
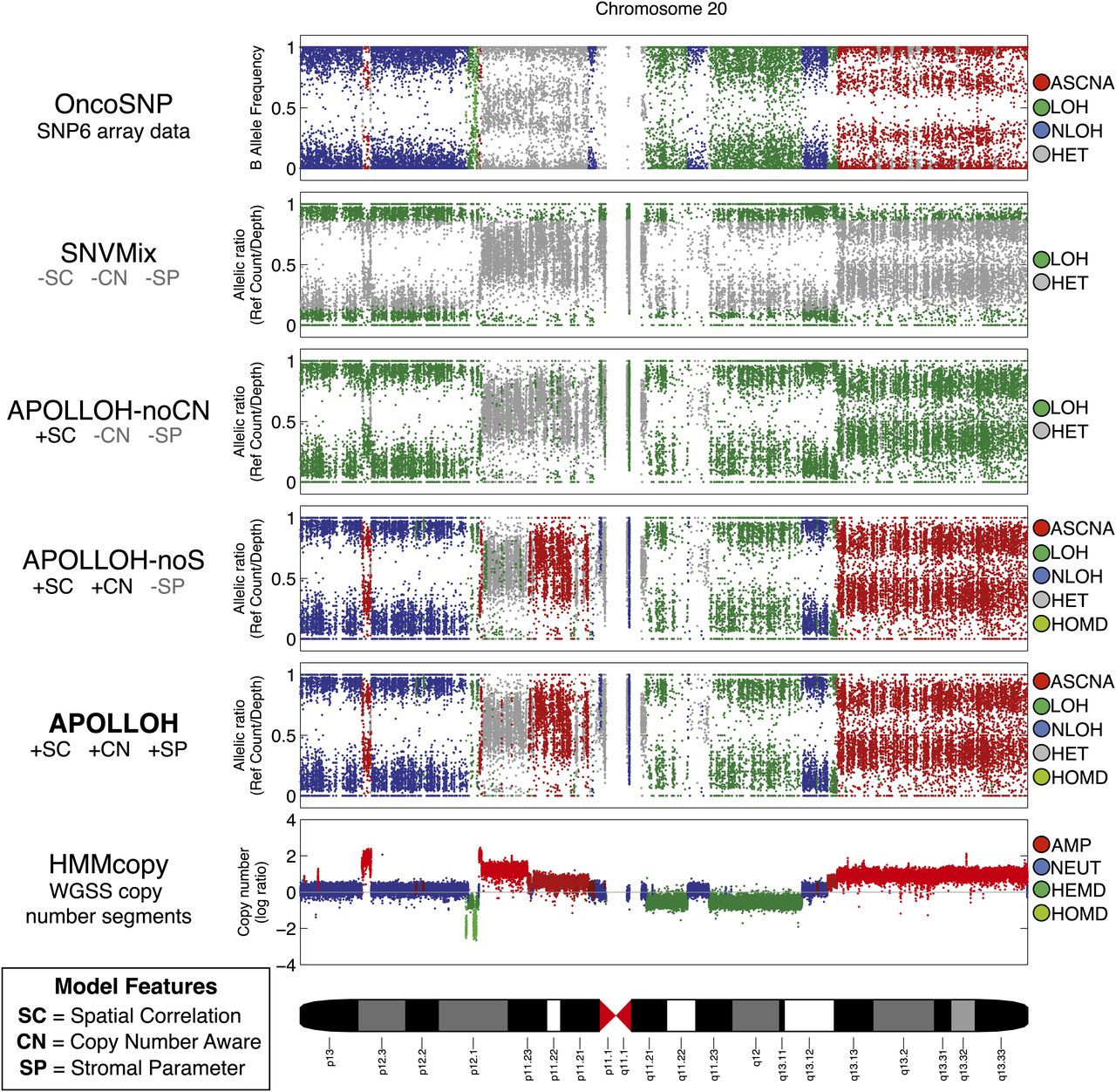
Systematic comparison of loss-of-heterozygosity (LOH) predictions for chromosome 20 of a triple-negative breast cancer genome (SA225). The OncoSNP software (Yau et al. 2010) was applied on an orthogonal platform, Affymetrix SNP6 arrays, and served as the ground truth data set for evaluation. SNVMix (Goya et al. 2010) was used to predict homozygous (LOH) and heterozygous (HET) genotypes on the whole-genome shotgun (WGSS) data to represent the independent, identically distributed (iid) model. APOLLOH is the full model that models copy number (CN) and normal contamination (SP). APOLLOH-noCN is a model variant of APOLLOH that analyzes WGSS without copy number or estimating normal contamination parameter, but models spatial correlation (SC) to predict only LOH and HET in a reduced state space. APOLLOH-noS models copy number but not normal cell proportion, predicting additional marginal states of allele-specific copy number amplification (ASCNA) in an expanded state space. Copy number results were predicted by HMMcopy (Supplemental Methods). Copy number states are amplification (AMP, four to five copies), neutral (NEUT, two copies), hemizygous deletion (HEMD, one copy), and homozygous deletion (HOMD).








