
Integrative analysis of genome-wide loss of heterozygosity and monoallelic expression at nucleotide resolution reveals disrupted pathways in triple-negative breast cancer
- Gavin Ha1,2,
- Andrew Roth1,2,
- Daniel Lai2,3,
- Ali Bashashati1,
- Jiarui Ding1,3,
- Rodrigo Goya2,4,
- Ryan Giuliany1,2,
- Jamie Rosner1,
- Arusha Oloumi1,
- Karey Shumansky1,
- Suet-Feung Chin5,
- Gulisa Turashvili1,
- Martin Hirst4,
- Carlos Caldas5,
- Marco A. Marra4,
- Samuel Aparicio1,6 and
- Sohrab P. Shah1,3,6,7
- 1Department of Molecular Oncology, British Columbia Cancer Agency, Vancouver, British Columbia V5Z 1L3, Canada;
- 2Bioinformatics Training Program, University of British Columbia, Vancouver, British Columbia V6T 1Z4, Canada;
- 3Department of Computer Science, University of British Columbia, Vancouver, British Columbia V6T 1Z4, Canada;
- 4Genome Sciences Centre, British Columbia Cancer Agency, Vancouver, British Columbia V5Z 1L3, Canada;
- 5Cancer Research UK, Cambridge Research Institute, Li Ka Shing Centre, Cambridge CB2 0RE, United Kingdom;
- 6Department of Pathology and Laboratory Medicine, University of British Columbia, Vancouver, British Columbia V6T 2B5, Canada
Abstract
Loss of heterozygosity (LOH) and copy number alteration (CNA) feature prominently in the somatic genomic landscape of tumors. As such, karyotypic aberrations in cancer genomes have been studied extensively to discover novel oncogenes and tumor-suppressor genes. Advances in sequencing technology have enabled the cost-effective detection of tumor genome and transcriptome mutation events at single-base-pair resolution; however, computational methods for predicting segmental regions of LOH in this context are not yet fully explored. Consequently, whole transcriptome, nucleotide-level resolution analysis of monoallelic expression patterns associated with LOH has not yet been undertaken in cancer. We developed a novel approach for inference of LOH from paired tumor/normal sequence data and applied it to a cohort of 23 triple-negative breast cancer (TNBC) genomes. Following extensive benchmarking experiments, we describe the nucleotide-resolution landscape of LOH in TNBC and assess the consequent effect of LOH on the transcriptomes of these tumors using RNA-seq-derived measurements of allele-specific expression. We show that the majority of monoallelic expression in the transcriptomes of triple-negative breast cancer can be explained by genomic regions of LOH and establish an upper bound for monoallelic expression that may be explained by other tumor-specific modifications such as epigenetics or mutations. Monoallelically expressed genes associated with LOH reveal that cell cycle, homologous recombination and actin-cytoskeletal functions are putatively disrupted by LOH in TNBC. Finally, we show how inference of LOH can be used to interpret allele frequencies of somatic mutations and postulate on temporal ordering of mutations in the evolutionary history of these tumors.
Segmental regions of loss of heterozygosity (LOH) are a common feature of tumor genomes. LOH can be measured by examination of heterozygous alleles in normal cells that have been rendered homozygous due to segmental aneuploidies or other mechanisms such as gene conversion, mitotic recombination, and mitotic nondisjunction. In numerous malignancies, tumor-suppressor genes such as PTEN, RB1, and TP53 often exhibit loss-of-function mutations coupled with LOH, thereby removing all wild-type alleles and rendering mutant alleles homozygous. Thus, genome-wide LOH is an essential feature to consider in the landscape of alterations of cancer genomes and has played a major role in analysis of recent large-scale genomic studies of cancer subtypes (Cancer Genome Atlas Research Network 2011). Whole-genome (and transcriptome [RNA-seq]) shotgun sequencing (WGSS) of patient tumor-derived DNA and RNA samples is now a common approach for interrogating cancer genomes and transcriptomes to simultaneously determine structural and nucleotide-level aberrations that underpin malignancies (Mardis and Wilson 2009; Shah et al. 2009, 2012; Stratton et al. 2009). The nucleotide resolution of these platforms allows the interrogation of all alleles in both the genomes and transcriptomes of tumors, enabling comprehensive analysis of genomic aberrations and, importantly, their consequent effect on transcription. Determination of the comprehensive nucleotide-level landscape of monoallelic expression (MAE) across all expressed single-nucleotide polymorphisms in genes associated with somatically induced LOH in the genome has not yet been undertaken in cancer. The impact of MAE is twofold in understanding and prioritizing candidate genes from the perspective of haploinsufficiency when alleles are lost (Berger et al. 2011) or oncogenic potential when alleles are specifically amplified (Jirtle 1999). Investigating MAE from the genomic-driven perspective via LOH can help to nominate genes whose expression of the remaining allele may have selective advantages for tumorigenesis and progression.
Ultimately, such characterizations are computational in nature, requiring efficient and robust algorithms for effective biological interpretation of the data. For LOH analysis, several algorithms have been developed for high-resolution genotyping arrays (Lin et al. 2004; LaFramboise et al. 2005; Beroukhim et al. 2006; Dutt and Beroukhim 2007; Staaf et al. 2008; LaFramboise 2009; Närvä et al. 2010); however, this platform is limited to interrogating fixed loci using hybridization intensities as a surrogate measure of nucleotide abundance. In the context of WGSS, which demands approaches for handling digital allelic count data, analysis of LOH is not yet fully explored. We set out to develop a principled probabilistic model for genome-wide, nucleotide resolution inference of LOH from paired tumor-normal sequence data. We examine the distribution of all germline heterozygous single-nucleotide polymorphisms (SNPs) inferred from normal DNA and probabilistically infer LOH in the corresponding loci of the tumor DNA using a hidden Markov model (HMM) approach. Our approach differs from previous methods that have predicted LOH in sequencing data by comparing allele frequencies independently for each site (Zhao et al. 2010) and for segmentation into regions for exome capture (Sathirapongsasuti et al. 2011) and whole-genome data (Boeva et al. 2011).
Several important challenges present themselves in this problem. First, heterozygous SNPs in the germline DNA are nonuniformly distributed across the genome; therefore, genomic distance between adjacent loci needs to be considered in the analysis. Second, the input data representing the observed allelic counts in the tumor DNA are discrete in nature and thus are not well suited to commonly used Gaussian or Student's-t distributions that are often used for the analogous problem in continuous array data. Third, the allelic count data from the tumor DNA will reflect the proportion of normal cells that are admixed with the tumor cells, consequently leading to the dilution of somatic alteration signals in the genome. Fourth, allelic skew due to allele-specific copy number amplifications (ASCNA) can often be erroneously interpreted as true loss of heterozygosity. Generally, ASCNA should still retain signal from the unamplified allele; however, the amplified allele can dominate the overall signal (LaFramboise et al. 2005; Dewal et al. 2011). Figure 1 demonstrates that the allelic distribution for region (iv) is shifted away from diploid heterozygosity but should not be confused as LOH. Thus, analytical approaches should consider somatic copy number changes when inferring LOH. We note that many of these challenges are similarly presented in the analysis of high-density genotyping arrays. Some of the solutions we propose below are inspired by work originally designed for arrays (LaFramboise et al. 2005; Bengtsson et al. 2010; Greenman et al. 2010; Van Loo et al. 2010; Yau et al. 2010; Li et al. 2011), although with specific application to the underlying distributional assumptions of digital allelic count data presented by genome sequencing.
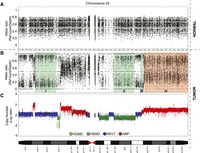
Illustration of empirical allelic ratios between tumor and normal genomic sequencing data from chromosome 20 of a triple-negative breast cancer genome (SA225), and effects of copy number. (A) Allelic ratio data of heterozygous loci in the normal genome are centered around 0.5, which represents the presence of two alleles. (B) At the same corresponding loci, allelic ratios in the tumor genome reveal four examples of somatically acquired segments of allelic imbalance in regions (i)–(iv). (C) The segmental copy number of the tumor helps give context to the allelic data: (i) copy neutral LOH (NLOH), AA/BB; (ii) deletion-induced LOH (DLOH), A/B; (iii) amplified LOH (ALOH), AAA/BBB; and (iv) allele-specific amplification (ASCNA), AAAB/ABBB. Allelic ratio value is defined as the reference read counts divided by total depth at a given position. A and B represent reference and nonreference alleles in the genotype, respectively.
To address these challenges, we developed a statistical approach called APOLLOH to infer regions of LOH from paired tumor-normal data (see Methods for details). Our approach relies on three inputs: (1) the set of genome-wide heterozygous SNP positions inferred from the normal genome, (2) the copy number profiles inferred from the tumor genome, and (3) the allelic counts of the tumor data for each heterozygous SNP position from 1. We fit a novel, nonstationary HMM (accounting for nonuniform distances between adjacent observations) to these allelic counts to map each SNP to heterozygous (HET), homozygous (LOH), or allele-specific amplification (ASCNA) marginal genotypes (Table 1), accounting for all somatic deviations away from heterozygosity. The model uses state-dependent binomial distributions to model the allelic counts and uses a two-component mixture to model the proportion of the observed signal expected to come from normal cells (Laframboise et al. 2007; Yau et al. 2010; Li et al. 2011). We applied the model to 23 triple-negative breast cancer (TNBC, defined by the absence of ER/PR receptor expression and the absence of ERBB2 gene amplification) patient samples whereby tumor and normal DNA was sequenced up to ∼30× coverage using whole-genome shotgun Illumina and SOLiD platforms. For all 23 samples, Affymetrix SNP 6.0 data—a standard, orthogonal technology commonly used to profile LOH in tumor genomes—were also acquired. These data served as a benchmark for systematic comparisons of accuracy of each of the novel aspects of the APOLLOH method against baseline methods. We include an in silico mixing experiment that establishes the relative merit of modeling normal contamination while determining the contamination levels that render tumor signals indistinguishable from normal signals at both 30× and 60×. We generated RNA-seq data from the tumor transcriptomes of 22 patients in order to permit studying the consequence of LOH predicted in the genome on allele-specific expression in the transcriptome. Our results therefore describe the first nucleotide-resolution genome/transcriptome-wide integrated analysis of LOH and MAE in a population of breast tumors and the landscape of allele-specific somatic structural alterations underpinning MAE in TNBC. Finally, we postulate on the merits of considering LOH when interpreting allelic distributions acquired from somatic point mutations for temporal ordering and subclonal inference.
APOLLOH model state representations of genotypes and zygosity status
Results
Application of APOLLOH to profile LOH in 23 deep-sequenced breast cancer genomes
APOLLOH can be summarized as a framework that progressively builds on the standard naive, independent, identically distributed (iid) binomial mixture model (Goya et al. 2010) with the addition of three features. First, the framework is an HMM that inherently accounts for spatial correlation. Next, copy number prior distribution is included to allow an expanded state space within amplified events and to distinguish ASCNA and LOH regions. Finally, the emission component explicitly accounts for normal cell contamination. Figure 2 illustrates the prediction improvements between model variants, cumulatively implementing each feature. The description of APOLLOH is outlined in the Methods, and full mathematical details are described in the Supplemental Methods, Supplemental Figure S1, and Supplemental Table S1.
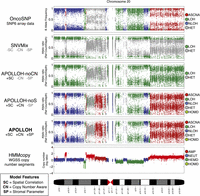
Systematic comparison of loss-of-heterozygosity (LOH) predictions for chromosome 20 of a triple-negative breast cancer genome (SA225). The OncoSNP software (Yau et al. 2010) was applied on an orthogonal platform, Affymetrix SNP6 arrays, and served as the ground truth data set for evaluation. SNVMix (Goya et al. 2010) was used to predict homozygous (LOH) and heterozygous (HET) genotypes on the whole-genome shotgun (WGSS) data to represent the independent, identically distributed (iid) model. APOLLOH is the full model that models copy number (CN) and normal contamination (SP). APOLLOH-noCN is a model variant of APOLLOH that analyzes WGSS without copy number or estimating normal contamination parameter, but models spatial correlation (SC) to predict only LOH and HET in a reduced state space. APOLLOH-noS models copy number but not normal cell proportion, predicting additional marginal states of allele-specific copy number amplification (ASCNA) in an expanded state space. Copy number results were predicted by HMMcopy (Supplemental Methods). Copy number states are amplification (AMP, four to five copies), neutral (NEUT, two copies), hemizygous deletion (HEMD, one copy), and homozygous deletion (HOMD).
We used whole-genome shotgun sequencing (WGSS) to generate 23 triple-negative breast cancer (TNBC) tumor-normal pairs from a cohort of patients described in a larger study (Shah et al. 2012). Seventeen and six patients were sequenced to generate a median of 78 Gb aligned per sample (∼26× sequence coverage) on the Applied Biosystems SOLiD (Life Technologies) and 86 Gb aligned per sample (∼29×) on the Illumina HiSeq sequencing platforms, respectively (Supplemental Table S2). Each genome was aligned to the reference genome using BioScope for SOLiD and BWA (Li and Durbin 2009) for Illumina data. The transcriptomes of 22 of these tumors were sequenced with RNA-seq on the Illumina GAii platform. The full analytical workflow for analysis of these data sets is presented in Supplemental Figure S2 and described in Methods.
Initial benchmarking of WGSS against genotyping arrays demonstrates that the platforms are correlated
We compared the APOLLOH results applied to the WGSS data with Affymetrix SNP6.0 data obtained from the same DNA extractions. We observed statistically significant positive correlation between the allelic ratios of predicted APOLLOH segments and the median B-allele frequency for overlapping SNP6 probes with each segment (Spearman's rho = 0.72, p < 0.001) (Supplemental Fig. S3; Supplemental Methods), demonstrating that WGSS is comparable to the SNP6 platform for analyzing allelic imbalance in cancer. The correlation coefficients across the cases were also significantly associated with the APOLLOH-estimated normal contamination (Spearman's rho = −0.71, p < 0.001) (Supplemental Fig. S4A; Supplemental Table S3), indicating that higher tumor content led to better platform agreement. Furthermore, the separation between predicted LOH, HET, and ASCNA clusters (Fig. 3A) were observed to vary over a dynamic range such that the distance between cluster centers was correlated with the proportion of normal content in the samples (Spearman's rho = −0.81, p < 0.001) (Supplemental Fig. S4B).
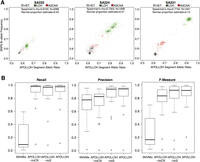
Comparison and evaluation of APOLLOH results using data from Affymetrix SNP6.0 genotyping arrays as the benchmark. (A) Initial benchmarking by comparing WGSS-derived allelic ratios and SNP6 B-allele frequencies. Three samples are shown with LOH clusters centered at locations reflecting APOLLOH normal contamination estimation. (B) For the 23 TNBC samples, precision, recall, and F-measure metrics were computed for LOH predictions from each APOLLOH model variant and SNVMix using OncoSNP (Yau et al. 2010) predictions (from SNP6 data) as the ground truth.
Evaluation of APOLLOH indicates model features systematically improve performance
Having established that WGSS and SNP6 allelic data were in general agreement, we examined the benefits of systematically modeling three key features of spatial correlation, copy number awareness, and normal cell contamination by comparing modular variations of the APOLLOH model (Fig. 2). Setting input copy number status to diploid for all positions reduced the framework to a standard HMM that did not model copy number (APOLLOH-noCN) or normal contamination. Setting stromal proportion s to zero in APOLLOH reduced the model to an HMM that modeled copy number but did not account for normal contamination (APOLLOH-noS). SNVMix (Goya et al. 2010) genotypes were used as the baseline naive i.i.d. binomial mixture model that did not account for the three features.
We evaluated LOH predictions for SNVMix, APOLLOH-noCN, APOLLOH-noS, and APOLLOH on the 23 TNBC WGSS samples using predictions from SNP6 array data analyzed by OncoSNP (Yau et al. 2010) as ground truth (Supplemental Methods). Precision, recall, and F-measure metrics were computed (Supplemental Methods) for each model variant and tumor sample (Fig. 3B; Supplemental Table S4A,B). SNVMix LOH predictions, determined by homozygous genotypes at each site independently using a global threshold on genotype probabilities, showed significantly lower sensitivity across all samples (median recall 0.09). APOLLOH-noCN had significantly higher recall (0.98, one-tailed Wilcoxon-signed-rank test, p < 0.001) and F-measure (0.83, p < 0.001) compared with SNVMix, establishing the benefit to modeling spatial correlation. APOLLOH-noS had significantly higher precision than APOLLOH-noCN (0.94 compared with 0.83, p < 0.001) due to the ability to distinguish LOH and ASCNA in amplified copy number regions, thereby reducing false-positive LOH calls as shown in the q-arm in Figure 2. The F-measure of APOLLOH-noS (0.92) was also significantly higher than APOLLOH-noCN (p < 0.01). The full APOLLOH model, which explicitly models normal contamination, also had a high F-measure with a median of 0.91, which was not significantly different from APOLLOH-noS (two-tailed Wilcoxon-signed-rank test, p = 0.11).
To assess the benefits of modeling copy number, we used 278,229 OncoSNP-predicted ASCNA positions as ground truth to evaluate performance in distinguishing LOH and ASCNA. APOLLOH-noCN correctly called only 6% (recall) as biallelic and had a precision of 0.39. In contrast, APOLLOH demonstrated median recall of 0.73 and precision of 0.82 (Supplemental Table S4C), firmly establishing that explicit consideration of copy number is essential for distinguishing LOH and ASCNA.
For five cases, we also evaluated performance of APOLLOH on an additional benchmark data set by applying the model to whole-exome sequence data published previously (Shah et al. 2012). Using SNP6 as truth, the median precision, recall, and F-measure were 0.85, 0.95, and 0.91, respectively (Supplemental Table S4D), drawing similarities to the WGSS evaluation. The agreement of LOH in these cases across three orthogonal data platforms provides an additional source of validation and demonstrates high confidence in the APOLLOH predictions.
Tumor-normal admixture simulation demonstrates performance maintained at 34% tumor content
We assessed the effectiveness of APOLLOH in predicting allelic imbalance and estimating normal proportion under varying proportions of tumor-normal content by using real data in a controlled in silico experiment. We sampled reads from a tumor sample (SA225) and its matched normal data to generate nine genome-wide data sets for 30× and 60× at proportions of 0.9 to 0.1 normal content. Based on the 13.8% original predicted normal contamination for this case, we conservatively used 15% to determine the following expected normal proportions: 0.915, 0.830, 0.745, 0.660, 0.575, 0.490, 0.405, 0.320, and 0.235. Figure 4A shows how the increased subsampling of normal proportion affects the signal of observable allelic imbalance in the 30× data. For 30× sampled coverage, APOLLOH accurately estimated the normal proportion parameter s for each mixture ≤0.745 with significant overall correlation (Spearman's rho = 0.92, p < 0.001) (Fig. 4B; Supplemental Table S5). The F-measure (Fig. 4C; Supplemental Table S5) for each mixture using SNP6 for ground truth comparison (from the original tumor DNA) indicated that high performance (F-measure = 0.94) was achieved at normal content of 0.58 and was maintained even at 0.66 (F-measure = 0.75). At high levels of contamination, inspection of the data clearly shows that allelic imbalance levels cannot be detected, because the contribution of heterozygous ratios from normal cells dominates the overall signal (Fig. 4A). At 60× coverage, the performance was consistent across all admixture levels, suggesting that sequencing genomes to such depths will likely lead to improved LOH prediction.
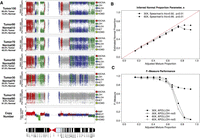
Tumor-normal sampling admixture experiment. Nine mixture proportions generated by sampling reads from the tumor and normal BAM files were analyzed (see Methods). (A) APOLLOH results are shown for chromosome 9 of mixture proportions of 0.09, 0.26, 0.43, 0.60, and 0.77 tumor reads sampled to 30×. (Tumour100) Results from the original tumor sample. (B) The normal proportion parameter s inferred by APOLLOH was significantly correlated (Spearman's rho = 0.92) with the mixture proportions of 0.1–1.0 (increments of 0.1) at 30× and 60×. (C) The F-Measure performance of APOLLOH and APOLLOH-noS (not accounting for normal contamination) for 30× and 60× admixtures was evaluated using Affymetrix SNP6.0 data as ground truth.
Comparison of performance of the full APOLLOH model to the APOLLOH-noS showed that modeling normal contamination modestly increased performance (Fig. 4C). Therefore, we suggest that the parameter estimation of the binomial even without direct inference of s adapts reasonably well to the altered distributions induced by normal contamination. In addition, we noted several anecdotal examples in which accuracy was improved in the full model over APOLLOH-noS (Supplemental Fig. S5). The estimate of the normal proportion of the full model has many additional benefits including informing case-specific stringency thresholds for somatic point mutation prediction and the depth of sequencing that would be needed to recover somatic point mutations. Taken together, these results establish the genome-wide estimation of normal contamination from APOLLOH as an effective indicator of normal cell admixture and provide a reasonable estimate of the upper bound of normal contamination where tumor signal can still be extracted from the data at 30× and 60× coverage.
Genomic landscape of allelic imbalance reveals widespread LOH in TNBC
To infer LOH profiles in the TNBC genomes, we ascertained the copy number profiles from the WGSS data (Fig. 5). The resulting copy number landscape resembled the landscape obtained from an external cohort of 118 basal-like (a subtype of TNBC) breast cancer samples profiled using SNP6 arrays (Curtis et al. 2012). Application of APOLLOH to the WGSS data from the 23 tumor TNBC samples then yielded a total of 37,204 LOH, 19,798 HET, and 2568 ASCNA segments (Supplemental Table S6A). LOH events were further characterized into 9447 (25%) deletion LOH (DLOH), 17,875 (48%) copy-neutral LOH (NLOH), and 9882 (27%) amplified LOH (ALOH). While the number of NLOH segments was higher than DLOH, the median length of a NLOH region was shorter (97 kb compared with 145 kb), and collectively covered, on average across the samples, less of the genome (16% compared with 23%). In contrast, HET regions were much larger with a median of 409 kb and accounted for >49% of the genome on average, compared with 46% by LOH events (Supplemental Fig. S6A; Supplemental Table S6B). The full list of APOLLOH-predicted segments is in Supplemental Table S7.
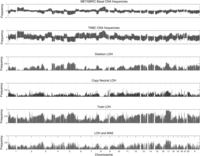
Genome-wide gene frequencies of APOLLOH predictions, copy number profiles from the current 23 cases, and an external (METABRIC) data set (Curtis et al. 2012), and monoallelic expression. From top to bottom, the first and second panels show copy number profiles for cohorts of 118 basal-like subtype breast cancer patients from METABRIC, analyzed on Affymetrix SNP6.0 arrays, and the 23 TNBC patients. Deletion gene frequency profiles (negated for display purposes) in both data sets show similar patterns to deletion LOH frequencies shown in the third panel. The fourth and fifth panels, respectively, show the profile of genes affected by copy neutral LOH and the profile of overall LOH events including genes found within deletions, copy neutral regions, and amplifications. The last panel shows the frequency profile of genes that are observed with monoallelic expression (MAE) as a consequence of genomic LOH events for 22 samples with available RNA-seq data.
LOH genes were determined by assessing complete overlap within predicted LOH segments. On average for each case within the genome, 3404 (16%), 2406 (12%), and 1072 (5%) genes within DLOH, NLOH, and ALOH segments were observed, respectively (Supplemental Fig. S6B; Supplemental Table S6C). The deletion-induced LOH accounted for the majority of the landscape; however, copy neutral LOH contributed substantially with notably higher gene frequencies within chromosomes 3p, 7, 8p, 10, 12, 14, 17, and 22 (Fig. 5). Regions with the highest frequency of amplified LOH were 1q and 17q (Supplemental Fig. S7). The most frequent large-scale event observed in the landscape of zygosity (Fig. 5) was the whole chromosome-level loss of heterozygosity of chromosome 17 in 18 cases (78%). The full list of gene-based LOH alteration frequencies is found in Supplemental Table S8.
For genes falling within amplified regions across the samples, the median proportion of LOH, ASCNA, and balanced CNA (BCNA) was 57%, 28%, and 10%, respectively (Supplemental Table S6D). Amplified and copy neutral LOH are consistent with the notion that segmental amplifications or duplications are the result of at least two copy number events in the evolutionary history of the tumor. We noted several examples, specifically on chromosome 17, that we speculate are regions whereby compound deletion–amplification events likely occurred in sequence (Supplemental Fig. S9). The distribution of the number of compound events across the 23 cases showed a wide variance, ranging from 471 to 2022 segments (Supplemental Fig. S9). This shows that at diagnosis these tumors have undergone varying degrees of complex genomic architecture evolution. We reported similar findings from analysis of somatic SNV mutation events in TNBC (Shah et al. 2012). Intriguingly, the number of compound events (NLOH and ALOH segments) did not correlate significantly (Spearman's rho = 0.22, p = 0.31) with the number of somatic missense and nonsense mutations for each sample. This suggests that the rate of accrual of complex genomic architecture aberrations is independent of the rate of point mutation accrual in the evolution of TNBCs. Thus, the relative contribution of distinct mutagenic mechanisms that shape the genomes of TNBC varies widely across the population. The significance of this observation in a clinical context is unknown; however, these results emphasize that genomic diversity in the form of complex CNAs should be considered alongside mutational profiling to assess the degree of clonal evolution in tumors.
Somatic inactivation of genes with germline stop codon mutations
We investigated the effects of LOH on genes that harbor heterozygous germline stop codon variants. We conservatively determined 1390 truncating variants that overlapped the normal heterozygous positions in our data set (Methods). Across the 23 cases, we found that LOH led to the loss of the amino acid coding allele in 291 positions, leaving only the stop codon allele that encodes a truncated protein (Supplemental Fig. S10; Supplemental Table S9A). In contrast, 582 events were observed to have the truncating variant lost due to LOH. Using 44,754 synonymous germline variants as a background distribution where 18,154 variant events (41%) were retained after LOH, we noted that the proportion of lost truncating variant alleles due to LOH was significantly enriched (χ2, p < 0.001). This suggests that selection on somatically driven LOH of a germline background of truncating polymorphisms may lead to removal of truncated genes. However, the 291 events still represent an intriguing upper bound on the possibility of partial or complete loss of function in the affected genes. The rate of occurrence (12.7 ± 6.4 per case) was comparable at the same order of magnitude to the number of genes affected by nonsynonymous coding mutations typically reported in epithelial cancer genomes (Ding et al. 2008; Shah et al. 2009, 2012; Pleasance et al. 2010a,b), indicating that somatic inactivation by LOH of germline truncating protein variants likely contributes meaningfully to the mutational landscape of TNBC. Moreover, this analysis outlines a genome-wide substrate composed of germline and somatic genetics upon which selection may be acting. Larger studies would be required to determine its implication in the pathogenesis of TNBC.
Analysis of LOH and somatic mutations reveals potential subclonality and temporal ordering
We next sought to interpret somatic point mutations in the context of their genomic architectures as defined by APOLLOH. We investigated 680 missense and 55 truncating (nonsense) mutations (Supplemental Table S9B) using previously validated data (Shah et al. 2012) and prediction tools (Methods) from the 23 cases used in this study. We observed that in 63 (9.3%) of the missense events, LOH rendered the mutation homozygous, which included mutations affecting TP53, PTEN, ERBB2, and PIK3CA. The mutation in PIK3CA was a canonical activating kinase domain mutation, H1047R, and was found in a region of ALOH, agreeing with previous findings (LaFramboise et al. 2005; Dewal et al. 2011) that the mutation was acquired early and was selectively amplified. In addition, mutations rendered homozygous due to LOH affected genes with roles in actin cytoskeleton and microtubule stabilization functions (KLHL1, ESPN, DIAPH1, CASC5), extracellular matrix (ECM) interactions (LAMA1), angiogenesis (BAI2), and cell division (CDC5L, CDCA7). In the truncating events, nine were homozygous for the stop codon (Supplemental Table S9C), leading to complete inactivation of genes such as RAD51C (involved in homologous repair), THSD4 (involved in ECM assembly), JAK1 (involved in the IFN-alpha/beta/gamma signal pathway), and CDK12 (a cyclin-dependent kinase involved in splicing) (Supplemental Table S9B).
For biallelic inactivation due to DLOH, temporal ordering of coincident mutation and the CNA deletion is challenging to ascertain. However, for mutations rendered homozygous that overlap NLOH and ALOH, the parsimonious explanation for the combined observations is that the mutation events likely arose first, and subsequent duplication or amplification of the remaining mutant allele followed. Thus, the resulting temporal ordering suggests that these are candidate tumorigenic mutations that were selected for throughout the evolutionary history of the tumor.
In contrast, 247 total missense and nonsense mutations in regions of LOH have allelic ratios that were skewed toward the wild-type allele (Supplemental Table S9B). These are more difficult to interpret, since there are competing explanations: (1) The events may be mutually exclusive, occurring independently in separate, individual cells; (2) in NLOH and ALOH regions, the mutation may have occurred subsequently to the LOH and amplification events, leading to the presence of the mutation in only a portion of the alleles. Whether subclonal or relatively late in the evolutionary process, these mutations were likely not early drivers of tumorigenesis. Ultimately, single-cell resolution would be required to adequately interpret their significance (see Discussion).
Monoallelic gene expression events associated with genomic LOH reveal disrupted pathways in TNBC
We investigated the association between APOLLOH results and transcriptome allelic ratio (TAR) by analyzing 22 TNBC patients for which tumor RNA-seq data were available (Supplemental Methods). For LOH-predicted segments, the corresponding TAR is expected to be monoallelic. In contrast, TAR for HET- and ASCNA-predicted segments may be observed as either balanced, skewed, or monoallelic depending on factors such as epigenetic modifications and mutations in regulatory elements (Pastinen and Hudson 2004). Across the cohort, we observed that the median TAR values for LOH, ASCNA, and HET were 0.83, 0.71, and 0.63, respectively (Fig. 6A). We also observed that the median TAR of APOLLOH-predicted LOH segments and the APOLLOH-estimated normal proportion parameter s showed statistically significant negative correlation (Spearman's rho = −0.91, p < 0.001) (Fig. 6B; Supplemental Table S3), explaining the observed overall deviation of the TAR distribution away from 1.0. Thus, the RNA-seq data corroborated the prediction of normal proportion from APOLLOH in addition to contributing to the accuracy of LOH calls. In contrast, the correlation for TAR within LOH regions and normal contamination predicted by OncoSNP was not as strong, but still significant (Spearman's rho = −0.85) (Supplemental Fig. S11; Supplemental Table S3).
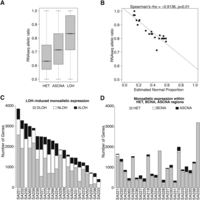
Integrative analysis of APOLLOH results and transcriptome RNA-seq expression. (A) The distribution of transcriptome RNA-seq symmetric allelic ratios that fall within HET, ASCNA, and LOH predicted regions are significantly different (pairwise Wilcoxon one-tailed test, p < 0.01). (B) The median symmetric allelic ratio of RNA-seq data within predicted LOH segments for each sample, represented as a point, strongly negatively correlated (Spearman's rho = −0.91) with estimated normal proportion parameter s (first principal component line is shown). (C,D) Distribution of the number of monoallelic expressed genes within genomic loss-of-heterozygosity (LOH), heterozygous (HET), and allele-specific copy number amplification (ASCNA) regions in 23 breast cancer samples. (C) The number of MAE genes established by LOH events are categorized into deletion (DLOH), copy neutral (NLOH), and amplification (ALOH) and sorted by total LOH in descending order. (D) The number of genes with MAE that overlapped genomic HET, balanced CNA (BCNA), and ASCNA regions are shown in same sorted order as in C.
The unbiased genome-wide coverage of WGSS nominated more normal heterozygous loci in each of the 23 cases compared with the full scaffold of probes on the SNP6 platform (Supplemental Table S10). Subsequently, the number of overlapping RNA-seq positions with available coverage was also approximately twofold more for WGSS (mean 108,778 ± 31,832) compared with SNP6 (mean 48,224 ± 13,570) (Supplemental Fig. S12). Moreover, the high resolution offered by genome sequencing enabled APOLLOH to predict 2021 LOH segments smaller than 3 kb, of which 1481 were not predicted by OncoSNP; these predictions were supported by similar RNA-seq allelic ratios (median of 0.83 and 0.80, respectively). In fact, 1020 of 1481 segments had boundaries located completely between or outside of the Affymetrix SNP6 probe scaffold (Supplemental Table S11). These results demonstrate that whole-genome sequence data are more suitable for comprehensively analyzing LOH and allelic expression at resolutions that are not attainable by SNP6.
Monoallelic expression (MAE) can arise as a result of genomic allelic loss via LOH events. To characterize the occurrence of this mechanism, we determined genes that exhibited MAE in the transcriptome established by co-occurring predicted LOH events (Supplemental Table S12; Methods). An average of 3137 genes per case exhibited MAE, of which 2017 (64%) were observed to be coincident with LOH (Fig. 6C). Deletion LOH gave rise to an average of 962 genes with MAE, whereas copy neutral and amplified LOH events led to an average MAE of 696 and 358 genes, respectively (Supplemental Table S6E). In contrast, there were far fewer instances of MAE of genes within HET, BCNA, and ASCNA regions, averaging 993, 29, and 98 per case, respectively. Only three (14%) cases had more genes implicated within these regions, than within regions of LOH (Fig. 6D). This suggests that genomic LOH explained the majority of MAE in TNBC and established a lower bound on the proportion of MAE that can be directly attributed to LOH. As a result, we suggest that only a minor proportion of MAE could be attributed to other modifications of the genome such as epigenetic factors and mutation. Moreover, we observed significant positive correlation between the abundance of MAE genes within HET, BCNA, and ASCNA regions and the predicted normal proportion (Supplemental Fig. S13), indicating that the MAE genes in these regions were likely germline (epigenetic) events whose signals became more detectable as normal cell content increased.
We next examined the genome-wide landscape of LOH-associated MAE. In general, the pattern of LOH-induced MAE closely mirrored the landscape of genomic LOH as shown in Figure 5. However, the absolute frequency of events was reduced, most likely due to lower expression of genes in deletion LOH regions, and our conservative approach for establishing MAE. Examination of the copy neutral frequencies also closely mirrors the shape across the genome of the LOH-associated MAE profile. Consistent with our observation from the genomic LOH landscape, the most frequent genes exhibiting LOH-associated MAE were found within chromosomes 3p, 5q, 8p, 10p, 14, and 17 (Fig. 5; Supplemental Fig. S14; Supplemental Table S8).
To further refine the interpretation of the LOH-induced MAE genes, we performed a pathway analysis to examine biological functions that could be modulated by these genes. Using the Reactome (Wu et al. 2010) database, we projected the genes onto a network of interacting proteins and clustered this network into highly connected modules (Methods). A total of 11 modules were identified, with seven having significantly enriched pathways (FDR < 0.05) (Fig. 7; Supplemental Table S13). In particular, Module 0 contained pathways involving cell-shape/motility, focal adhesion and integrin signaling; Module 2 contained M-Phase genes; Module 3 contained homologous recombination (HR); and Module 4 contained Wnt and cadherin signaling, and chromatin-remodeling complexes. Haploinsufficiency in HR genes is known to lead to chromosome fragmentation and genome instability (Thacker and Zdzienicka 2003; Date et al. 2006), and Wnt, cell cycle, and focal adhesion are all known from functional studies to modify tumor initiation and/or tumor progression and furthermore have been specifically associated with breast cancer pathogenesis. Intriguingly, genes in Module 1 nominated functionally enriched gene sets that are linked along a chain of related oncogenic pathways. Notably, integrin signaling, regulation of actin cytoskeleton, focal adhesion, and Wnt signaling exhibit considerable cross talk with growth factor signaling (Turner 2000) due to EGFR and PI3 kinase, both of which are known oncogenic drivers in breast cancer. Our results now implicate a genomic mutational mechanism for disrupting the normal function of these pathways, in the form of LOH-associated MAE, that has been underappreciated in the literature. The identification of these core pathways in our analysis indicates that LOH-associated MAE contributes a measurable component of the somatic mutational landscape which also includes CNAs, point mutations, insertions/deletions, and epigenetic changes that collectively modulate biological function.
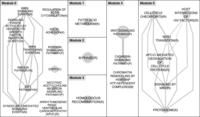
Pathway enrichment analysis of genes with monoallelic expression (MAE) established by loss-of-heterozygosity (LOH) events. Gene networks were inferred using Reactome Functional Interaction software (Wu et al. 2010) within the Cytoscape (Smoot et al. 2011) plug-in. LOH-induced MAE genes were used in the analysis and subsequently clustered into modules. At a false discovery rate (FDR) of 0.05, significantly enriched pathways included Modules 0–5. Shown are the Enrichment Map (Merico et al. 2010) networks generated for the significant pathways (Supplemental Table S13), highlighting the interactions between pathways identified within each of the six modules.
Discussion
We have described a probabilistic framework for predicting regions of LOH in genome sequencing data of cancers, and implemented the model as a nonstationary HMM called APOLLOH. The algorithm models discrete, digital allelic count, taking advantage of the base-pair resolution quality offered in sequencing data. The experimental workflow allows the analysis to be performed at an unprecedented number of possible heterozygous sites in the normal and, in contrast to genotyping arrays, are unrestricted to fixed loci. We applied the algorithm to 23 triple-negative breast cancer genomes sequenced to ∼30× coverage on two massively parallel sequencing platforms. We also investigated the extent of LOH in affecting allele-specific expression by analyzing matching tumor transcriptome RNA-seq data. Its application to this data set constitutes the largest study for analyzing a sequenced cancer cohort with the aim of examining loss of heterozygosity and its role in monoallelic expression.
The performance of the variants of the APOLLOH framework shows progressively improved results when features are incrementally added in order: spatial correlation, copy number data inclusion, and normal contamination modeling (full model). The model benefits from copy number particularly in regions of amplifications where false-positive LOH predictions are reduced and instead attributed as being a signal for allele-specific amplification. One caveat with using OncoSNP for the basis of the evaluation is the inclusion of germline LOH regions in the truth set; these regions will be devoid of data in the APOLLOH analysis due to the inclusion of only informative heterozygous positions (Fig. 2 at 20q11.22-23). This may suggest that the observed recall (sensitivity) rates should, in fact, be even higher.
Accounting for normal cell contamination did not significantly improve accuracy in our benchmarking analysis; however, we noticed that there were specific instances in which incorporation of the s parameter allowed APOLLOH to be more sensitive to LOH (Supplemental Fig. S5). Moreover, the full model has the advantage of providing the normal proportion estimate for each sample, which is useful not only for confirming the general validity of the LOH predictions but also aides in interpretation of other somatic alterations (e.g., point mutations) in the context of cellularity. This also provides pathologists with objective, quantitative estimations of cellularity that may be more accurate than manual inspection of histological slides, which is the current standard practice.
We used LOH results to interpret somatic point mutations in the context of temporal ordering of genomic aberrations and subclonality. The presence of complex clonal populations and tumor heterogeneity was recently shown when inferring the mutational profiles of TNBC (Shah et al. 2012). While APOLLOH explicitly accounts for normal cell contamination, it does not yet inherently model subclonality and heterogeneity for LOH prediction. The presence of subclonal allelic imbalance signals, among other tumor cells admixed with normal cells, is more difficult to detect, potentially leading to false negatives with the current model. This is an exciting and on-going subject of future extensions and presents a challenging task particularly enabling the analysis of the interplay between chromosomal architecture, such as sequential compound copy number events, and subclonal somatic mutations in the context of LOH. Motivation for reconstructing the temporal sequence of genomic aberrations can be drawn from a recent study of primary and cell line breast cancer SNP6 array data (Greenman et al. 2012). Ultimately, the establishment of single-cell sequencing technologies (Navin et al. 2011; Hou et al. 2012; Xu et al. 2012) will drive development of reliable solutions to help deconvolute the complexities of profiling tumors with subclonal and tumor-normal admixture cell populations.
We report the landscape of allelic imbalance across 23 whole triple-negative cancer genomes, surveying genes that are affected by LOH predicted segments. The strongest signal resides in chromosome 17, which is observed, in 78% of the cases, to have nearly complete chromosomal level LOH. Despite the majority of LOH events being induced by deletions in chromosome 17, nearly 20% of cases show substantial copy neutral LOH, which would have otherwise been overlooked if allelic-specific imbalance was not considered. This result is similar to those previously reported in another breast cancer cohort (Van Loo et al. 2010) and in a high-grade serous ovarian data set (Cancer Genome Atlas Research Network 2011), reinforcing the suggested genomic link between TNBC and ovarian high-grade serous cancers (Bowtell 2010).
This is the first and largest sequencing study aimed at analyzing genome and transcriptome data in combination to determine LOH and its effects on allelic expression, particularly MAE. We provided an analysis of MAE that investigates only the genomic-driven perspective via LOH, providing a verification of APOLLOH predictions and helping to nominate allelic imbalanced genes whose expression may have biological impact to the progression and state of the tumor. Indeed, pathway analysis of the genes affected by LOH-associated MAE revealed core oncogenic pathways and therefore implicates LOH with coincident MAE as an important mechanism of pathway abrogation that complements copy number, point mutation, and epigenetic analysis. Interestingly, the results show that a minority of MAE is associated with diploid regions. This implies that either LOH is specifically targeting regions of the genome with preexisting MAE or, more likely, that the majority of MAE in TNBC is explained by fixed genome aberrations, rather than epigenetic regulation. Full integration of all of these molecular views of tumor landscapes are likely to reveal yet additional insights into tumor biology.
This study provides a framework for analysis of allelic imbalance in tumor-normal genome sequencing experiments. The analysis of 23 TNBC genomes shows that LOH is a prominent feature of TNBC somatic aberrations and modulates a significant portion of the transcriptome in the form of monoallelic expression. These results indicate that analysis of LOH is an integral component to the comprehensive interpretation of cancer genomes, and we conclude that APOLLOH will complement the growing arsenal of computational tools designed for cancer-focused sequencing studies.
Methods
APOLLOH workflow overview
A full representation of the APOLLOH framework as a probabilistic graphical model is given in Supplemental Figure S1, and all mathematical details of the method are described in the Supplemental Methods. Biospecimen collection, histopathological review, and library construction are also described in the Supplemental Methods. Application of the method to the 23 tumor/normal data set was carried out as follows.
APOLLOH analyzes positions 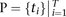 that are heterozygous SNPs in the normal genome. We obtained these using GATK (McKenna et al. 2010), which predicted between ∼1 and 2.2 million positions genome-wide per patient (Supplemental Table S10). Restricting the
analysis to positions where both alleles are present in the matched normal sample reduces the dimensionality of the analysis
to T loci and ensures that detected homozygosity will be somatic events. From the tumor genome data, the read counts mapping to
the reference base (A allele), read counts mapping to the nonreference base (B allele), and total depth at all positions in P were extracted using SAMtools (Li et al. 2009) and represented as a1:T, b1:T, and N1:T, respectively.
that are heterozygous SNPs in the normal genome. We obtained these using GATK (McKenna et al. 2010), which predicted between ∼1 and 2.2 million positions genome-wide per patient (Supplemental Table S10). Restricting the
analysis to positions where both alleles are present in the matched normal sample reduces the dimensionality of the analysis
to T loci and ensures that detected homozygosity will be somatic events. From the tumor genome data, the read counts mapping to
the reference base (A allele), read counts mapping to the nonreference base (B allele), and total depth at all positions in P were extracted using SAMtools (Li et al. 2009) and represented as a1:T, b1:T, and N1:T, respectively.
If the alleles are observed as equally likely, showing no skew toward one particular allele, then the genotypes can be treated
symmetrically (e.g., AA and BB or AAB and ABB are treated the same) using the symmetric reference count, 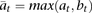 . APOLLOH is flexible to use
. APOLLOH is flexible to use  or a; however, in this study, we modeled the alleles separately and therefore will describe the asymmetric version of the model
throughout.
or a; however, in this study, we modeled the alleles separately and therefore will describe the asymmetric version of the model
throughout.
APOLLOH uses copy number information that is provided as biologically interpretable classes of segmental copy number changes in the tumor sample: homozygous deletion (no copies), hemizygous deletion (one copy), neutral (two copies), one copy gain (three copies), and two and three copy amplifications (four and five copies). Copy number status c1:T is then assigned to all positions in P based on its overlap within the corresponding copy number segment. Copy number profiling of the tumor genome aligned reads was performed using an in-house HMM-based approach called HMMcopy (Supplemental Methods; http://compbio.bccrc.ca/software/hmmcopy/).
APOLLOH performs inference and segmentation of genotypes G1:T given the input data a1:T, N1:T, c1:T from the tumor. Subsequently, the genotypes at each position are encoded into the corresponding zygosity status ZS1:T of LOH, HET, and ASCNA, which are divided into groups of states based on copy number (Table 1). APOLLOH is implemented as an HMM that simultaneously provides classification of regions into biologically interpretable discrete genotype states and segments input data. The tumor allelic ratio data are modeled using a mixture of binomial distributions that also considers the proportion of normal cell contamination in the sample. The HMM models spatial dependency using state transition probabilities that account for the distance between adjacent positions (Colella et al. 2007), and deterministically informed by copy number status ct at each t ∈ P.
Tumor-normal sampling mixture experiment
Nine whole-genome BAM files were generated and compiled by sampling reads from the tumor and normal BAM files of SA225 at mixture proportions of 0.1 increments. For each chromosome and each mixture combination, the total number of reads was set to be the same as the normal BAM file. This resulted in ∼30.5× coverage or 91 Gb of aligned reads for each genome-wide BAM file. We repeated this for nine more genome samplings at ∼60×. APOLLOH hyperparameter settings for the Beta prior distribution of the normal proportion parameter s were assigned uniform settings, αs = 5000 and βs = 5000. We used the copy number results from the original tumor BAM file for APOLLOH analysis of all nine mixtures in 30× and 60× samplings.
Truncating variant and mutation analysis
For germline truncating variants, normal heterozygous positions for each sample were used. For somatic truncating mutations in the SOLiD genomes, the published set of validated mutations (Shah et al. 2012) was used; for the Illumina genomes, a set of mutations was predicted using JointSNVMix (Roth et al. 2012) and filtered by the classifier MutationSeq (Ding et al. 2012). The positions for each sample were annotated using snpEff (Cingolani 2012) (hg36.54), and positions with codon effects “STOP_LOST” (germline only) and “STOP_GAINED” were extracted. The remaining alleles following LOH were assigned as WT and MUT if the tumor allelic ratio was >0.5 or <0.5, respectively; for the validated mutations, the ultra-deep amplicon sequencing allelic read counts were used.
For nonsynonymous mutations, positions with the codon effect “NON_SYNONYMOUS_CODING” were used.
Analysis of monoallelic expression
We used SNVMix to generate genotypes for all transcriptome positions intersecting loci used in the APOLLOH analysis. Parameters for SNVMix were set using the two-component mixture, s × 0.5 + (1 − s)μg, where μaa = 1, μab = 0.5, μbb = 0, and s is inferred by APOLLOH on the genomic data. We compared these parameters to the distributions of transcriptome allelic ratios (TAR) and found them appropriate (Supplemental Fig. S15). A gene g was determined to have MAE status if the genotypes for all positions xg ∈ P overlapping g had a marginal posterior probability of being homozygous (paa + pbb) greater than heterozygous (pab).
Reactome FI (Wu et al. 2010) analysis was performed using the Cytoscape v2.8.1 (Smoot et al. 2011) plug-in. Genes that had LOH-MAE frequencies of 10 or greater were used in the analysis. Significant pathways (FDR < 0.05) in Modules 0–5 were analyzed using EnrichmentMap (Merico et al. 2010) analysis to determine relationships between pathways within the module. For this analysis, we used gene sets, in GMT format, as described in Shah et al. (2012).
Data access
The genome and transcriptome sequencing files can be downloaded at the European Genome-phenome Archive under accession number EGAS00001000132. The source code for APOLLOH can be accessed at http://compbio.bccrc.ca/software/apolloh.
Acknowledgments
This study was funded by the Canadian Breast Cancer Foundation. S.P.S. is supported by the Michael Smith Foundation for Health Research. G.H. is supported by the Natural Sciences and Engineering Research Council of Canada.
Author contributions: S.P.S. provided project conception and oversight. G.H., S.P.S., and S.A. wrote the manuscript. G.H., S.P.S., and A.R. completed the algorithm design and implementation. G.H. carried out all analytical experiments. D.L. and G.H. performed copy number analysis. A.B., J.D., Ro.G., J.R., Ry.G. and K.S. performed whole genome and RNA-seq data generation, analysis, and discussions. A.O., S.F.C., and G.T. were responsible for sample preparation and histopathological review. M.H. performed library construction and sequencing. C.C. contributed tumor specimens from the Addenbrookes (Cambridge UK) Tumour bank. S.A., M.A.M., and C.C. performed oversight of sequencing data generation and were the TNBC sequencing study project leaders.
Footnotes
-
↵7 Corresponding author
E-mail sshah{at}bccrc.ca
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.137570.112.
Freely available online through the Genome Research Open Access option.
- Received January 11, 2012.
- Accepted May 22, 2012.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see http://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 3.0 Unported License), as described at http://creativecommons.org/licenses/by-nc/3.0/.








