
Genome-wide mapping of alternative splicing in Arabidopsis thaliana
- Sergei A. Filichkin1,
- Henry D. Priest1,
- Scott A. Givan1,
- Rongkun Shen1,3,
- Douglas W. Bryant1,2,
- Samuel E. Fox1,
- Weng-Keen Wong2 and
- Todd C. Mockler1,4
- 1 Department of Botany and Plant Pathology and Center for Genome Research and Biocomputing, Oregon State University, Corvallis, Oregon 97331, USA;
- 2 Department of Electrical Engineering and Computer Science, Oregon State University, Corvallis, Oregon 97331, USA
Abstract
Alternative splicing can enhance transcriptome plasticity and proteome diversity. In plants, alternative splicing can be manifested at different developmental stages, and is frequently associated with specific tissue types or environmental conditions such as abiotic stress. We mapped the Arabidopsis transcriptome at single-base resolution using the Illumina platform for ultrahigh-throughput RNA sequencing (RNA-seq). Deep transcriptome sequencing confirmed a majority of annotated introns and identified thousands of novel alternatively spliced mRNA isoforms. Our analysis suggests that at least ∼42% of intron-containing genes in Arabidopsis are alternatively spliced; this is significantly higher than previous estimates based on cDNA/expressed sequence tag sequencing. Random validation confirmed that novel splice isoforms empirically predicted by RNA-seq can be detected in vivo. Novel introns detected by RNA-seq were substantially enriched in nonconsensus terminal dinucleotide splice signals. Alternative isoforms with premature termination codons (PTCs) comprised the majority of alternatively spliced transcripts. Using an example of an essential circadian clock gene, we show that intron retention can generate relatively abundant PTC+ isoforms and that this specific event is highly conserved among diverse plant species. Alternatively spliced PTC+ isoforms can be potentially targeted for degradation by the nonsense mediated mRNA decay (NMD) surveillance machinery or regulate the level of functional transcripts by the mechanism of regulated unproductive splicing and translation (RUST). We demonstrate that the relative ratios of the PTC+ and reference isoforms for several key regulatory genes can be considerably shifted under abiotic stress treatments. Taken together, our results suggest that like in animals, NMD and RUST may be widespread in plants and may play important roles in regulating gene expression.
Alternative pre-mRNA splicing is an essential mechanism for increasing transcriptome plasticity and proteome diversity in eukaryotes. In some cases, alternatively spliced pre-mRNAs may yield thousands of splice variants for one gene. Alternative splicing (AS) has been studied extensively at the protein and functional level in animal species (for review, see Black and Gravely 2006; Blencowe 2006; Soller 2006). In contrast to the case in animals, studies of the functional significance of AS at the protein level in plants are scarce. Nevertheless, AS is predicted to impact proteome diversity by generating multiple protein isoforms. For example, in Arabidopsis and rice, an estimated 50% and 19% of the alternative first exons, respectively, could alter the N-terminal sequences of the affected proteins (Chen W-H et al. 2007). Another example involves the numerous near-full length proteins encoded by multiple mRNA isoforms of the serine/arginine splicing factors in Arabidopsis (Palusa et al. 2007; for review, see Reddy 2007). Alternative splicing can be spatially and developmentally regulated and is frequently associated with environmental stress (Brett et al. 2002; Palusa et al. 2007; for review, see Reddy 2007; Mazzucotelli et al. 2008). In addition to increasing proteome diversity, alternative splicing plays a role in regulating the level of functional transcripts via a mechanism termed regulated unproductive splicing and translation (RUST) (Lewis et al. 2003; Lareau et al. 2007).
Genome-wide studies of alternative pre-mRNA splicing in a variety of organisms have relied on Sanger sequencing of expressed sequence tags (ESTs) and cDNAs, high-density DNA microarrays (for review, see Mockler et al. 2005) and most recently ultrahigh-throughput RNA sequencing (RNA-seq) approaches (Mortazavi et al. 2008; Pan et al. 2008; Sultan et al. 2008; Wang et al. 2008). Global alternative splicing has been investigated in plants using traditional Sanger EST and cDNA data (Zhu et al. 2003; Iida et al. 2004; Alexandrov et al. 2006; Campbell et al. 2006; Wang and Brendel 2006; Chen F-C et al. 2007; Ner-Gaon et al. 2007), but such data are biased against low-abundance transcripts and toward transcript termini due to the preponderance of end-sequence reads. Several studies using pyrosequencing-based high-throughput sequencing (HTS) for transcriptome analysis have been reported for plants (Cheung et al. 2006; Barbazuk et al. 2007; Emrich et al. 2007; Weber et al. 2007). Estimates have suggested that ∼22%–30% of Arabidopsis intron-containing genes are alternatively spliced (Campbell et al. 2006; Wang and Brendel 2006; Chen F-C et al. 2007; Barbazuk et al. 2008).
Recently developed HTS technologies (for review, see Shendure and Ji 2008) enable a more efficient approach for cataloging alternative splice variants by providing remarkable sequencing throughput. These technologies include sequencing-by-synthesis (SBS) approaches, such as 454 Life Sciences (Roche) pyrosequencing (Margulies et al. 2005; Rothberg and Leamon 2008; http://www.454.com/), Illumina's (formerly Solexa) SBS technology (http://www.illumina.com/), and sequencing-by-ligation (Applied Biosystems SOLiD System; http://www3.appliedbiosystems.com/). HTS offers much higher throughput and lower costs than conventional Sanger sequencing. Illumina-based RNA sequencing (RNA-seq) has been used to study alternative splicing in mammals (Mortazavi et al. 2008; Pan et al. 2008; Sultan et al. 2008; Wang et al. 2008). These RNA-seq studies have exploited the exceptional sequencing depth provided by the Illumina platform to show that up to 95% of human genes (Pan et al. 2008; Wang et al. 2008) are alternatively spliced.
Despite numerous studies, the extent and complexity of alternative splicing in plants is not well characterized. Here, we have used the Illumina RNA-seq approach to catalog constitutive and alternative splicing in the model plant Arabidopsis thaliana under normal physiological conditions and different abiotic stress treatments. In contrast to other methods, RNA-seq provides broad and deep sequencing of the transcriptome at single-base resolution, allowing accurate empirical determination of splice junctions and alternative splicing events with low false discovery rates. Our data provide an unprecedented and unbiased evaluation of alternative splicing in Arabidopsis, the premier model plant. RNA-seq confirmed most annotated introns and known splice variants and identified thousands of novel alternatively spliced mRNA isoforms. We predict that at least ∼42% of intron-containing Arabidopsis genes are alternatively spliced. This estimate is significantly higher than previous estimates based on cDNA/EST sequencing. Surprisingly, a substantial proportion of novel introns detected by RNA-seq carried nonconsensus terminal dinucleotide splice signals. Our analyses suggest that a high percentage of alternatively spliced mRNA isoforms contain premature termination codons (PTCs). Many of these transcripts are likely to be targeted by nonsense mediated mRNA decay (NMD) surveillance pathway (for review, see Chang et al. 2007) or could be regulated by unproductive splicing and translation (RUST) mechanism, as proposed for human SR spliceosomal proteins (Lareau et al. 2007). We show that for some essential regulatory genes, the relative abundance of unproductive isoforms can be regulated by abiotic stress. Thus, RUST may play a significant role in regulating gene expression in plants. Our survey confirms that the repertoire of alternative splicing in plants and animals is different and that the complexity and extent of alternative splicing in plants has been significantly underestimated.
Results
Mapping of the Arabidopsis transcriptome
To achieve a nonbiased and complete analysis of the Arabidopsis transcriptome, we utilized two approaches: cDNA libraries were prepared using either oligo(dT) or random priming methods (Fig. 1A). In the first approach we used poly(A)+ RNA and oligo(dT) primed reverse transcription (RT) to generate full-length enriched double-stranded cDNA (Zhu et al. 2001). In the second protocol, we used highly purified [at least two consecutive cycles of purification on an oligo(dT) affinity column] poly(A)+ RNA template that was essentially free of nonpolyadenylated RNA and random hexamer primed RT. The RNA-seq data generated using both methods represented Arabidopsis tissues at different developmental stages and time points of the diurnal cycle as described in Methods. For each abiotic stress treatment condition RNA-seq libraries were prepared and sequenced individually. The double-stranded RNA-seq cDNA libraries from both protocols were prepared as previously described (Fox et al. 2009) and subjected to HTS using the Illumina 1G platform (for review, see Quail et al. 2008; Shendure and Ji 2008). Three technical replicates (i.e., cDNA samples sequenced on different Illumina flow cells) for the full-length enriched oligo(dT) primed cDNA experiment generated 28.6, 17.4, and 20.6 million trimmed 32-mer Illumina reads, respectively. Three technical replicates for the random-primed cDNA experiment generated 51.2, 90.4, and 69.3 million trimmed 32-mer Illumina reads, respectively. Technical replicates for the two approaches were in close agreement (Supplemental Fig. 1). Pooled data from the two sequencing approaches yielded ∼271 million trimmed and filtered 32-mer reads. These microreads were aligned to perfectly matching locations in the reference genome using the HashMatch tool (HD Priest, DW Bryant, SA Givan, CM Sullivan, and TC Mockler, in prep.; http://mocklerlab-tools.cgrb.oregonstate.edu/). Approximately 84 million reads perfectly matched the Arabidopsis genome or matched annotated splice junctions (The Arabidopsis Information Resource, TAIR 8 database release; http://www.arabidopsis.org). All Illumina RNA-seq data can be searched and visualized in a genome viewer at http://athal.cgrb.oregonstate.edu/ or downloaded from the NCBI Short Read Archive (accession no. SRA009031).
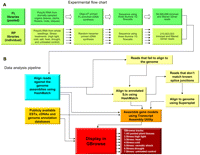
Flow of experiments and data analysis. (A) Design of Arabidopsis RNA-seq experiments and methods of preparation of cDNA libraries for HTS. FL, full length enriched oligo(dT) primed cDNA libraries; RP, randomly primed cDNA libraries. (B) Computational pipeline for HTS data analyses.
Plotting the alignments of unique single-copy microread matches along the five Arabidopsis chromosomes revealed extensive transcriptional activity in the genome (Fig. 2). The chromosome coverage profiles were similar for the full-length and random-primer-generated cDNA libraries (Supplemental Fig. 2), thus we conclude that either approach is suitable for a broad survey of the transcriptome. We compared the chromosomal profiles of transcript-derived RNA-seq coverage with the cytosine methylation distribution (Zilberman et al. 2007; data set GEO GSE5974). As expected, RNA-seq reads matching multiple locations in the genome mapped predominantly to repeat-rich chromosomal regions consistent with an association of repetitive elements with intensively methylated regions such as centromeres and heterochromatic knobs (Supplemental Fig. 3).
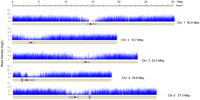
Transcription profile of the A. thaliana genome. Distribution of RNA-seq microread density along chromosome length is shown. Each vertical blue bar represents log2 of the frequency of unique single-copy cDNA-derived microreads plotted against chromosome coordinates. A schematic drawing of the chromosome and its features is shown below the microread density. Approximate boundaries of Arabidopsis centromeres (Kotani et al. 1999; The Arabidopsis Genome Initiative 2000; Tabata et al. 2000; Kumekawa et al. 2001; Copenhaver 2003) are depicted in gray. Red circles indicate unsequenced centromeric gaps. Heterochromatic knobs are denoted by violet ellipses. Chromosome portions corresponding to the telomeres and nucleolar organizing regions are not shown.
Comparison of the RNA-seq data to the annotated Arabidopsis genome (version TAIR8; http://www.arabidopsis.org) revealed that ∼95% of the reads matching the genome mapped to annotated genic regions, whereas only ∼5% mapped to intergenic regions. This is consistent with the exceptional quality of the Arabidopsis genome assemblies and annotation (Fig. 3A). The depth of read matches to intergenic regions and annotated gene features, such as introns, coding sequence (CDS), splice junctions (SJs), 5′- and 3′-untranslated regions (UTRs) by perfect matching microreads is illustrated in Figure 3B. As expected, the depth of coverage of intergenic regions and introns was lower than that for exonic features. The depth of coverage along the length of transcripts decreased toward the 5′ termini for RNA-seq data derived from the full-length enriched (FL) cDNA libraries (Fig. 3C), presumably reflecting the 3′ bias introduced during oligo(dT)-primed reverse transcription. Consistent with this bias, we identified RNA-seq reads matching 93.4% of annotated 3′ UTRs in the FL data as compared to 72.9% of annotated 5′ UTRs (data not shown). In contrast, coverage by the randomly primed library was more evenly distributed (Fig. 3C). The sharp decrease in coverage depth at the termini of transcripts is an artifact of our matching standard, which required 32-mer reads to perfectly match a target sequence over the entire length of the microread. Analysis of the RNA-seq coverage over the lengths of all TAIR8 annotated cDNAs (Fig. 3D) demonstrated that the coverage profiles were similar for the two types of libraries, and overall 50% of TAIR8 annotated cDNAs had at least 93.5% of their sequence lengths represented by Illumina RNA-seq reads.
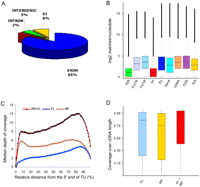
Depth and coverage of annotated gene features. (A) Distribution of the RNA-seq microreads along annotated Arabidopsis annotated genomic features. Among reads perfectly matching the Arabidopsis genome, there were 71.3 million matches to annotated exons, 6.7 million matches to annotated splice junctions, 4.4 million matches to annotated intergenic regions, and 1.4 million matches to annotated introns. Of the remaining 187 million reads, ∼20% matched the Arabidopsis genome allowing for up to two mismatches and the remaining ∼50% aligned with more than two mismatches or did not match at all. (B) Box-and-whisker plots of log2-transformed numbers of microread matches at each nucleotide position for TAIR annotated intergenic regions (IGR), 5′ untranslated regions (5′UTR), 3′ untranslated regions (3′UTR), introns (Int), exons (Ex), genes (Gene), cDNAs (cDNA), coding sequences (CDS), and splice junctions (SJs). The bottom and top of the box represent the 25th and 75th quartiles, respectively, and the middle line is the median. Black filled circles show outliers. (C) Distribution of the RNA-seq microread coverage along the length of the transcriptional unit. The median depth of coverage along the length of each individual cDNA was calculated as described in the Supplemental Material and plotted against the relative length of the transcriptional unit (cDNA) for full-length enriched oligo(dT)-primed libraries (blue diamonds) and randomly primed libraries (yellow circles). The combined data from the two libraries are depicted by red triangles. (D) Coverage over the length of TAIR8 annotated cDNAs. Perfect match 32-mer Illumina reads were mapped to the TAIR8 annotated cDNAs for nuclear genes using HashMatch (http://mocklerlab-tools.cgrb.oregonstate.edu/). Illumina read coverage along the predicted sequence features was calculated using a Perl script. Box-and-whisker plots depict the Illumina coverage calculated as the percentage of bases along the length of the cDNA sequence that was supported by Illumina reads from the FL, RP, and combined FL + RP data sets. The bottom and top of the box represent the 25th and 75th quartiles, respectively. The black line is the median and the red diamonds are the mean.
Prediction and validation of alternative splicing events
A prerequisite for a comprehensive survey of alternative splicing is the ability to reliably detect expressed exons and splice junctions. Overall we detected 92.6% and 80.1% of annotated exons and splice junctions, respectively; with 87.5% of exons and 76.7% of splice junctions detected in both the full-length and randomly primed libraries (Fig. 4A). Next, we assessed our ability to detect known alternative splicing events. We mined our RNA-seq data to identify reads that specifically detected previously known splice variants included in the TAIR annotation (Fig. 4A; Supplemental Fig. 4). We detected 79.3% of annotated alternative exons, 69.2% of annotated alternative splice junctions, and 72.5% of annotated alternative introns. Collectively our results validated 86.3% of previously known splice variants in Arabidopsis.
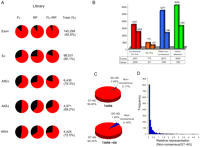
Survey of constitutive and alternative splicing in Arabidopsis. (A) Detection of annotated gene features and alternative splicing events by RNAseq. Annotated gene features (Exon, SJ) and alternative splicing events, including alternative splicing at both acceptor and donor splice sites (AltEx), an alternative splice junction (AltSJ) and alternative intronic sequences (AltInt) were identified by aligning RNA-seq microreads as described in the Supplemental material. Pie charts depict the proportions of the annotated features in full-length (FL), randomly primed (RP), and combined (FL + RP) cDNA libraries detected by at least one perfect match RNA-seq read. Total (%) indicates the total number and percentage of annotated features detected in the combined (FL + RP) data. (B) Distribution of novel splicing events among annotated genes. The histogram depicts the numbers of novel alternative splicing events and alternatively spliced genes containing consensus GT-AG and other nonconsensus intron splice signal dinucleotides and introns retention events within TAIR8-annotated genes. (C) Pie charts depict the proportions of consensus and nonconsensus intron terminal dinucleotide classes among annotated TAIR8 introns (TAIR8; upper panel) and the combined TAIR8 + supersplat-predicted introns (TAIR + SS; lower panel). (D) The histogram depicts the relative representation of consensus and nonconsensus splice junctions as a frequency distribution. The relative representation was calculated (the average number of reads spanning nonconsensus splice junctions/the average number of reads spanning constitutive consensus splice junctions in the same gene) for 1539 genes that contain both consensus and nonconsensus introns.
We used supersplat to search for novel introns (DW Bryant, R Shen, HD Priest, W-K Wong, and TC Mockler, in prep.). The potential novel supersplat-predicted introns were filtered using a series of ad hoc selection criteria as described in the Methods. In brief, supersplat-predicted splice junctions were retained if supported by at least two distinct independent Illumina reads, each originating from a different biological sample, and aligning within an annotated (TAIR8) genic region, with different overlap lengths (a minimum overlap length of six bases) on the flanking exons that were themselves supported by additional RNA-seq reads. This analysis identified 3307 novel consensus GT-AG splice junctions occurring in 2299 genes (Fig. 4B), including alternative splicing events of different types: Alternative donor and/or acceptor splice sites, novel introns, novel terminal 5′ and 3′ exons (Supplemental Fig. 4). In addition, 775 novel introns contained weak nonconsensus 5′ terminal GC donor site splice signals (Fig. 4C). Together with previously annotated GC-AG introns these newly identified SJs increased the genome-wide proportion of introns with GC-AG signal dinucleotides ∼1.6-fold. Among these novel introns we identified a small proportion of conserved splice signals typical for minor U12 type introns (Alioto 2006; Supplemental Fig. 5; Supplemental Table 1).
We also used supersplat to search for novel introns containing rare nonconsensus terminal dinucleotide pairs. This genome-wide analysis predicted the existence of 5273 additional introns with nonconsensus splice signals; these introns were found in 2146 genes (Fig. 4B). Although this absolute number represents a large increase in the number of introns with nonconsensus terminal dinucleotide signals as compared to previous estimates, it is important to note that these nonconsensus introns represent a relatively small fraction (∼4%) of all introns in Arabidopsis (Fig. 4C). To assess the relative abundance of these novel nonconsensus introns, we compared the average number of RNA-seq reads spanning nonconsensus splice junctions to the average number of reads spanning constitutive consensus splice junctions in the same gene (Fig. 4D). This analysis demonstrated that the nonconsensus splice junctions typically represent low abundance splicing events with >88% of nonconsensus splice junctions being supported by fewer reads than the consensus splice junctions in the same gene. We also mined the RNA-seq data to identify retained introns and novel exons within annotated introns. In addition to confirming 72.5% (Fig. 4A) of annotated intron retention events, this search identified 6000 novel events within the introns of 3120 genes (Fig. 4B). These results are consistent with previous studies suggesting that intron retention is the most prevalent form of alternative splicing in plants (Wang and Brendel 2006).
To construct empirical transcription unit assemblies using our RNA-seq data and the publicly available cDNA/EST data, we used the Transcription-unit Assembly Utility (TAU; HD Priest, DW Bryant, SA Givan, CM Sullivan, and TC Mockler, in prep.; http://mocklerlab-tools.cgrb.oregonstate.edu) that rapidly and accurately assembles transcript data into alternatively spliced (when applicable) transcript models. The reference guided assembly of the RNA-seq and public EST/cDNA data sets using the TAU algorithm produced a total of 89,994 transcript models mapping to 31,703 loci at an arbitrary minimal gene length cutoff value of 200 nucleotides (nt). A percentage (70.3%) of gene models (22,302) contained at least one predicted intron. The TAU models, each of which represents one possible mRNA isoform, served as an input for our estimates of the extent of alternative splicing in Arabidopsis. When combined with the previously annotated (TAIR8) alternative splicing events not included in the supersplat/TAU predictions, alternatively spliced genes comprised 41.9% (9273 genes) of spliced nuclear genes (excluding single exon models). This is a conservative estimate because it omits several thousand potential intron retention events (Fig. 4B) that are excluded from the TAU transcript assemblies in cases where the RNA-seq data does not completely cover every base of an intron sequence. Including all potential intron retention events detected in the RNA-seq data suggests that up to 56% of intron-containing Arabidopsis genes could be alternatively spliced. Collectively, 59,774 out of 76,699 (77.9%) of the alternatively spliced TAU models introduced in-frame PTCs. These PTCs resided more than 55 nt upstream of an exon/exon junction, thus indicating that the corresponding transcripts could be potential candidates for NMD (Nagy and Maquat 1998; Maquat 2004; Hori and Watanabe 2007).
To validate novel alternative splicing events predicted by the RNA-seq data, we used RT-PCR, quantitative real-time RT-PCR (qRT-PCR), and Sanger sequencing. We randomly selected 168 splice junctions predicted from the RNA-seq data, including 91 consensus GT-AG and 77 nonconsensus introns representing 12 different intron terminal dinucleotide combinations (Supplemental Tables 2, 3). These validation experiments confirmed 95% and 88% (on average among all tested dinucleotide groups) of the tested events with consensus and nonconsensus splice signals, respectively (Supplemental Table 3). We were not able to distinguish whether the relatively small proportion of unconfirmed events represented false supersplat discoveries or were the result of suboptimal primer design. The validated alternative splicing events represented all major classes, including intron retention (IntronR), exon skipping, alternative donor and/or acceptor splice site selection, introns overlapping a constitutive intron, but differing in both donor and acceptor site positions, and novel terminal exons (summarized in Supplemental Fig. 7; Supplemental Tables 2, 3).
Differences in the RNA-seq coverage of specific mRNA isoforms were observed under various abiotic stress conditions, presumably reflecting regulation of these alternative splicing events. To identify differentially expressed gene features (e.g., exons, introns, splice junctions) we analyzed data sets corresponding to individual stress treatments. We used database queries to identify the numbers of reads normalized for overall gene expression levels matching particular TAIR8 annotated gene features. The resulting normalized and featurized RNA-seq read counts were analyzed using the “stats” package in R and “complete linkage” clustering, also known as the maximum or furthest-neighbor method. This method calculates the distance between clusters, which is defined as the greatest distance between a member in one cluster and a member in the other cluster. This clustering analysis revealed several groups of Arabidopsis exons, introns, and SJs that were coordinately expressed under various abiotic stress conditions (Fig. 5A). A closer examination of individual events under specific abiotic stress conditions confirmed that certain alternative splicing events are stress associated. Illustrative examples including cold-induced intron retention in the OUTER ENVELOPE PROTEIN 16 transcript (Fig. 5B), stress-regulated exon skipping and cassette exon events in the ACCLIMATION OF PHOTOSYNTHESIS TO ENVIRONMENT 2 transcript (Fig. 5C) and, novel introns in transcripts of the splicing factor AT1G02840 (SRP34) (Fig. 5D), are shown in Figure 5. An example of stress modulation of splicing of a C2H2-type zinc finger protein is shown in Supplemental Figure 9. Other examples of stress associated intron retention are provided in Supplemental Figure. 8.
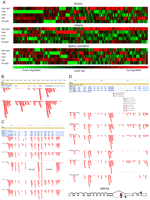
Identification of stress-associated alternative splicing. (A) Exons, introns, and splice junctions were identified by the changes in expression levels (i.e., by the normalized number of the RNA-seq microreads encompassing each feature) under different abiotic stress conditions relative to untreated control. The “Exons” panel represents 807 differentially expressed exons; change in expression level ranged from −30- to +82-fold normalized to untreated control. The “Introns” panel represents 1230 differentially expressed introns with expression changes ranging from −54- to +263-fold. The “Splice Junctions” panel features 1093 exon–exon splice junctions (with changes in normalized expression from −22- to +46-fold). Gene clusters were computed by the default settings of heatmap.2 in the R “gplots” package as described in the Methods. Up- and down-regulated features are shown in red and green, respectively; black corresponds to no change relative to the untreated control. (B) Cold-induced intron retention (bracketed) in the OUTER ENVELOPE PROTEIN 16 (AT2G28900) transcript. Changes in microread density coverage are indicated by a horizontal bracket. (C) Stress-regulated exon skipping (brackets) and cassette exon (arrow) events in the ACCLIMATION OF PHOTOTSYNTHESIS TO ENVIRONMENT 2 (AT5G46110) transcript. (D) Detection and validation of novel SJs in transcripts of splicing factor SRP34 (AT1G02840). SJs corresponding to the untreated control, high light, heat, and dehydration treatments are shown in gray, yellow, red, and brown, respectively. Position of alternatively spliced intron 10 is bracketed. A previously undetected splice isoform containing a poison cassette exon (red rectangle) is illustrated in the bottom panel. Locations of reference and premature termination codons are indicated by red (top) and black (bottom) stars, respectively.
An analysis of CIRCADIAN CLOCK ASSOCIATED 1 (CCA1), a MYB-related transcription factor that functions in the circadian clock of Arabidopsis, provides an example of an evolutionarily conserved IntronR event in an essential plant regulatory gene (Fig. 6). The TAU-generated A. thaliana CCA1 mRNA models predicted two distinct alternative splicing events yielding two CCA1 splice isoforms: One retaining a full length intron 4, another containing retained intron 4 with spliced nested intron 4a (Fig. 6A; Supplemental Fig. 6). Both splicing events result in unproductive CCA1 isoforms with premature termination codons (Fig. 7B; Supplemental Figure 6B) that could be potential targets for nonsense mediated mRNA decay (for review, see Maquat 2004; Isken and Maquat 2008). Sanger sequencing and the depth of coverage of the intron 4a SJ by RNA-seq reads confirmed the structures of both transcripts (Supplemental Fig. 6C,D). Dense microread coverage of the reference intron 4 in the CCA1 transcript contrasted with the low coverage of other introns, suggesting that intron 4 may be retained in some portion of mature CCA1 transcripts (Fig. 6C). We used IntronR4-specific and exon-flanking primers to show that a significant proportion of the mature CCA1 transcripts indeed retain intron 4 (Supplemental Fig. 6A). The gene structure and primer design strategy are illustrated in Supplemental Figure 6B. Sanger sequencing of individual CCA1 isoforms (Supplemental Fig. 6C) confirmed the IntronR4-containing transcript models generated by TAU (Fig. 6A). We then evaluated the levels of Arabidopsis CCA1 transcript variants under different environmental stresses using qRT-PCR and isoform-specific primers. Accumulation of the IntronR4-containing transcripts increased approximately sixfold under a high light treatment (Fig. 6D). The drastic increase of this PTC+ isoform was accompanied by a moderate increase in the level of the mRNA encoding the full-length isoform. In contrast, cold treatment stimulated a fivefold increase in levels of the reference isoform and a drastic decrease in the level of the intron 4-containing mRNA (Fig. 6D). These data indicate that different types of stress can differentially regulate CCA1 isoform levels. To determine whether the retention of intron 4 in CCA1 is conserved among other phylogenetically diverse plant species, we also investigated the splicing of the CCA1-like genes of the monocot grasses Brachypodium and Oryza, and the dicot tree Populus. The intron retention event was readily detectable in Brachypodium distachyon; a comparison of the gene structures and the microread data for Arabidopsis and Brachypodium is shown in Figure 6C. Analysis of the CCA1 homolog mRNAs using isoform-specific RT-PCR confirmed that this intron retention event is conserved among the four plant species tested (Fig. 6E).

Intron retention and novel splice junction events in the CCA1 locus. (A) Empirical CCA1 gene models (orange) generated by the TAU tool using RNA-seq data. (B) Predicted polypeptides are shown schematically with the DNA binding MYB domain shown by a red box. (C) Gene models of homologous CCA1/LHY loci in A. thaliana, Oryza sativa, Brachypodium distachyon, and Populus trichocarpa. cDNA microread coverage is shown for Arabidopsis and Brachypodium. SJs of intron 4 and 4a splicing in Arabidopsis and Brachypodium are marked by brown broken lines. (D) Quantification of the IntronR4 event by qRT-PCR under different abiotic stress conditions. Lanes labeled Hi Light, Heat, Cold, Salt, and Drought correspond to high light, heat, cold, salt, and dehydration treatments, respectively. Relative expression was estimated using −ΔΔCt method (Livak and Schmittgen 2001) and EF-1-ALPHA mRNA as an internal housekeeping gene control. (E) RT-PCR confirmation of CCA1 IntronR4 in rice, poplar, and Brachypodium. IntronR4-specific primers were designed as described for A. thaliana (as shown in panel B). RT-PCR products corresponding to the retained intron 4 (if downstream intron 5 is spliced) are denoted by an asterisk (*); pre-mRNAs are indicated by a dash (–). Sanger sequencing of gel-purified amplified DNA fragments confirmed the sequence of all RT-PCR products. The predicted fragment sizes are 492, 573, and 782 bp for rice (Os, Oryza sativa, ssp. Japonica, locus ID: LOC_Os08g06110), Brachypodium (Bd, Brachypodium distachyon, locus ID: Bradi3g16510), and poplar (Pt, Populus trichocarpa, locus ID: estExt_Genewise1_v1.C_LG_XIV1950), respectively.
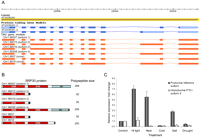
Stress-regulated alternative splicing of Arabidopsis splicing modulator ATSRP30. (A) Known isoforms (blue) and TAU-predicted variants (orange) of the ATSRP30 gene. (B) Predicted ATSRP30 protein domain structures (RRM, RNA recognition motif; SR, serine/arginine rich domain) and polypeptide sizes. (C) Quantification of accumulation of full-length ATSRP30 (reference isoform 1) and a PTC-containing isoform (4) by qRT-PCR under various abiotic stresses. Note the significant shift in the relative isoform ratio under high light, heat, and salt treatments. Relative expression levels were calculated using −ΔΔCt method (Livak and Schmittgen 2001) and EF-1-ALPHA mRNA as an internal reference control.
Alternative splicing of pre-mRNAs of serine/arginine splicing factors
Serine/arginine (SR) splicing factors are essential components of the spliceosome and regulate both constitutive and alternative splicing in plants (for review. see Reddy 2007; Barta et al. 2008). Pre-mRNAs of SR proteins are themselves alternatively spliced and this splicing is under tight spatial, temporal, and developmental control (Lopato et al. 1999; Lorkovic et al. 2000; Kalyna et al. 2003; Isshiki et al. 2006; Kalyna et al. 2006; Ali et al. 2007; Palusa et al. 2007; Tanabe et al. 2007). SR proteins may control splicing of their own transcripts both in mammals (Lejeune et al. 2001; Jumaa and Nielsen 1997) and plants (Lopato et al. 1999; Kalyna et al. 2003; Ali et al. 2007; Reddy 2007). Splicing events that lead to accumulation of PTC+ isoforms has been observed for mammalian SR genes; these isoforms are thought to sustain a negative feedback loop to regulate SR protein production by coupling alternative splicing and nonsense mediated decay (Lareau et al. 2007). Mutations in plant SR proteins alter relative levels of both SR splice variants and other pre-mRNAs (Kalyna et al. 2003, 2006; Ali et al. 2007; Reddy 2007).
Analysis of the Arabidopsis SR gene family by RNA-seq detected a total of 133 SJs representing 122 previously annotated and 13 novel introns (Supplemental Table 4). Twelve out of the 13 novel introns detected in our RNA-seq data were validated in vivo by RT-PCR (Supplemental Table 4). Roughly one-third of the predicted alternative splicing events in the Arabidopsis SR genes result in apparently aberrant transcripts. Most of these aberrant mRNAs are potential targets for NMD because of the position of an introduced PTC relative to an exon–exon junction (for review, see Maquat 2004; Chang et al. 2007; Isken and Maquat 2008). To assess the extent of accumulation of PTC+ transcripts and the association between unproductive transcript isoforms and abiotic stress, we examined the plant homologs AT1G09140 (ATSRP30) and SRP34/SR1 (Lopato et al. 1999) of human splicing factor SFRS1 (also known as SF2/ASF). The Arabidopsis ATSRP30 and SRP34 genes were associated with a particularly large number of alternatively spliced transcript models (Figs. 5D, 7; Supplemental Fig. 10). Of the ATSRP30 transcript models predicted by TAU (Fig. 7A), at least four correspond to previously confirmed splice variants (Lopato et al. 1999; Palusa et al. 2007). The reference isoform 1 of ATSRP30 encodes the full-length protein (Fig. 7B). Use of an alternative donor splice site in the tenth intron of isoform 4 would introduce a PTC located more than 55 nt upstream of an exon–exon junction and therefore, this mRNA may be a target for down-regulation by NMD (Nagy and Maquat 1998; Maquat 2004). We experimentally validated several novel SJs predicted in ATSRP30 and SRP34 transcripts using isoform-specific qRT-PCR. The relative levels of the full-length ATSRP30 isoform increased sevenfold and fivefold under high light and heat stress treatments, respectively (Fig. 7C; Supplemental Fig. 10D). In contrast, the relative abundance of unproductive isoform 4 decreased under heat stress and remained unchanged under light stress, demonstrating that the relative levels of isoforms significantly shift in a stress-dependent manner. RNA-seq data suggested at least two alternative splicing events within the tenth intron of the SRP34 gene (Fig. 5D) consistent with a “poison cassette exon” (Lareau et al. 2007) in isoform 2. Analysis using isoform-specific primers (Supplemental Fig. 10C) confirmed the existence of this isoform in vivo (Supplemental Fig. 10E). Poison cassette exons occur frequently in mammalian SR transcripts (however, not in the human ASF/SF2 homolog of SRP34/ATSRP30) and introduce an early in-frame stop codon (Lareau et al. 2007). We also identified an alternative donor site that led to the 5′ extension of the downstream exon, producing PTC+ isoform 3. SJ-specific primers confirmed that this SRP34 PTC+ isoform also occurs in vivo (Supplemental Fig. 10E).
Discussion
Deep sequencing of the Arabidopsis transcriptome using the Illumina RNA-seq approach revealed that alternative splicing in plants is considerably more extensive and more complex than previously predicted. Previous cDNA/EST assessments estimated that from 20% (Wang and Brendel 2006; Chen F-C et al. 2007; Barbazuk et al. 2008) to 30% (Campbell et al. 2006) of Arabidopsis genes are alternatively spliced. Our empirical RNA-seq analysis combined with previously annotated (TAIR8) alternative splicing indicates that at least 42% of intron-containing genes in Arabidopsis are subject to alternative splicing. Among novel introns, our analyses reveal a surprisingly high number of introns with rare nonconsensus terminal dinucleotide splice signals. As expected, the consensus GT-AG introns constitute the highest proportion of intron terminal dinucleotide signals among detected AS events, and GC-AG introns represent the second largest class. Together with the TAIR-annotated introns, empirically predicted novel GC donor splice sites increased genome-wide proportion of GC-AG introns ∼1.6-fold. This finding supports a notion that alternatively spliced genes in Arabidopsis (Campbell et al. 2006), mammals (Thanaraj and Clark 2001), and worms (Farrer et al. 2002) are enriched in weak 5′ terminal GC splice signals. Accumulation of GT to GC conversion events might have been evolutionary driven by selection for alternative splicing (Churbanov et al. 2008). The previous underestimation of nonconsensus introns likely reflects biases in favor of GT-AG and to a lesser extent GC-AG introns among most ab initio gene prediction algorithms and the commonly used tools for alignment of EST/cDNA sequences to genomic sequences. Experimental validation of selected putative novel splicing events suggests that many of these unusual splicing events indeed occur in vivo. Our survey also underscores the superior detection capability of the RNA-seq method over the conventional cDNA/EST approach, especially for low abundance transcript variants. The Illumina RNA-seq approach for the study of alternative splicing will be of even greater utility with recent technological advances including longer reads, lower sequencing error rates, and increased throughputs.
Our estimates based on transcription unit modeling using the TAU algorithm suggest that a high proportion of alternatively spliced transcript isoforms harbor in-frame PTCs. We found that ∼78% of alternatively spliced TAU models contained at least one PTC located 55 nt or more upstream of an exon/exon junction. Such mRNAs generally elicit an NMD response in animals (for review, see Maquat 2004; Chang et al. 2007) and plants (Hori and Watanabe 2007). We acknowledge that the number of PTC+ TAU models could be overestimated in our analysis because the TAU algorithm predicts all possible combinations of nonmutually exclusive alternative splicing events affecting the same gene. However, our estimate is in general agreement with cDNA/EST-supported predictions of a high proportion of in-frame PTC+ transcripts introduced by alternative splicing in Arabidopsis and rice (Campbell et al. 2006; Wang and Brendel 2006) and in humans (Lewis et al. 2003; Saltzman et al. 2008). The coupling of alternative splicing with nonsense-mediated decay appears to be widespread among eukaryotes (for review, see Muhlemann et al. 2008). In recent years it has become increasingly clear that alternative splicing, in addition to generating proteome diversity, plays a role in regulating the level of functional transcripts by regulated unproductive splicing and translation (RUST) (Lewis et al. 2003; Lareau et al. 2007). It has been estimated that up to 72% of alternative splicing events in human spliceosomal factors introduce in-frame PTCs (Saltzman et al. 2008), while more than one-third of PTC+ splicing isoforms could be targeted for NMD (Lewis et al. 2003). Lareau et al. (2007) proposed that RUST regulates nonsense-mediated decay of human PTC+ SR transcripts. Our survey suggests that the majority of all alternatively spliced transcript isoforms in Arabidopsis contain at least one PTC at a distance greater than 55 nt upstream of an exon/exon junction and therefore, could be potentially detected as aberrant by the NMD surveillance machinery. The debate is ongoing with regard to how many of these aberrant PTC+ isoforms are functional and may be involved in homeostatic regulation through the RUST mechanism and how many may represent stochastic noise of the splicing machinery (Pan et al. 2006). Studies of AS in Arabidopsis mutants deficient in the key components of the NMD surveillance mechanism may directly address these questions. For example, a recent study using tiling arrays showed that expression of many coding and noncoding transcripts was up-regulated in NMD-impaired Arabidopsis up-frameshift (upf) mutants (Kurihara et al. 2009). Deep transcriptome sequencing of upf mutants, such as upf3 (Hori and Watanabe 2005) would provide a detailed catalog of NMD-targeted aberrant transcripts.
Among the different classes of alternative splicing events in Arabidopsis, intron retention was most prevalent and was frequently associated with specific abiotic stresses. Since most of the intron retention events in Arabidopsis result in PTC+ isoforms, it is possible that intron retention, as well as other nonsense-generating splicing events, may play a significant role in RUST-regulated gene expression in plants. This hypothesis is in agreement with our observation of highly conserved intron retention events detected in essential regulatory genes. Despite low sequence homology in the retained intron, an example of such an event is the conserved IntronR4 in the CCA1/LHY mRNAs of Arabidopsis, Populus, Oryza, and Brachypodium; mono- and dicotyledonous species that diverged from a common ancestor ∼120–170 million years ago (Lynch and Conery 2000; Tuskan et al. 2006). This intron retention event resulted in accumulation of high levels of PTC+ transcripts under specific abiotic stress conditions. Other specific stress treatments also led to a dramatic shift in the relative ratio of the full-length versus the PTC+ CCA1 transcript variants, suggesting that splicing of this pre-mRNA may be tightly regulated by stress. Similar to CCA1, A. thaliana circadian clock-associated protein AtGRP7 is subjected to alternative splicing yielding PTC+ transcripts. AtGRP7 binds its own pre-mRNA and regulates its abundance by production of PTC+ transcripts via a negative autoregulatory feedback loop (Schoning et al. 2007). Concomitantly, NMD-impaired mutants accumulate high levels of the PTC+ AtGRP7 transcripts. In another example, Song et al. (2009) showed that alternatively spliced PTC+ transcripts of another clock-regulated gene, SUPPRESSOR OF OVEREXPRESSION OF CO1 (SOC1), may play an important regulatory role. The authors demonstrated that aberrant SOC1 transcripts with a retained sixth intron are direct binding targets of the EARLY FLOWERING9 (ELF9) protein and ELF9 possibly regulates SOC1 expression through an NMD pathway. Interestingly, our RNA-seq-based transcriptional unit models also suggest that SOC1 transcripts retain the sixth intron and that this IntronR event may be associated with specific abiotic stress conditions (Supplemental Fig. 11). Our RNA-seq analysis of the Arabidopsis transcriptome suggests numerous instances of other genes with specific splicing events that may be linked to individual stress treatment(s) (Supplemental Figs. 9, 10; data not shown).
A close examination of transcripts of SR splicing factors by RNA-seq validated a majority of the previously predicted ATSRP30 isoforms (Lopato et al. 1999; Palusa et al. 2007) and revealed several novel splice variants. By quantifying individual splice forms represented in the RNA-seq data, we demonstrated that the levels of ATSRP30 isoforms shifted dramatically under various abiotic stress conditions. Because ATSRP30 alters splicing of its own pre-mRNA (Lopato et al. 1999; Kalyna et al. 2003) and regulates alternative splicing of other plant pre-mRNAs (Lopato et al. 1999; Ali and Reddy 2006; Kalyna et al. 2006), this shift may be responsible for adaptive changes in the plant transcriptome due to stress.
In summary, this genome-wide analysis of alternative splicing in Arabidopsis reveals a high degree of splicing plasticity, including under different abiotic stress conditions. It also suggests the possibility of widespread usage of a regulatory splicing mechanism similar to RUST in animals. Future studies are needed to elucidate the detailed molecular mechanisms underlying the network of splicing factors and their transcript targets, as well as the functional consequences of individual alternative splicing events.
Methods
Plant materials, growth conditions, and tissue collection
Arabidopsis thaliana (accession Col-0) was used in all experiments. Three-wk-old Col-0 plants were grown at 22°C in long days (16:8 h day/night; at light intensity 200 μmol m−2 sec−1). Rosette leaves, inflorescences, siliques, and roots were collected at time points 0 (dawn), 4, 8, 12, 16, (dusk), 20, and 24 h. Tissues were collected at each time point, flash-frozen in liquid nitrogen and ground to a fine powder using a Retsch Mixer Mill 301. For RNA isolation pulverized tissues from each time point were pooled together in equal proportions.
Stress treatments
Abiotic stresses included high light, heat, cold, salt, and dehydration treatments. Arabidopsis seeds were treated for 10 min in 70% ethanol followed by sterilization in 50% bleach/0.1% Tween 20 and rinsed three times in sterile water. Sterilized seeds were plated on 15-cm diameter Petri dishes containing 20 mL of Murashige-Skoog (MS) agar (Murashige and Skoog 1962) supplemented with 1.5% sucrose. Prior to germination, seeds were incubated for 4 d in the dark at 4°C. Seedlings were grown on MS agar plates at 21°C in long- ays (16:8 h day/night; light intensity of 130 μmol m−2 sec−1). Twelve-d-old seedlings were stress-treated under various conditions. All treatments were initiated in the middle of the light cycle and continued for 24 h. Tissues (whole seedlings, including roots) were collected 1, 2, 5, 10, and 24 h post-stress induction. Approximately 70 seedlings were collected at each time point for each treatment condition, instantly flash-frozen, and pooled together in equal proportions. For the heat and cold stress conditions seedlings were grown at 42°C and 4°C, respectively. For salt stress, 20 mL of 0.5 M sodium chloride was added to each Petri dish with the seedlings. Drought was simulated by dehydration treatment with 20 mL of 25% polyethylene glycol (PEG 6000, Sigma-Aldrich). In the high-light condition, plates with seedlings were transferred onto the surface of water at 22°C in a white light-reflective container and exposed to the continuous light at intensity 1000 μmol m−2 sec−1 in a Conviron PGR15 growth chamber. Plants grown under the default conditions as described above were used as an untreated control.
RNA isolation
Total RNA was isolated using modified protocol previously described (Filichkin et al. 2007). First, RNA was extracted using The Plant RNA Reagent (Invitrogen). RNA was treated for 10 min at 65°C with RNAsecure reagent (Ambion). To eliminate genomic DNA amplification, all RNA preparations were treated for 15 min at 37°C with RNase-free Turbo DNase (Ambion). Next, total RNA was further purified using the RNAeasy Mini RNA kit (Qiagen) according to the manufacturer's RNA clean up protocol. Isolation of mRNA essentially free of ribosomal and other nonpolyadenylated RNAs was critical for generation of nonbiased randomly primed (RP) libraries. For the RP libraries the poly(A) mRNA was isolated by two consequent cycles of purification on oligo(dT) cellulose using the Micro-PolyA-Purist kit (Ambion). Concentration, integrity, and extent of contamination by ribosomal RNA were monitored using ND-1000 spectrophotometer (Thermo Fisher Scientific) and Bioanalyzer 2100 (Agilent Technologies).
Full-length enriched (FL) cDNA libraries
FL cDNA libraries were prepared essentially as described (Fox et al. 2009). Libraries were generated using the SMART method (Zhu et al. 2001). cDNA (5–7 μg in a 50-μL volume) was mixed with 750 μL of Illumina nebulization buffer and fragmented for 7 min in a nebulizer (Invitrogen) using compressed nitrogen at 35 psi. The sheared cDNA was purified using the QIAquick PCR Purification kit (Qiagen) and eluted into 32 μL of water.
Randomly primed (RP) cDNA libraries
For the RP cDNA libraries, the first cDNA strand was synthesized using 1 μg of poly(A) mRNA essentially free of rRNA, random hexamer primers (300 ng per microgram of RNA), and Superscript III reverse transcriptase (RT) (Invitrogen). The second strand of cDNA was synthesized using DNA polymerase I (Klenow fragment) by combining 20 μL of the first strand reaction, 8 μL of 10× Klenow Buffer (NEB), 1 unit of RNase H (Invitrogen), 68.8 μL of water, and 30 units of DNA polymerase I (NEB). The reaction was incubated for 90 min at 15°C and cDNA was purified using the QIAquick PCR clean up kit.
Preparation of cDNA for Illumina 1G genome analyzer
FL or RP cDNA (30 μL) was combined with 10 μL of 10 mM ATP in 5× T4 DNA ligase buffer (Invitrogen), 4 μL of 10 mM dNTPs mix, 2.5 μL of T4 DNA polymerase (3 U/μL), 1 μL of Klenow DNA polymerase (5 U/μL), and 2.5 μL of T4 polynucleotide kinase (10 U/μL, NEB). After incubation for 30 min at 20°C the DNA was purified using QIAquick PCR Purification kits. To add dA to the termini, 32 μL DNA from the prior step was mixed with 5 μL of 10× Klenow buffer, 10 μL of 1 mM dATP, and 3 μL of Klenow exo-polymerase (3′ to 5′ exo minus, 5 U/μL, NEB) and incubated for 30 min at 37°C. DNA was purified using a QIAquick MinElute Reaction Clean-up kit (Qiagen) and eluted into 12 μL of water. To ligate Illumina adapters, 10 μL of cDNA from the prior step was mixed with 5 μL of 5× T4 DNA ligase buffer, 6 μL of adapter oligo mix, 4 μL of T4 DNA ligase (NEB), and incubated for 15 min at 25°C. DNA was purified using a QIAquick MinElute PCR Purification kit. cDNA was size-fractionated (typically with average fragment length of 170 base pairs [bp]) on 3.5% (w/v) NuSieve GTG agarose. The fractionated libraries were PCR amplified using Phusion Hot Start High-Fidelity DNA polymerase (NEB) and the following PCR protocol: 30 sec at 98°C, then 10 sec at 98°C, 30 sec at 65°C, and 30 sec at 72°C for 18 cycles, followed by a 10-min extension step at 72°C. Prior to cluster generation, DNA was diluted to a final concentration of 5–10 pM to generate 25,000 to 40,000 clusters per tile of the flow cell.
Oligonucleotide primer design and RT-PCR amplification of SJs
To eliminate the possibility of amplification of genomic DNA, all RNA preparations were treated with DNase I and “no reverse transcriptase” negative controls were performed for each PCR reaction. The first cDNA strand was synthesized using 1 μg of poly(A) mRNA, and random hexamer primers and Superscript III reverse transcriptase (RT) from the Invitrogen first-strand cDNA synthesis kit according to the manufacturer's protocol. The first-strand cDNA reaction was diluted 10-fold prior to PCR amplification and ∼50 ng of cDNA was used as the PCR template.
Forward SJ-specific oligonucleotide primers were designed to partially overlap predicted SJs with the 3′ terminus of the oligonucleotide spanning into the adjacent exon ∼5–7 nt. The melting temperature of SJ-specific primers was selected in the range of 58–62°C. The reverse primer was designed within the sequences of the closest adjacent exon. SJ-specific and flanking primer designs are depicted in detail in Supplemental Figures 6 and 7. Both SJ-specific and exon-flanking primer pairs were selected using Primer3 (http://primer3.sourceforge.net/). The annealing temperature during PCR amplification was selected to be high enough to prevent potential mispriming by each of SJ-overlapping oligonucleotide segments alone. The PCR amplification was carried out using Phusion polymerase and a “touch down” PCR protocol as follows: 98°C for 10 sec, 68°C for 30 sec, and 72°C for 1 min for seven cycles with annealing temperature decreasing 1°C at each consequent cycle, followed by 24 cycles of 95°C for 10 sec, 60°C for 30 sec, 72°C for 1 min, and a final extension at 72°C for 10 min. The PCR products were separated in 2% agarose (SFR; MidSci) gels and stained with ethidium bromide prior imaging according to standard procedures (Sambrook et al. 1989).
Sanger sequencing
RT-PCR products were sequenced using standard procedures on an Applied Biosystems 3730 capillary sequencer.
Quantitative real-time PCR
Quantitative real time PCR (qRT-PCR) was performed as previously described (Mockler et al. 2004). All qRT-PCR reactions were run on a Bio-Rad CFX96 real-time PCR detection system using SYBRgreen. Expression levels of splice variants relative to untreated control were calculated using the ΔΔCt method (Livak and Schmittgen 2001) and with the housekeeping gene EF-1-ALPHA mRNA as a control.
Bioinformatics resources and tools
The Arabidopsis genome sequence, annotation and annotated sequence features were downloaded from TAIR (TAIR 8 database release; ftp://ftp.arabidopsis.org/home/tair/Sequences/). RNA-seq reads were mapped to the Arabidopsis genome using Illumina's ELAND (AJ Cox, unpubl.), HashMatch (HD Priest, DW Bryant, SA Givan, CM Sullivan, and TC Mockler, in prep.), and supersplat (DW Bryant, R Shen, HD Priest, W-K Wong, and TC Mockler, in prep.; http://mocklerlab-tools.cgrb.oregonstate.edu/). Alternative splicing events and differentially expressed gene features were identified using database queries. Microread coverage along the annotated transcription units was calculated using HashMatch data corresponding to the TAIR8-annotated cDNAs. Microread coverage and DNA methylation plots were generated in R using the CairoPNG library package (http://www.R-project.org). The box-and-whisker plots were generated using log2 transformed output from RGA and the boxplot function in R. Clustering of abiotic stress-associated gene features was performed using the default settings with “heatmap.2” in the “gplots” package of R. The distance was calculated by the “dist” function using the “euclidean” method from the “stats” package in R. This method calculates the usual square distance between two data sets. The “hclust” function with the “complete” agglomeration method from “stats” package in R was used for clustering. Arabidopsis genome annotations and microread matches were visualized (http://athal.cgrb.oregonstate.edu) using GBrowse version 1.69.
Mapping RNA-seq microreads
Raw Illumina microreads were obtained after base calling in the Solexa Pipeline version 0.2.2.6. We removed microreads matching SMART adapters, Solexa sequencing adapters and microreads of low quality (containing ambiguous nucleotide calls), and then the low quality bases at the 3′ ends of reads were trimmed. Microreads were truncated to the first 32 bases and only reads with a length of exactly 32 bases were retained for subsequent analysis using HashMatch and supersplat. These reads were termed “valid” microreads and used for all subsequent analysis.
We downloaded the Arabidopsis chromosome sequences, TAIR8 annotations, and sequence files for individual annotated genome features (genes, cDNAs, 3′ UTRs, 5′ UTRs, introns, exons, intergenic regions) from TAIR (ftp://ftp.arabidopsis.org/home/tair/Sequences/). A Perl script was used to generate 32-mer HashMatch reference databases containing all possible 32-mers from the Arabidopsis chromosome sequences and the TAIR8-annotated sequence features. A Perl script was used to generate all possible SJ-spanning 32-mers based on the TAIR8 gene model annotations. Individual 32-mer HashMatch databases for gene features (e.g., SJs, exons, introns) were mined to identify “informative” 32-mers unique to specific annotated alternative splice variants.
HashMatch (http://mocklerlab-tools.cgrb.oregonstate.edu/) was used to rapidly align perfectly matching microreads against the databases of reference sequences of the same length. HashMatch is optimized for fixed-length microreads (e.g., 25-mers, 32-mers) and exact matches, and identifies reads that match perfectly to a genome or any annotated feature within a genome, including splice junctions, exons, introns, UTRs, and intergenic regions. Valid microreads were traversed through each feature-specific database of 32-mers using HashMatch. The resulting alignments represent perfect matches to locations in the genome or annotated genomic features.
Discovery of splice junctions
For discovery of reads mapping to potentially novel splice junctions (SJs), we used supersplat (DW Bryant, R Shen, HD Priest, W-K Wong, and TC Mockler, in prep.). The program supersplat predicts splice junctions from microread data by exhaustively aligning microreads against a reference sequence assuming a gapped alignment, which allows a microread to span an intron. We first removed valid microreads that aligned anywhere in the genome or matched TAIR-annotated SJs. A filter similar to DUST (Morgulis et al. 2006) and implemented in Perl (http://bioperl.org/pipermail/bioperl-l/1999-November/003313.html) to remove reads containing low-complexity stretches. The reads remaining after the DUST filter were aligned to the Arabidopsis genome using supersplat. In this analysis we used the default supersplat setting that identifies potential splice junctions representing all possible ITDNs. The resulting supersplat output was subjected to post-processing using a series of shell and Perl scripts. Potential novel introns containing nonconsensus ITDNs predicted by supersplat were filtered to retain only those supported by at least two distinct independent RNA-seq reads with different overlap lengths on each side of the predicted intron (i.e., the portions of the reads aligning to the predicted exons), a minimum overlap length of six bases on one exon, additional support of at least one microread matching each of the predicted flanking exonic sequences, a minimum predicted intron length of 20, a maximum predicted intron length of 4000, and as a surrogate for biological replicates, requiring each predicted intron to be supported by reads originating in RNA-seq libraries representing completely independent biological samples [e.g., the oligo(dT) primed libraries and the random-primed libraries, or different stress treatments].
Empirical transcription-unit modeling
TAU (HD Priest, DW Bryant, SA Givan, CM Sullivan, and TC Mockler, in prep.) is an algorithm for rapidly assembling gene models derived completely from empirical transcript evidence. Briefly, TAU uses alignments of transcript data (RNA-seq, Sanger, or 454) to a reference sequence to construct a reference-guided assembly of spliced transcript models. Contiguous regions of transcript sequence alignments represent putative exons. Spliced RNA-Seq reads (found by supersplat) and spliced ESTs/cDNAs define splice junctions. TAU joins adjacent exons via splice junctions to construct spliced multiexon gene models. Due to the nature of the algorithm, TAU not only finds spliced gene models, but also combinatorially assembles all possible alternatively spliced gene models that can be supported by the available sequence data. The TAU algorithm also calculates an average per-base sequencing depth for the generated gene models in order to rank the models from a given locus by their relative abundances. The TAU model that possesses the highest average transcript sequence depth is reported as the primary variant and so on. For these analyses, TAU omitted all assembled models shorter than 200 nt, and did not assemble more than 50 models per genomic locus.
Intron classifications
The U12 intron matches were identified by matching to the consensus “NNATCCTN” sequence directly downstream of the 5′ splice site of a given intron. For U2 introns the splice site consensus of the last 3 nt of an exon and the first 6 nt of the intron at donor site should be MAG|GTRAGT. Four possible consensus sequences (AAGGTAAGT, CAGGTAAGT, AAGGTGAGT, and CAGGTGAGT) were matched to the donor site sequence using a Hamming distance. The Hamming distance counts the number of differences between the reference sequence (in this case, the sequence from the predicted donor splice site) to the four possible consensus sequences. If the Hamming distance was five or less (e.g., five or fewer mismatches), the intron was categorized as U2 type.
Estimation of false discovery rate for splice junctions
The rate of false positive discovery (FDR) of splice junctions was estimated by mapping simulated error-containing 32-mer reads to known TAIR8 splice junctions. Three hundred million 32-mer sequences were randomly sampled from the Arabidopsis genome. To simulate Illumina sequencing errors, a Perl script was used to randomly introduce nucleotide changes to these 32-mers using a per-base error rate of 3.6%, which was the average error rate in the real Illumina data used in this study. These error-containing simulated reads were mapped to 121,526 TAIR8 annotated Arabidopsis splice junctions that could be unambiguously interrogated using perfect match 32mers. The number of introns evaluated differs from the actual number of introns in the TAIR8 annotation because a small percentage of exon–exon junctions cannot be detected using the criteria of exact 32-mer matches over the splice junction. The rate of false positive splice junction discovery was assessed using different minimum flanking sequence lengths on each side of the splice junction. Using a minimum splice junction flanking sequence length of six bases, 934 Arabidopsis splice junctions were detected. Therefore, we infer an FDR for detection of novel splice junctions of ∼934/121,526 or ∼0.77%. For comparison, using a minimum splice junction flanking sequence length of four bases, 6280 splice junctions were detected, which corresponds to an estimated FDR of ∼6280/121,526 or ∼5.17%.
Depth of microread coverage
Microread coverage along the annotated transcription units was calculated using HashMatch data corresponding to the TAIR-annotated cDNAs. First, the length of each cDNA was divided into 100 equal segments (bins) for all annotated cDNAs. For example, for a cDNA 1500 nt long, the 51st segment corresponds to nucleotides 751–765. Then, the mean number of 32-mer RNA-seq microreads perfectly matching each segment of each individual cDNA was assigned to the corresponding bin. Finally, a median number (i.e., the depth) for each bin was calculated and the resulting values were plotted against segment number. The cDNA coverage was computed as the percentage of nucleotides represented by microreads and the frequency distribution plotted as the total number of genes in each bin.
Box-and-whisker plots
Single-base density microread match profiles were produced by generating a total number of microread matches at each nucleotide position of the reference sequence corresponding to each annotated genomic feature (i.e., intergenic regions, 5′ UTRs, 3′ UTRs, introns, exons, genes, cDNAs, CDS, SJs, and nonannotated novel genes using RGA (http://rga.cgrb.oregonstate.edu/). For each annotation feature, all density profiles were concatenated into a single output file. The data from this single RGA output file were log2 transformed and plotted in box-and-whisker graph using the R “boxplot” function (R Development Core Team 2008).
Chromosomal transcript coverage and DNA methylation plots
Microread coverage and DNA methylation plots were generated in R using the CairoPNG library package (http://www.R-project.org). The log2 of frequency of the unique cDNA microreads matching to genomic sequence was plotted along chromosomal coordinates. The methylation plots were produced using Arabidopsis genome tiling array data (Zilberman et al. 2007) deposited at NCBI Gene Expression Omnibus (GEO record GSE5974 and data set GSM138296). The graph was generated by plotting the log2 signal ratio of the immunoprecipitated methylated DNA array signal divided by the input control signal (total DNA) from data set GSM138296 (described in detail in Zilberman et al. 2007) in the same set of chromosomal coordinates as described above for the microread density plot.
Clustering of stress-associated gene features
Microread matches against gene features (genes, exons, introns, and splice junctions) were calculated using HashMatch data and loaded into a MySQL database. Differentially expressed features were identified using database queries and the following criteria: normalized gene expression (GE) levels of at least 50 read hits per gene model for both the treatment and control samples; >10 read hits per feature for treatment or control; fold change of normalized GE for both treatment and control <10; fold change of normalized read hits for the feature >5. Gene feature (exons, introns, splice junctions) clusters were computed by the default settings of heatmap.2 in gplots package of R. Clustering of abiotic stress-associated gene features was performed using the default settings with “heatmap.2” in the “gplots” package of R. The distance was calculated by the “dist” function using the “euclidean” method from the “stats” package in R. This method calculates the usual square distance between two data sets. The “hclust” function with the “complete” agglomeration method from “stats” package in R was used for clustering. The “complete linkage” clustering, also known as maximum or furthest-neighbor method, is a method of calculating distance between clusters in hierarchical cluster analysis. The distance between clusters is defined as the greatest distance between a member in one cluster and a member in the other cluster.
Visualization
Arabidopsis genome annotations and microread matches were visualized using GBrowse version 1.69.
Acknowledgments
We thank Mark Dasenko for assistance with Illumina sequencing, Anne Marie Dougherty, Jennifer Long, and Kate Peremyslova for assistance with RNA preparation, and Chris Sullivan and Steve Drake for computational support. This work was funded by Oregon State University startup funds to T.C.M., and partially supported by NSF Plant Genome Grant DBI 0605240 to T.C.M. and USDA NRI Grant 2008-01077 to T.C.M. and S.A.F. H.D.P. was supported by a Computational and Genome Biology Initiative Fellowship from Oregon State University.
Author contributions: T.C.M. and S.A.F. conceived the experimental design. S.A.F. collected tissues, prepared mRNAs, cDNA libraries for Illumina sequencing, developed and conducted RT-PCR and qRT-PCR validation assays of predicted splice isoforms. H.D.P., D.W.B., R.S., S.A.G., and T.C.M. coded the bioinformatic tools and conducted the analyses. W.K.W. consulted on algorithm design. S.A.G. developed and implemented web-based visualization tools. S.E.F. assisted with tissue collection and preparation of mRNA and cDNA libraries. S.A.F., H.D.P., and T.C.M. wrote the paper. All authors read and approved the manuscript.
Footnotes
-
↵4 Corresponding author.
E-mail tmockler{at}cgrb.oregonstate.edu; fax (541) 737-3573.
-
[Supplemental material is available online at http://www.genome.org. The Arabidopsis RNA-seq data used in this study has been deposited at the NCBI Short Read Archive (http://www.ncbi.nlm.nih.gov/Traces/sra/sra.cgi) under accession no. SRA009031. These data are also available in a genome viewer at http://athal.cgrb.oregonstate.edu and are available for download at http://athal-files.cgrb.oregonstate.edu/. The TAU, HashMatch, and supersplat software tools used in this study are available at http://mocklerlab-tools.cgrb.oregonstate.edu/.]
-
Article published online before print. Article and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.093302.109.
-
- Received March 1, 2009.
- Accepted October 5, 2009.
- Copyright © 2010 by Cold Spring Harbor Laboratory Press
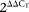 method
method







