
Genome-wide determination of RNA stability reveals hundreds of short-lived noncoding transcripts in mammals
- Hidenori Tani1,
- Rena Mizutani1,
- Kazi Abdus Salam1,
- Keiko Tano1,
- Kenichi Ijiri1,
- Ai Wakamatsu2,
- Takao Isogai2,
- Yutaka Suzuki3,4 and
- Nobuyoshi Akimitsu1,4
- 1Radioisotope Center, The University of Tokyo, 2-11-16 Yayoi, Bunkyo-ku, Tokyo 113-0032, Japan;
- 2Graduate School of Pharmaceutical Sciences, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-0033, Japan;
- 3Department of Medical Genome Sciences, Graduate School of Frontier Sciences, The University of Tokyo, 5-1-5 Kashiwanoha, Kashiwa-shi, Chiba 277-8562, Japan
Abstract
Mammalian genomes produce huge numbers of noncoding RNAs (ncRNAs). However, the functions of most ncRNAs are unclear, and novel techniques that can distinguish functional ncRNAs are needed. Studies of mRNAs have revealed that the half-life of each mRNA is closely related to its physiological function, raising the possibility that the RNA stability of an ncRNA reflects its function. In this study, we first determined the half-lives of 11,052 mRNAs and 1418 ncRNAs in HeLa Tet-off (TO) cells by developing a novel genome-wide method, which we named 5′-bromo-uridine immunoprecipitation chase–deep sequencing analysis (BRIC-seq). This method involved pulse-labeling endogenous RNAs with 5′-bromo-uridine and measuring the ongoing decrease in RNA levels over time using multifaceted deep sequencing. By analyzing the relationship between RNA half-lives and functional categories, we found that RNAs with a long half-life (t1/2 ≥ 4 h) contained a significant proportion of ncRNAs, as well as mRNAs involved in housekeeping functions, whereas RNAs with a short half-life (t1/2 < 4 h) included known regulatory ncRNAs and regulatory mRNAs. The stabilities of a significant set of short-lived ncRNAs are regulated by external stimuli, such as retinoic acid treatment. In particular, we identified and characterized several novel long ncRNAs involved in cell proliferation from the group of short-lived ncRNAs. We designated this novel class of ncRNAs with a short half-life as Short-Lived noncoding Transcripts (SLiTs). We propose that the strategy of monitoring RNA half-life will provide a powerful tool for investigating hitherto functionally uncharacterized regulatory RNAs.
Whole transcriptome analyses using tiling microarrays (Bertone et al. 2004) and deep sequencing (Nagalakshmi et al. 2008) have revealed huge numbers of novel transcripts, including long and short noncoding RNAs (ncRNAs). The ratio of noncoding to protein-coding genomic regions increases as a function of developmental complexity (Mattick 2004), suggesting that revealing the functions of ncRNAs transcribed from noncoding genomic regions is important for understanding genome function in higher organisms. The ncRNAs can be roughly classified into two groups: small transcripts, such as microRNAs and piwi-interacting RNAs (piRNAs), and long transcripts (Prasanth and Spector 2007). Although the biological importance of small ncRNAs has been documented in recent years, the physiological functions of long ncRNAs (lncRNAs) are poorly understood. Recently, significant efforts have been applied to reveal the function of lncRNAs. Several approaches have succeeded in identifying dozens of functional lncRNAs (Guttman et al. 2009). However, the biological functions of the vast majority of lncRNAs remain unclear. Thus, novel properties that can distinguish functional ncRNAs from transcriptional noise are required.
Numerous studies of mRNAs have revealed that changing the abundance of transcripts by regulated RNA degradation is a critical step in the control of various biological pathways (Keene 2010). It has been estimated that the mRNA abundance of 5%–10% of human genes is controlled through the regulation of RNA stability (Bolognani and Perrone-Bizzozero 2008). It has been proposed that the specific half-life of each mRNA is closely related to its physiological function (Lam et al. 2001; Yang et al. 2003; Raghavan and Bohjanen 2004; Sharova et al. 2009; Rabani et al. 2011; Schwanhäusser et al. 2011). Although mRNAs of most housekeeping genes have long half-lives, mRNAs of many regulatory genes, which encode proteins that are required for only a limited time in the cell—such as cell cycle regulators, factors responsible for responses to external stimuli, and regulators of growth or differentiation—often have short half-lives. Moreover, most transcriptionally inducible genes are disproportionately classified into the group of genes with rapid mRNA turnover. It is possible, therefore, that the RNA stability of noncoding transcripts also reflects their functions.
Traditionally, RNA decay has been assessed by blocking global transcription with transcriptional inhibitors, e.g., actinomycin D (ActD), and subsequently monitoring ongoing RNA decay over time. However, inhibitor-mediated global transcriptional arrest has a profoundly disruptive impact on cellular physiology and interferes with the precise determination of the RNA degradation rate (Blattner et al. 2000; Friedel et al. 2009). Here, we present a novel inhibitor-free method (5′-bromo-uridine immunoprecipitation chase, BRIC) that enables measurement of RNA decay under nondisruptive conditions. Determination of the half-lives of whole transcripts by BRIC, combined with multifaceted deep sequencing (BRIC-seq), suggest that there is a relationship between the stability of ncRNAs, as well as mRNAs, and their physiological functions.
Results
Genome-wide determination of RNA half-lives by BRIC-seq
The BRIC method labels endogenous transcripts with 5′-bromo-uridine (BrU) by its addition to cell culture media (Fig. 1A). Total RNAs containing BrU-labeled RNAs (BrU-RNAs) are then isolated from cells at sequential time points after removal of BrU from the culture medium, and BrU-RNAs are recovered by immunopurification (Fig. 1A; Supplemental Fig. S1A,B). The recovery ratios of BrU-RNAs to nonlabeled RNAs were 10–100:1 when cells were labeled with 150 μM BrU, indicating that most RNAs were effectively labeled and were suitable for assaying decreasing levels (Supplemental Fig. S1A). We also confirmed that BrU-RNAs were recovered within a linear range under the experimental conditions used (Supplemental Fig. S1B). The isolated BrU-RNAs can be used as templates for reverse transcriptase–mediated cDNA synthesis for subsequent quantification of RNA, because the BrU-RNA template does not cause misincorporation by reverse transcriptase (Core et al. 2008). Importantly, BrU did not cause harmful effects compared with other uridine analogs, such as 4-thiouridine (Friedel et al. 2009) and 5-ethynyluridine (Fig. 1B; Supplemental Fig. S1C; Jao and Salic 2008). Moreover, BrU did not alter RNA localization, in contrast to ActD, which significantly affects the nuclear speckle localization of MALAT1, a nuclear lncRNA (Fig. 1C). Thus, BrU is a suitable agent to label RNAs for monitoring RNA degradation under physiological conditions. To test whether the BRIC method can determine RNA decay under nondisruptive conditions, we compared the RNA decay rates of MALAT1 (Tano et al. 2010) in HeLa Tet-off (TO) cells using the BRIC method and the Tet-off system, which does not use transcription inhibitors. The half-life of MALAT1 measured by the BRIC method and reverse transcription–quantitative real-time polymerase chain reaction (RT-qPCR) (t1/2 = ∼7.6 h) was similar to that measured by the Tet-off system and RT-qPCR (t1/2 = ∼7.0 h) (Fig. 1D). In contrast, ActD inhibited the degradation of MALAT1 (t1/2 > 12 h) (Supplemental Fig. S2). Thus, the BRIC method allowed us to measure RNA decay under physiologically undisturbed conditions in vivo.
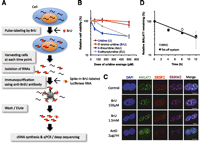
5′-Bromo-uridine immunoprecipitation chase (BRIC) for measuring RNA decay in physiologically undisturbed conditions in vivo. (A) Overview of BRIC. (B) A549 cells were treated with various uridine analogs at the concentrations indicated for 48 h. The number of viable cells was counted using the Cell Counting Kit-8 (Wako). The relative abundance (100% in untreated cells) is shown in the graph. Values represent mean ± SD obtained from four replicate experiments. (C) A549 cells were treated with 5′-bromo-uridine (BrU) or Actinomycin D (ActD) at the concentrations indicated for 7 h. RNA-FISH was used to detect MALAT1 (green). Nuclear speckle proteins, SRSF2 (red) and SRRM2 (magenta), were assayed by immunostaining. Cells were counterstained with DAPI (scale bars, 10 μm). (D) Decay rates of MALAT1 were determined by BRIC and RT-qPCR (gray line) or by the Tet-off system and RT-qPCR (black line) in HeLa TO cells. Relative quantitative values at time 0 h were arbitrarily adjusted to 100%. Values represent mean ± errors obtained from duplicate experiments. The half-lives of MALAT1 using BRIC or the Tet-off system were 7.6 h and 7.0 h, respectively.
We investigated the half-lives of mRNAs and ncRNAs to validate the assumption that RNA half-life is associated with the function of the RNA. We analyzed more than 5 million RNA-seq tags obtained from BRIC–deep sequencing analysis (BRIC-seq) at several time points after BrU pulse-labeling of HeLa TO cells. We calculated genic representations using “reads per kilobase of exon model per million mapped reads” (RPKM) as a means of normalizing for gene length and depth of sequencing (Mortazavi et al. 2008). We then mapped the sequencing tags to the human reference genome sequence (hg18) using Eland as a mapping software, allowing no mismatch settings. We used several public databases for annotation (see Supplemental Tables S1–S7; Methods). Comparison of RNA-seq tags from two independent BRIC-seq experiments showed high reproducibility for transcripts with an RPKM >1 (Supplemental Fig. S3). Transcripts were selected for analysis that had an RPKM value >1 at time 0 h and determined the half-lives for 11,052 mRNAs (average half-life: ∼6.9 h) and for 1418 annotated ncRNAs (average half-life: ∼7.0 h). The actual number of ncRNAs in our analysis was less than the number of ncRNAs assumed previously, because we selected highly and moderately expressed transcripts with an RPKM > 1. Thus, the analysis is biased toward highly and moderately expressed transcripts. Typical mapping data of RNA-seq tags are shown in Figure 2. BRIC-seq allows us to monitor the degradation of the RNAs lacking a poly(A) tail, such as histone mRNAs (Supplemental Fig. S4A,B). Moreover, we could determine the stability of pre-miRNAs (Supplemental Fig. S4C,D). The relative distribution of ncRNA half-lives was similar to that of mRNA half-lives (Fig. 2D), suggesting that the stabilities of ncRNAs are regulated, as is the case with mRNAs.
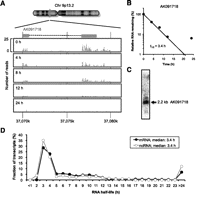
Typical BRIC-seq data. (A) Typical mapping data of RNA-seq tags obtained from BRIC-seq using a Genome Analyzer (Illumina). The chromosomal locus of AK091718 is shown. (B) The relative level of AK091718 RNA remaining was determined by BRIC through deep sequencing. The numbers of sequencing tags at the various time points are shown in A. The relative quantitative values at time 0 h were arbitrarily adjusted to 100%. (C) The expression of AK091718 in HeLa TO cells assessed by Northern blot hybridization. (D) Distribution of RNA half-lives determined by BRIC-seq. BRIC-seq determined the half-lives of 11,052 mRNAs and 1418 ncRNAs expressed in HeLa TO cells. (Black circles) The fraction of mRNAs; (white circles) the fraction of ncRNAs. Transcripts with a half-life >24 h are included in the category “>24.”
We then performed an analysis of Gene Ontology (GO) terms, the association of functional categories with differences in mRNA half-life distribution (Fig. 3; Table 1). GO categories associated with regulatory functions were significantly represented in the short-lived mRNA group (t1/2 < 4 h). In contrast, GO categories associated with housekeeping functions were disproportionately represented in the long-lived mRNA group (t1/2 ≥ 4 h). These results indicate that mRNA half-lives are correlated with the functions of genes, consistent with previous reports (Lam et al. 2001; Yang et al. 2003; Raghavan and Bohjanen 2004; Sharova et al. 2009; Rabani et al. 2011; Schwanhäusser et al. 2011). We further analyzed the correlation between GO terms and mRNA half-lives without splitting them into two groups in terms of RNA half-life. We confirmed that several GO terms, such as regulation of transcription, are associated with short-lived mRNA. In contrast, several GO terms, such as translation, are associated with long-lived mRNAs (Supplemental Fig. S5). We then investigated the relationship between ncRNA half-lives and functional categories (Table 2). We found that several ncRNAs (tRNAs, small nucleolar RNAs [snoRNAs], and small Cajal body–specific RNAs [SCARNAs]) (Jády and Kiss 2001) that are involved in housekeeping functions have long half-lives (t1/2 ≥ 4 h). In contrast, well-known regulatory lncRNAs, such as CDKN2B-AS1 (Yap et al. 2010; Kotake et al. 2011), HOTAIR (Gupta et al. 2010), TUG1 (Khalil et al. 2009; Yang et al. 2011), and GAS5 (Kino et al. 2010) have short half-lives (t1/2 < 4 h). We hypothesize that the half-lives of ncRNAs correlate with functional characteristics and that lncRNAs with regulatory functions may be enriched in the short-lived ncRNA group. We designated this novel class of ncRNAs as Short-Lived noncoding Transcripts (SLiTs), and we identified 795 SLiTs.
GO categories significantly associated with mRNAs of different stabilities
Relationship between functions and stabilities of ncRNAs
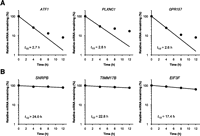
Representative data of mRNA stabilities. Relative levels of mRNA remaining were determined by BRIC-seq. The relative quantitative values at time 0 h were arbitrarily adjusted to 100%. (A) GO categories associated with regulatory functions. (Left) ATF1 is involved in transcription factor activity; (center) PLXNC1 is involved in multicellular organismal development; and (right) GPR157 is involved in G-protein-coupled receptor protein signaling pathway. (B) GO categories associated with housekeeping functions. (Left) SNRPB is involved with the spliceosome; (center) TIMM17B is involved with the mitochondrion; and (right) EIF3F is involved in translation.
Bioinformatic analysis of SLiTs
To further characterize the SLiTs, we performed a series of bioinformatic analyses. Tissue profiling for SLiTs was performed, and the Z-score was calculated using the Adult Tissue TSSseq database (Supplemental Fig. S6; Supplemental Table S8; Yamashita et al. 2010, 2011). The Z-score averages of SLiTs and long-lived ncRNAs were 0.75 and 0.74, respectively. The results suggest that the tissue specificities of SLiTs are almost the same as those of long-lived ncRNAs. We also examined whether the half-life of an ncRNA correlates with other factors as follows. (1) Evolutionary sequence conservation was examined using the public database from http://hgdownload.cse.ucsc.edu/goldenPath/hg18/phastCons44way/vertebrate/ (Supplemental Fig. S7; Supplemental Table S9). The average phastCons scores of SLiTs and long-lived ncRNAs were 0.08 and 0.29, respectively, and the P-value was <0.001. The results suggest that the evolutionary sequence conservations of SLiTs are lower than those of long-lived ncRNAs. The low conservation of SLiTs may be the result of recent and rapid adaptive selection. (2) The absolute expression levels between SLiTs and long-lived ncRNAs were examined. The averages of the absolute expression levels of SLiTs and long-lived ncRNAs were 42.8 and 544.7 RPKMs, respectively, and the P-value was below 0.001. These results indicate that the expression levels of SLiTs were lower than those of long-lived ncRNAs. This is reasonable, because the SLiTs are expected to show low abundance because of their fast degradation. (3) The frequencies of the AU-rich element (ARE), which is the most well-known determinant of mRNA stability and whose consensus core sequence is AUUUA, were examined in SLiTs and in long-lived ncRNAs (Supplemental Table S10). The frequencies of AREs in SLiTs and long-lived ncRNAs were 86% and 77%, respectively, and the P-value was below 0.001. The frequency of AREs in SLiTs is potentially higher than that in long-lived ncRNAs. Thus, we found unique characteristics of SLiTs compared with long-lived ncRNAs. These unique characteristics may reflect the regulatory functions of SLiTs.
Identification of novel lncRNAs involved in cell proliferation among SLiTs
We investigated the function of a number of SLiTs in an attempt to identify important regulatory ncRNAs based on RNA half-life. We first selected 36 uncharacterized SLiTs (t1/2 < 4 h) that did not overlap regions containing known protein-coding or microRNA-containing genes. Some of these SLiTs might have been nonsense-mediated decay (NMD) targets (Mendell et al. 2004), because NMD is a mechanism for eliminating aberrant mRNAs harboring premature termination codons and other genomic noise, such as inactive transposons and pseudogenes (He et al. 2003; Baker and Parker 2004) that are likely to be nonfunctional. Put simply, transcripts that are degraded by NMD have a low potential to be bona fide functional ncRNAs. Based on this hypothesis, we excluded NMD-targeted transcripts from later analysis. To identify transcripts that were degraded through NMD, we quantified SLiT expression in cells with an abrogated NMD pathway by introducing a UPF1 siRNA (Supplemental Fig. S8A; Supplemental Table S11) and judged that a given transcript was targeted by NMD when its level was increased by >200% in the UPF1 knockdown cells (levels of the endogenous NMD-targeted ncRNA, SNHG1 [Smith and Steitz 1998], were increased by 258%). Ten of the 36 transcripts were judged to be NMD-targeted transcripts and were excluded from further analysis. We then determined the cellular localization of the remaining SLiTs. Among these 26 transcripts, 15, two, and nine transcripts were localized to the nucleus, cytoplasm, or both, respectively (Supplemental Fig. S8B; Supplemental Table S12). RNAs retained in the nucleus are the least likely to be translated; therefore, we judged that they were likely to be bona fide ncRNAs. Transcripts localized in the cytoplasm were enriched in the polysome fraction (data not shown) and were excluded. As a result, we identified 15 uncharacterized SLiTs that were likely to be bona fide ncRNAs (Supplemental Table S13). These 15 SLiTs are longer than 200 nt, fulfilling an established criterion for lncRNA classification. The degradation and abundance of three SLiTs—BX648321, NR_024586, and BX537481—were altered by retinoic acid, a typical physiological stimulant involved in cellular proliferation and differentiation (Fig. 4; Ross et al. 2000). The abundance of all three SLiTs increased to >150% after retinoic acid treatment. Interestingly, their half-lives were very short (t1/2 < 2 h). ActD, which influences the stability of mRNA, also affects 76% of SLiTs (Supplemental Figs. S9, S10; Supplemental Table S14). These results suggest that the stability of SLiTs is tightly regulated and supports the notion that short half-life indicates tight regulation and the possession of a particular biological function.
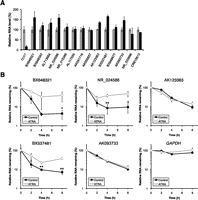
Degradation of several SLiTs was altered by all-trans retinoic acid (ATRA) in HeLa TO cells. (A) Altered abundance of SLiTs by ATRA except BC018860, whose level was below the detection limit. HeLa TO cells were untreated (gray bar) or treated with 10 mM ATRA (black bar). Values represent mean ± errors obtained from duplicate experiments. (B) Decay rates of 5 SLiTs, whose levels were increased >135% in ATRA-treated cells compared with control cells in A and of GAPDH were determined by BRIC and RT-qPCR in control cells (solid circle and black bar) and in ATRA-treated cells (open circle and gray bar). The relative quantitative values at time 0 h were arbitrarily adjusted to 100%. Values represent mean ± SD obtained from triplicate experiments [(**) P < 0.01; (*) P <0.05, Student's t-test].
Functions of the 15 SLiTs were assessed by depleting each transcript using siRNAs and determining the number of viable cells (Fig. 5A). We could only significantly reduce the levels of five transcripts (Supplemental Table S13), because siRNA tends to be less effective on nuclear RNAs compared with cytoplasmic RNAs (Ideue et al. 2009). Cell growth was significantly reduced by depletion of NR_015389, AK091718, and AK055657 with two different siRNAs (Fig. 5A). The knockdown experiment with two different siRNAs can minimize the possibility of the off-targeting on the phenotype. Consistent with the expectation from the knockdown experiment, cell growth was significantly increased by overexpression of NR_015389, AK091718, and AK055657. These results show that the three SLiTs are involved in cell growth. Importantly, no floating cells (dead cells) were seen by depletion of NR_015389, AK091718, and AK055657 under microscopic observation (data not shown), suggesting that these SLiTs are modifying factors but are not essential for cell proliferation. Biochemical fractionation indicated that NR_015389, AK091718, AK055657, AK123363, and NR_033895 were predominantly enriched in the heavy fractions of the sucrose density-gradient analysis, along with nuclear lncRNAs, such as NEAT1 (Clemson et al. 2009; Sasaki et al. 2009; Sunwoo et al. 2009) and MALAT1, which are components of nuclear bodies (Fig. 5B; Supplemental Fig. S11; Hutchinson et al. 2007; Miyagawa et al. 2012). These five SLiTs may localize to specific nuclear domains by forming a dense RNA–protein complex, similar to NEAT1 and MALAT1.
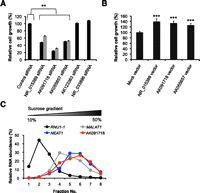
Functional analysis of SLiTs. (A) HeLa TO cells were treated with a control siRNA or with siRNAs targeting indicated SLiTs. The number of viable cells was counted using the Cell Counting Kit-8. Two siRNAs were used for NR_015389, AK091718, and AK055657 to rule out the possibility of an off-target effect of siRNA (black or gray bars indicate the first or second siRNA, respectively). The relative abundance (100% in control cells) is shown in the graph. Values represent mean ± SD obtained in four replicate experiments [(**) P < 0.01, Student's t-test]. (B) HeLa TO cells were treated with plasmid vectors as indicated. The number of viable cells was counted using the Cell Counting Kit-8. The relative abundance (100% with mock vector) is shown in the graph. Values represent mean ± SD in six replicate experiments [(***) P < 0.001, Student's t-test]. (C) The relative amount of RNA was quantified by RT-qPCR from nuclear fractions obtained by sucrose gradient centrifugation.
Discussion
In this study, we report a novel method for determining the half-life of an entire transcriptome. Moreover, by analyzing the relationship between RNA half-lives and functional category, we found that the half-lives of ncRNAs correlate with functional characteristics. We propose a new category of ncRNAs that have short half-lives. We named this novel class of ncRNAs, Short-Lived noncoding Transcripts (SLiTs). To the best of our knowledge, this is the first report to propose a new category of ncRNAs according to their half-life. Bioinformatic analyses revealed the unique characteristics of SLiTs compared with long-lived ncRNAs. We suspect that the unique characteristics of SLiTs may reflect their regulatory functions. Finally, using SLiT criteria, we identified three novel nuclear ncRNAs that are involved in cell proliferation and three novel nuclear lncRNAs with stabilities regulated by retinoic acid.
The nucleoside analog, 4-thiouridine (4sU), was previously used to label endogenous RNAs and mRNAs, and half-lives were indirectly estimated by comparing the preexisting mRNAs and purified 4sU-containing mRNAs (Friedel et al. 2009; Rabani et al. 2011; Schwanhäusser et al. 2011). According to these reports, the median half-life of mRNAs in mammalian cells varied (40 min to ∼9 h). This discrepancy may be due to inherent limitations in indirect estimation of RNA degradation rates by comparing purified 4sU-containing mRNAs with preexisting mRNAs. Indeed, the yield of purification of 4sU-containing mRNAs would dramatically alter the estimation. In this regard, directly chasing decreasing levels of labeled RNA would be more suitable for determining RNA turnover.
SLiTs are different from previously identified groups of ncRNAs, such as TSSs (transcription start site-associated RNAs) (Seila et al. 2008), PASR (promoter-associated short RNAs), PALR (promoter-associated long RNAs), or TASR (termini-associated short RNAs) (Affymetrix/Cold Spring Harbor Laboratory ENCODE Transcriptome Project 2009), because SLiTs do not flank active promoters or terminators of genes. SLiTs are clearly detectable without depleting the nuclear RNA degradation pathway; therefore, they are different from CUT (cryptic unstable transcripts) and PROMPT (promoter upstream transcripts) (Wyers et al. 2005; Preker et al. 2008), which are only detectable in the absence of the nuclear RNA degradation pathway. The majority of SLiTs differ from lincRNAs defined in previous reports (Guttman et al. 2009; Khalil et al. 2009), because the expression levels of most lincRNAs are very low, whereas the expression levels of most SLiTs are significant, perhaps reflecting their importance in biological processes.
In this study, several nuclear SLiTs were silenced by siRNAs. Indeed, previous reports stated that nuclear-localized ncRNA such as RN7SK (Robb et al. 2005; Berezhna et al. 2006), MALAT1 (Tano et al. 2010; Miyagawa et al. 2012), and NEAT1 (Clemson et al. 2009) could be silenced by siRNAs. There are at least two possibilities for silencing nuclear-localized RNA by siRNA: (1) The component of siRNA-induced silencing localized in the nucleus can function to cleave endogenous nuclear target RNAs in the nucleus (Robb et al. 2005). (2) The cytoplasmic RISC cleaves endogenous nuclear target RNAs at the M phase of the cell cycle when the nuclear envelope disassembles and nuclear components, including nuclear ncRNAs, are contacted by cytoplasmic components such as the RISC complex.
We propose that SLiTs are a new classification of regulatory lncRNAs, and we suggest that ncRNA half-life is a key parameter for regulating the function of lncRNAs and for identifying uncharacterized functional noncoding transcripts.
Methods
Cell culture and drug treatments
A549, HeLa TO (Clontech), and HEK293T cells were grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and antibiotics at 37°C in a humidified incubator with 5% CO2. Cells were treated with uridine (Sigma-Aldrich), 5′-bromo-uridine (BrU; Wako), 4-thiouridine (4sU; Sigma-Aldrich), or 5-ethnyluridine (EU; Invitrogen) for 48 h, and the number of viable cells in a 96-well plate were counted using a Cell Counting Kit-8 (Dojindo), in accordance with the manufacturer's instructions.
Plasmid constructs
To construct pGEM-Luc2, the cDNA encoding firefly luciferase 2 (Luc2) was amplified using the primer sets listed in Supplemental Table S15 and pGL 4.13 SV40 as a template. The cDNA was then cloned into pGEM-T (Promega), according to the manufacturer's instructions. To construct the tetracycline-dependent transcriptional repression system of MALAT1, the cDNA encoding human MALAT1 was amplified using a human cDNA library as a template. The cDNA was then cloned into the pGEM-T vector (Promega), as described by the manufacturer. The resulting cloned human MALAT1 cDNA spanned nucleotides 493–8443, corresponding to the reference sequence (RefSeq) accession number NR_002819. The tet response element (TRE) spliced to firefly luciferase 2 (Luc2) cDNA was amplified using the pTRE-Luc2 vector as a template. The pTRE-Luc2 vector was constructed using the pTRE promoter region, which was amplified using a pTRE vector (Clontech) as the template. This fragment was inserted at blunted EcoRV and StuI sites, located upstream of the luciferase cDNA in pGL 4.13 SV40 (Promega). The amplified TRE+Luc2 fragment was digested with ApaI and ligated at the ApaI site, upstream of MALAT1, in pGEM-T-MALAT1. The resultant plasmid (pTRE-Luc2-MALAT1) expressed MALAT1 spliced to Luc2 cDNA at its 5′ end (Luc2-MALAT-1) whose transcription was repressed by tetracycline or doxycycline. To construct the expression vectors for NR_015389 and AK091718, the cDNAs encoding human NR_015389 and AK091718 were amplified using a human cDNA library as a template. The cDNAs were cloned into pcDNA3.1/hygro(+) (Invitrogen), according to the manufacturer's instructions. The AK055057/FLJ31095/IMR321000193 cDNA clone was purchased from NBRC, NITE. Additional information can be provided upon request.
BRIC
For pulse-labeling of newly transcribed RNA with BrU, typically, cells were incubated for 24 h at 37°C in the presence of 150 μM BrU in a humidified incubator with 5% CO2. At indicated time points after the removal of BrU-containing medium, cells were harvested for RNA preparation using RNAiso Plus (TaKaRa), and total RNA was isolated.
To normalize the isolation of BrU-labeled transcripts between time points, BrU-labeled luciferase RNA was used as a spike-in control to serve as an internal standard. The BrU-labeled luciferase RNA was transcribed with 0.3 mM BrUTP, 7.5 mM rNTPs, and 1.6 μg of linearized p-GEM Luc2 using the T7 RiboMax Express Large Scale RNA Production System (Promega). The BrU-labeled luciferase RNA was purified using the QIAGEN RNeasy MinElute Cleanup Kit (QIAGEN), in accordance with the manufacturer's instructions.
Anti-BrdU monoclonal antibody (mAb)–coated protein G-Sepharose beads were prepared as follows. Protein G-Sepharose 4 Fast Flow (GE Healthcare) slurry (20 μL per sample) was washed three times with 1 mL of 0.1% bovine serum albumin (BSA) in phosphate-buffered saline (PBS). The final pellet was resuspended in 300 μL of 0.1% BSA in PBS, and 1 μg of the anti-BrdU mAb (MBL), which also recognizes BrU, was added per sample. The mixture was incubated with rotation for 1 h at room temperature. The beads were washed three times with 1 mL of 0.1% BSA in PBS. The final pellet was resuspended in 200 μL of 0.1% BSA in PBS containing 100 U of RNasin plus RNase inhibitor (Promega).
Two micrograms (for RT-qPCR) or 12 μg (for deep sequencing) of BrU-labeled total RNA, or 0.8 ng of BrU-labeled luciferase RNA as a spike-in control, were prepared as described above, and dissolved in buffer A (25 mM Tris-HCl at pH 7.4, 6.25 mM EDTA). Each mixture was denatured by heating for 1 min at 80°C and then added to the anti-BrdU mAb-conjugated beads. The mixture was incubated for 1 h at room temperature with rotation. Beads were washed three times with 1 mL of 0.1% BSA in PBS. The final pellet was resuspended in 100 μL of buffer B (10 mM Tris-HCl at pH 7.4, 50 mM NaCl), and then 300 μL of ISOGEN LS (Nippon Gene) was added, followed by RNA isolation in accordance with the manufacturer's instructions. The isolated RNA was used for RT-qPCR or deep sequencing.
Deep sequencing and data analysis
Using 1 μg of RNA, an RNA-seq library was constructed using the mRNA Seq Sample Preparation Kit, according to the manufacturer's instructions (Illumina). Thirty-six base-pair single-end-read RNA-seq tags were generated using an Illumina GA sequencer, according to the standard protocol. The fluorescent images were processed to sequences using the analysis Pipeline supplied by Illumina. RNA-seq tags were mapped to the human reference genome sequence (hg18) using Eland as a mapping software and allowing no mismatch settings. Splitting RNA-seq tags representing exon–exon junctions were rescued according to Ref-seq extracted from the UCSC Genome Browser. We calculated genic representations using “reads per kilobase of exon model per million mapped reads” (RPKM) as a means of normalizing for gene length and depth of sequencing (Mortazavi et al. 2008). RNA-seq tags were assigned to corresponding RefSeq transcripts or to lncRNAs when their genomic coordinates overlapped. We then analyzed two replicates of HeLa TO cells expressing BrU-incorporated RNAs using the Illumina high-throughput sequencing platform. The origin and the orientation of the RNAs were documented genome-wide by mapping the reads to the reference human genome. In this study, we used criteria such that data >1 RPKM can be used to determine the half-lives of transcripts. We determined the half-lives of the RNAs by calculating the time when the RNA-seq value reached half of the initial RNA-seq value (0 h time point) (Supplemental Fig. S12). Data from time points after the time point at which the RNA-seq value reached half that of the initial value were omitted from the estimation. For instance, the 24-h time-point value of the A RNA model and the 12- and 24-h time-point values of the B RNA model in Supplemental Figure S12 were omitted from our analysis procedure. By following this strategy, we could obtain the most accurate estimation, because the RNA-seq values of later time points with relatively inaccurate data were omitted from the calculation. To clarify the errors associated with RNA half-life measurements, we determined the error value, calculated (ave2 − ave1)/ave1 values for each transcript, and drew the distribution of these values in a graph of abundance frequency (Supplemental Fig. S13).
Databases
We used mRNA sequences available from public databases: 18,001 RNAs from NM of RefSeq (Sayers et al. 2010). We selected used uncharacterized ncRNA candidates available from several public databases: 2829 RNAs from NR of RefSeq (Sayers et al. 2010), 1738 RNAs from FLJ full-length human cDNAs (Ota et al. 2004; Wakamatsu et al. 2009), 36 RNAs from the Mammalian Gene Collection (MGC Project Team et al. 2009); Deutsches Krebsforschungszentrum (DKFZ); Kazusa DNA Research Institute (KIAA), and 261 RNAs from human large intergenic noncoding RNAs (Khalil et al. 2009). The sum of these RNAs was 5507, and 1418 out of 5507 RNAs passed the cutoff of RPKM ≥ 1. We examined the long ncRNA databases, 831 RNAs from human long noncoding RNAs (Jia et al. 2010), and 223 out of 831 RNAs passed the cutoff. We also examined the other lncRNA database, 5445 RNAs from the lncRNA database (http://www.lncrnadb.org), and 1386 out of 5445 RNAs passed the cutoff. (See also Supplemental Tables S1–S7.)
Reverse transcription–quantitative real-time polymerase chain reaction (RT-qPCR)
The isolated RNA was reverse-transcribed into cDNA using the Quantitect Reverse Transcription Kit (QIAGEN) or the PrimeScript RT Master Mix (Perfect Real Time; TaKaRa). The cDNA was amplified using the primer sets listed in Supplemental Table S17. Luciferase or GAPDH was used for normalization in BRIC or in other experiments, respectively. SYBR Premix Ex Taq II (Perfect Real Time; TaKaRa) was used in accordance with the manufacturer's instructions. Quantitative real-time reverse transcription PCR analysis was performed using a Thermal Cycler Dice Real Time System (TaKaRa).
Functional analysis
To identify functional groups significantly over-represented among short- or long-lived transcripts, we compared the overall distribution of RNA half-lives against the distribution of RNA half-lives for specific functional categories. For this purpose, GO terms were associated to each gene using loc2GO at NCBI. Half-lives of the genes that were associated with a particular GO term were calculated for each group. The statistical significance of the difference in the half-lives between different gene groups was evaluated by Wilcoxon's signed rank test.
Tet-off system
HeLa TO cells were transfected with plasmid using Lipofectamine 2000 (Invitrogen), according to the manufacturer's instructions. Briefly, plasmid DNA was used at a final concentration of 1 μg/mL. The cells were incubated for 24 h at 37°C in a humidified incubator with 5% CO2. After the addition of 1 μg/mL doxycycline (tetracycline analog), cells were harvested at the indicated time points. Total RNA was isolated according to the method of Chomczynski and Sacchi (1987). To distinguish Tet-off promoter-driven exogenous MALAT1 from endogenous MALAT1, the amount of luciferase cDNA (as a tag sequence) conjugated to the 5′ end of MALAT1 was measured by qPCR.
RNA fluorescence in situ hybridization (FISH) and immunocytochemistry
To prepare template cDNA for synthesis of the RNA probe, a 3.3-kb fragment of MALAT1 was amplified using the primer sets listed in Supplemental Table S17. The amplified MALAT1 fragment was cloned into pGEM-T (Promega). A digoxigenin (DIG)–labeled MALAT1 RNA probe was prepared using DIG RNA labeling mix (Roche) and T7 RNA polymerase (Promega), according to the manufacturers' instructions. The transcribed DIG-labeled RNA probes were partially digested in alkaline buffer (60 mM Na2CO3, 40 mM NaHCO3 at pH 10.2) (Cox et al. 1984). FISH was performed to visualize endogenous RNAs according to a previously described protocol (Sone et al. 2007). The hybridized probes were detected with standard immunocytochemical procedures, using the following antibodies: anti-DIG sheep monoclonal antibody (Roche) and Alexa Fluor 488-conjugated anti-sheep IgG polyclonal goat antibody (Invitrogen). To visualize nuclear proteins, we used the following antibodies: anti-SRSF2 mouse mAb (Sigma-Aldrich) and anti-SRRM2 rabbit polyclonal antibody (kindly donated by Dr. Blencowe, University of Toronto). The secondary antibodies used were Alexa Fluor 594–conjugated anti-mouse IgG polyclonal goat antibody (Invitrogen) and Alexa Fluor 647–conjugated anti-rabbit IgG polyclonal goat antibody (Invitrogen). For the detection of DNA, samples were incubated with DAPI (Wako). Fluorescent images were obtained using a confocal microscope TCS SP5 with a 63×/1.4 NA HC PL APO CS lens (Leica). Acquired confocal images were pseudocolored and merged using the image analysis software LAS AF (Leica). All images shown are representative of several repeated experiments.
siRNA treatments
The sequences of the siRNAs used are listed in Supplemental Table S16. These siRNAs were transfected into cells using Lipofectamine RNAiMAX (Invitrogen), according to the manufacturer's instructions. Briefly, siRNA duplexes were used at a final concentration of 10 nM. When single transfection was performed, cells were harvested 48 h or 72 h after transfection. When double transfections were performed, cells were retransfected 24 h after the first transfection, and harvested 72 h or 96 h after the first transfection. The qPCR analysis was used to determine whether RNA interference achieved significant depletion of each target sequence.
Overexpression of SLiTs
The expression vectors were transfected into cells using Lipofectamine 2000 (Invitrogen), according to the manufacturer's instructions. Briefly, the expression vectors were used at a final concentration of 1 μg/mL. The number of viable cells in a 96-well plate was counted 48 h after transfection using a Cell Counting Kit-8 (Dojindo) in accordance with the manufacturer's instructions. Cells were also harvested 48 h after transfection. qPCR analysis was used to determine the overexpression levels of the SLiTs.
Cell fractionation
We followed the protocol of the Hirose Laboratory with slight modifications. HeLa TO cells (∼1 × 107 cells) were collected using a cell scraper and centrifuged at 500g for 5 min. The cell pellet was washed in ice-cold RSB150 buffer (10 mM Tris-HCl at pH 7.4, 150 mM NaCl, 2.5 mM MgCl2) and centrifuged. The cell pellet was resuspended in 800 μL of ice-cold RSB150 buffer. The cells were divided into two tubes for total or nuclear/cytoplasmic fractions. For the total fraction, cells were resuspended in 0.5% Triton X-100 and placed on ice. For the nuclear/cytoplasmic fractions, 0.25 mg/mL digitonin was added to the cells and incubated for 5 min on ice. The cells were centrifuged at 3000g for 1 min at 4°C, yielding the cytoplasmic (supernatant) and nuclear (pellet) fractions. The supernatant was reserved on ice as a source of the cytoplasmic fraction. The nuclear pellet was washed twice in ice-cold RSB150 buffer and centrifuged. The nuclear pellet was resuspended in 400 μL of ice-cold RSB150 buffer and 0.5% Triton X-100. The obtained fractions were subjected to RNA extraction using ISOGEN LS (Nippon Gene), in accordance with the manufacturer's instructions. Samples were eluted with 40 μL of RNase-free water for each fraction. The RNA solutions were treated with DNase I (TaKaRa); then a 10% RNA solution from each fraction was reverse-transcribed into cDNA and quantified by qPCR.
Northern blot analysis
Total RNA (10 μg) was resolved using 1% formaldehyde agarose gel electrophoresis under denaturing conditions and then transferred to a nylon membrane. RNA blots were stained with methylene blue to check for equal loading and transfer. Hybridization was performed with ULTRAhyb (Ambion) using random primed [α-32P]dCTP-labeled cDNA probes. Hybridization signals were visualized and quantified with an FLA-9000 (FUJIFILM).
Sucrose gradient centrifugation
For cytosolic fractionation, HeLa TO cells (∼5 × 106 per 10-cm dish) were washed twice with ice-cold PBS containing 100 μg/mL cycloheximide and lysed directly on the plate by the addition of hypotonic lysis buffer (100 μg/mL cycloheximide, 1 mM dithiothreitol, 200 U/mL RNase inhibitor [Promega], 1.5 mM KCl, 2.5 mM MgCl2, 5 mM Tris-HCl at pH 7.4, 1% Triton X-100, and 1% deoxycholate). The lysate was collected and treated as described (Jefferies et al. 1994). Briefly, 500-μL aliquots were layered onto linear sucrose gradients (10%–50% in 80 mM NaCl, 5 mM MgCl2, 20 mM Tris-HCl at pH 7.4, and 1 mM dithiothreitol) and centrifuged at 50,000 rpm for 90 min at 4°C. The gradient was then fractionated using a Piston Gradient Fractionator (BioComp). RNA from each fraction was isolated using RNAiso Plus (TaKaRa) and analyzed by RT-qPCR, as described. For nuclear fractionation, isolated nuclei were disrupted by brief sonication, followed by centrifugation. The nuclear lysate was layered onto linear sucrose gradients (10%–50% in 80 mM NaCl, 5 mM MgCl2, 20 mM Tris-HCl at pH 7.4, 1 mM dithiothreitol) and centrifuged at 50,000 rpm for 90 min at 4°C. The fractionation of the sucrose gradient and isolation of RNA are described above.
Data access
The short-read sequence archive data that appear in this paper are registered in GenBank (http://www.ncbi.nlm.nih.gov/genbank) /DDBJ (http://ddbj.sakura.ne.jp). The data of two independent BRIC-seq experiments, with or without ActD, at three time points are registered as follows: the accession numbers DRA000357 (0 h, without ActD), DRA000358 (6 h, without ActD), DRA000360 (12 h, without ActD), DRA000361 (6 h, with ActD), and DRA000359 (12 h, with ActD). The data for determining the RNA half-lives at five time points are registered as follows: accession numbers DRA000345 (0 h), DRA000346 (4 h), DRA000347 (8 h), DRA000348 (12 h), and DRA000350 (24 h).
Acknowledgments
We thank Dr. J.A. Steitz (Yale University), Dr. A. Vilborg (Yale University), and Dr. A.H. Fox (University of Western Australia) for critical reading of the manuscript; Dr. S. Nakagawa (Riken) for instruction in FISH analysis; Dr. T. Hirose (AIST) and Dr. Y. Tomari (The University of Tokyo) for fruitful discussions; and MBL Co., Ltd for donating the anti-BrdU antibody. This work was financially supported by the Takeda Science Foundation, the Naito Foundation, Grants-in-Aid for Scientific Research, and Research Fellowship of the Japan Society for the Promotion of Science. This research was supported by the Funding Program for World-Leading Innovative R&D on Science and Technology of the Japan Society for the Promotion of Science.
Authors' contributions: H.T., Y.S., and N.A. conceived and designed the experiments. A.W. and T.I. prepared the FLJ human cDNA data set and performed data analysis. H.T., R.M., K.S., and K.T. performed the experiments and data analysis. H.T., K.I., T.I., Y.S., and N.A. analyzed the data and wrote the manuscript.
Footnotes
-
↵4 Corresponding authors.
E-mail ysuzuki{at}k.u-tokyo.ac.jp.
E-mail akimitsu{at}ric.u-tokyo.ac.jp.
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.130559.111.
- Received August 12, 2011.
- Accepted February 8, 2012.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see http://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 3.0 Unported License), as described at http://creativecommons.org/licenses/by-nc/3.0/.








