidates for functional studies.
Methods
Construction of plasmids
Plasmids expressing CFP (pY10TC) or YFP (pY10TY) were constructed from a yeast integration vector pRS306 with a selectable URA3 marker (Sikorski and Hieter 1989). The following regions were inserted into the pRS306 multiple cloning sites: a 447-bp TRP1 homologous fragment for chromosomal integration, a 468-bp ADH1 site, a 630-bp GAL1-10 regulatory region, a yECFP or a yVYFP fluorescent protein-coding site (from pJRL2 plasmid derivatives, kindly provided by Dr. E. O'Shea) (Raser and O'Shea 2004), and a 291-bp 3′ UTR from the ACT1 site.
Mutation library construction
Random mutagenesis on the GAL1-10 regulatory region was performed through error-prone PCR, using the GeneMorph II random mutagenesis kit (Stratagene). Plasmid pY10TY was used as the template for PCR amplification, and a PCR primer pair was designed to flank the GAL1-10 region of the pY10TY plasmid. The linear PCR mutagenesis was preceded by a two-step process: First, a typical error-prone PCR reaction was set up as described in the manual (Stratagene) with two modifications: (1) only the forward PCR primer was added to the reaction; and (2) PCR cycles were extended to 50 rounds. Second, the reverse PCR primer was added, and the reaction was completed by one more cycle of PCR, followed by a 10-min PCR extension.
Site-directed mutagenesis was performed as described by Dieffenbach and Gabriela (2003). PCR products were amplified by Jumpstart Taq DNA Polymerase (Sigma-Aldrich) using plasmid pY10TY as a template. A total of 140 PCR primer pairs, each of them containing one mismatched nucleotide from the GAL1-10 wild-type sequence, were designed.
Mutagenic PCR products replaced the wild-type GAL1 sequence in vector pY10TY. E. coli GC10 chemical competent cells (Gene Choice) were used for transformation. Clones were picked and submitted for sequencing.
DNA sequencing
Forward PCR primer 5′-CCTAAAGTAGTGACTAAGGTTGGC-3′ and reverse PCR primer 5′-GGTGTGTATTTTATGTCCTCAGA-3′ were designed to flank the GAL1-10 regulatory region of the E. coli construct. To sequence a construct, we first amplified the plasmid DNA using a TempliPhi DNA amplification kit (GE Healthcare). We then sequenced each construct four times. Mutagenic constructs were sequenced at the Washington University Genome Sequencing Center.
All sequencing reaction was prepared by Big Dye mix v3.1 (Applied Biosystems). Sequences from the same construct were analyzed, assembled, and viewed using phred, phrap (Ewing and Green 1998), and consed (Gordon et al. 1998).
A single-nucleotide mutation was assigned using a customized Perl script based on the following formula: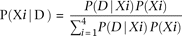 where D are the observed sequences; Xi are the mutated bases i = {A, C, G, T}; and P(Xi) is the mutation rate over the entire library.
where D are the observed sequences; Xi are the mutated bases i = {A, C, G, T}; and P(Xi) is the mutation rate over the entire library.
Strains
A yeast haploid strain BY4742 (MATα his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 ARG) and a derivative of a haploid strain BY4741 with a YHR018C gene deletion (Mata his3Δ1 leu2Δ0 ura3Δ0 met15Δ0 arg4Δ0 LYS) have been described previously (Brachmann et al. 1998) and were kindly provided by Dr. M. Johnston (University of Colorado, Denver). A wild-type GAL1 construct expressing CFP and mutagenic GAL1 constructs expressing YFP were integrated at the TRP1 locus of the yeast strain BY4742 and BY4741 derivative, respectively. Diploids were obtained by mating and further selection on synthetic lysine and arginine double-dropout media.
Yeast transformation
Plasmids pY10TC and pY10TY that contained either wild-type or mutagenic GAL1 sites were amplified using a TempliPhi DNA amplification kit (GE Healthcare). The rolling cycle products were digested by the AscI restriction enzyme (New England Biolabs), followed by the yeast transformation (Gietz and Woods 2002).
Each mutagenic plasmid was transformed individually, and six clones from each transformation event were selected for further analysis. To confirm the integration event of an E. coli construct into a yeast strain, we sequenced at least one yeast colony for each yeast transformation event. The PCR products were then treated with ExoSAP-IT (USB) for clean-up and submitted for sequencing.
Measurement of reporter gene expression
Each yeast clone was grown in 2-mL 96-well plates overnight at 30°C in 600 μL of YPD media. Yeast cultures (5 μL) were transferred to 600 μL of synthetic uracil dropout media with 2% raffinose and were grown to an OD600 of <0.5. To induce the GAL1-10 regulatory region, cultures (30 μL) were transferred to 600 μL of synthetic uracil dropout media containing 2% raffinose and 2% galactose. To induce the 21 randomly selected genes, yeast strains were cultured in 2% glucose media.
Fluorescence measurement was performed on a Beckman Coulter Cell Lab Quanta SC after 4 h and 8 h of induction. For each well, the fluorescence intensities of both CFP and YFP were measured simultaneously for all 15,000 cells. The expression level in each well was calculated by averaging the YFP-versus-CFP ratio for 15,000 cells. Each plate contained eight control strains with both CFP and YFP fluorescence proteins driven by a wild-type GAL1 construct. To control the plate variation, the expression value of each well was then normalized to the average of the control samples on the same plate. Induction and measurement were replicated for each plate.
Comparison of genes with uAUGs between S. cerevisiae and S. paradoxus
Sequence alignment between S. cerevisiae and S. paradoxus was downloaded from http://www.genetics.wustl.edu/jflab/data4.html (genome alignments of three strains combined with S. paradoxus). We estimated S. paradoxus 5′ UTR sequence based on the RNA-seq report for S. cerevisiae (Nagalakshmi et al. 2008). To examine whether the overlapped genes with uAUGs is significant between two species, we performed hypergeometric tests. We applied the same test to all 64 trinucleotides and ranked the P-value of these tests.
RNA isolation and quantitative real-time PCR assays
RNA samples were isolated using a standard TRIzol method (Invitrogen). Purified RNA was reversely transcribed into cDNA using a SuperScript III kit (Invitrogen) with random hexamers (IDT). Real-time quantitative RT-PCR analyses were performed on a Bio-RAD CFX96 real-time PCR detection system (Bio-Rad) using an ABsolute Blue QPCR SYBR Green kit (Thermo Scientific).
Data access
The sequence data from this study have been submitted to GenBank (http://www.ncbi.nlm.nih.gov/ genbank) under accession numbers JQ676216–JQ676818; accession numbers are given in Supplemental Table S4.
Acknowledgments
We thank Dr. Gary D. Stormo for his insightful advice regarding all of the experiments. We thank Dr. Justin C. Fay and Dr. Ting Wang for their creative suggestions and helpful discussions. We thank Kay D. Tweedy and Xuhua Chen for the preparation of experimental materials. We also thank Jim Dover and Linda Riles for their support on yeast molecular technology. We thank the reviewers for several important suggestions that substantively improved the paper. Finally, we thank the Genome Center of Washington University for providing sequences. This work was supported by the Children's Discovery Institute.
Footnotes
-
↵1 Corresponding author.
E-mail rmitra{at}genetics.wustl.edu.
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.117366.110.
- Received November 2, 2011.
- Accepted March 14, 2012.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see http://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 3.0 Unported License), as described at http://creativecommons.org/licenses/by-nc/3.0/.








