
In-depth characterization of the microRNA transcriptome in a leukemia progression model
- Florian Kuchenbauer1,4,
- Ryan D. Morin2,4,
- Bob Argiropoulos1,
- Oleh I. Petriv3,
- Malachi Griffith2,
- Michael Heuser1,
- Eric Yung1,
- Jessica Piper1,
- Allen Delaney2,
- Anna-Liisa Prabhu2,
- Yongjun Zhao2,
- Helen McDonald2,
- Thomas Zeng2,
- Martin Hirst2,
- Carl L. Hansen3,
- Marco A. Marra2,5,6, and
- R. Keith Humphries1,5,6
Abstract
MicroRNAs (miRNAs) have been shown to play important roles in physiological as well as multiple malignant processes, including acute myeloid leukemia (AML). In an effort to gain further insight into the role of miRNAs in AML, we have applied the Illumina massively parallel sequencing platform to carry out an in-depth analysis of the miRNA transcriptome in a murine leukemia progression model. This model simulates the stepwise conversion of a myeloid progenitor cell by an engineered overexpression of the nucleoporin 98 (NUP98)–homeobox HOXD13 fusion gene (ND13), to aggressive AML inducing cells upon transduction with the oncogenic collaborator Meis1. From this data set, we identified 307 miRNA/miRNA* species in the ND13 cells and 306 miRNA/miRNA* species in ND13+Meis1 cells, corresponding to 223 and 219 miRNA genes. Sequence counts varied between two and 136,558, indicating a remarkable expression range between the detected miRNA species. The large number of miRNAs expressed and the nature of differential expression suggest that leukemic progression as modeled here is dictated by the repertoire of shared, but differentially expressed miRNAs. Our finding of extensive sequence variations (isomiRs) for almost all miRNA and miRNA* species adds additional complexity to the miRNA transcriptome. A stringent target prediction analysis coupled with in vitro target validation revealed the potential for miRNA-mediated release of oncogenes that facilitates leukemic progression from the preleukemic to leukemia inducing state. Finally, 55 novel miRNAs species were identified in our data set, adding further complexity to the emerging world of small RNAs.
MicroRNAs (miRNAs) are short RNA molecules, 19–25 nucleotides (nt) in length, recently identified to play key roles in regulating gene expression by inhibiting translation and/or triggering degradation of target mRNAs (Bartel 2004). Their maturation from a primary miRNA transcript (pri-miRNAs) to pre-miRNA hairpins and finally short double-stranded RNA duplexes is regulated by the nucleoplasmic enzyme RNASEN and its cytoplasmic counterpart DICER1 (Lund et al. 2004). Based on thermodynamic stability, one of the mature strands is thought to be preferentially incorporated into the RNA inducing silencing complex (RISC) protein complex, producing a biologically active miRNA, whereas the other is considered as inactive strand called miRNA* (star) or passenger strand (O’Toole et al. 2006). The mature miRNA comprises a “seed region”, including the nucleotides 2–7 of the 5′ end (Grimson et al. 2007). The seed region primarily defines the specificity of a miRNA toward the 3′ UTR of its target mRNAs (Grimson et al. 2007; Jongen-Lavrencic et al. 2008). Each miRNA generally has a few hundred predicted target mRNAs, but only a small set of these interactions have been experimentally validated. Thus far, 678 human miRNA sequences have been cataloged (miRBase, release 11, 2008) and identified by either cloning or computational prediction.
The emerging awareness of the large number of miRNAs, their complex expression patterns, and broad range of potential targets has triggered major interest in understanding their possible regulatory functions. Indeed it is now clear that miRNAs play critical roles in physiological (Looijenga et al. 2007; Park et al. 2007; Tang et al. 2007; Thatcher et al. 2007; Wang et al. 2007) as well as multiple malignant processes (Bandres et al. 2007; Hernando 2007; Jay et al. 2007; Looijenga et al. 2007; Lui et al. 2007; Negrini et al. 2007; Porkka et al. 2007; Sevignani et al. 2007; Shell et al. 2007; Tran et al. 2007; Yu et al. 2007b). Specifically in the context of hematologic malignancies, seminal studies from the group of Carlo Croce have strongly linked miRNAs to lymphoma development (Calin et al. 2002, 2004, 2005). Recent findings indicate miRNA expression profiling as a useful tool for classification and prognostic purposes in acute myelogenous leukemia (AML) (Debernardi et al. 2007; Mi et al. 2007; Garzon et al. 2008; Isken et al. 2008) and point to involvement of specific miRNAs like miR-223 and the miRNA 17-106a cluster in myeloid regulatory networks such as CEBPA and the receptor for CSF1 (also known as M-CSF) (Fazi et al. 2005, 2007; Fontana et al. 2007). These initial findings encourage further efforts directed at obtaining a comprehensive and quantitative picture of the miRNA transcriptome to gain further insights into the multistep process of AML development.
Such efforts to date have principally relied on methods to detect single miRNAs or on a larger scale to profile the miRNA transcriptome using real-time PCR or microarray platforms. These methods are limited as they are restricted to the detection and profiling of known miRNA sequences previously identified by sequencing or homology searches (Griffiths-Jones 2006). These approaches feature reliable reproducibility and facilitate clustering of samples by similar miRNA expression profiles (Davison et al. 2006; Porkka et al. 2007). Alternative sequenced-based methods for miRNA profiling, while initially complex and expensive due to laborious cloning techniques (Aravin and Tuschl 2005; Pfeffer et al. 2005), are now becoming practical due to the development of “next generation” sequencing strategies. In addition to enabling the detection of miRNA variation in mature miRNA length, as well as enzymatic modification of miRNAs such as RNA editing (Kawahara et al. 2007) and 3′ nucleotide additions (Ruby et al. 2006; Landgraf et al. 2007), these newer high-throughput strategies permit high-resolution views of expressed miRNAs over a wide dynamic range of expression levels. In-depth miRNA profiling by sequencing has already been realized in several tissues from different organisms including Caenorhabditis elegans (Ruby et al. 2006), Arabidopsis thaliana (Margulies et al. 2005), primates (Berezikov et al. 2006b), and human embryonic stem cells (Morin et al. 2008) using massively parallel sequencing.
In an effort to gain further insights into the role of miRNAs in AML, we have applied the Illumina massively parallel sequencing platform to carry out an in-depth, quantitative comparative analysis of miRNA expression in a murine model of leukemia progression (Pineault et al. 2005). This leukemia model simulates the stepwise conversion of a nonleukemic myeloid progenitor cell, induced from normal mouse bone marrow by engineered overexpression of the nucleoporin 98 (NUP98)–homeobox gene HOXD13 fusion gene (ND13), to a highly aggressive AML inducing cells upon transduction with the potent oncogenic collaborator Meis1 (Pineault et al. 2005; Pineault et al. 2003). Our results provide a comprehensive view of the miRNA transcriptome in a well-defined leukemia progression model and reveal both a striking repertoire of expressed miRNAs, including identification of 55 novel miRNAs, and a remarkable range of expression levels spanning some five orders of magnitude. Interestingly, few miRNAs were detected that were uniquely expressed in the preleukemic versus leukemic state, but multiple differentially expressed miRNAs were identified, thus suggesting that the functional role for miRNAs in leukemic transformation may be highly complex. Adding to this complexity, we show that almost all miRNAs exhibited isoforms of variable length and thus potentially distinct function.
Results
All experiments were carried out using a Hox-based leukemia progression model as previously described (Pineault et al. 2005). To model the preleukemic state, we used a murine bone marrow–derived cell line generated by transduction with the ND13 fusion gene. These cells are growth factor dependent and are transplantable, giving rise to short-term myeloid restricted repopulation without evolution to leukemia over extended in vivo follow-up (Pineault et al. 2005). As a model of progression to the leukemic state, we transduced the ND13 cell line with the Hox cofactor Meis1, thus generating a cell line that remains growth factor dependent but induces aggressive and rapidly fatal myeloid leukemia upon transplantation (Pineault et al. 2003, 2005). Both cell lines exhibit stable, homogenous, and almost identical immune phenotypes of primitive hematopoietic progenitors and stable differential in vivo functional properties of preleukemic versus leukemic cells (for details, refer to Methods and Supplemental Fig. 1).
Sequencing and annotation of small RNAs
Small RNAs were isolated from the preleukemic progenitor line, hereafter referred to as ND13, and the leukemic line, hereafter referred to as ND13+Meis1, and processed to allow deep sequencing on the Illumina platform (previously known as Solexa sequencing). A total of 9.56 × 107 and 7.23 × 107 reads were sequenced from the myeloid progenitor ND13 and leukemic ND13+Meis1 cell lines, respectively, producing (after removal of ambiguous reads), 3.4 × 106 (ND13) and 2.6 × 106 (ND13+Meis1) unique 27 nt reads. After mapping the sequences to the mouse genome (NCBI Build 37), a total of 3.90 × 105 (ND13) and 2.96 × 105 (ND13+Meis1) unique small RNA sequences remained. Each of these sequences was classified either as a known class of small RNAs, genomic repeats, degradation fragments of larger noncoding RNAs, known mRNA sequences, or small RNAs deriving from unannotated intergenic regions (Fig. 1A). The most abundant (based on read count) RNA species in both libraries were classifiable as known miRNAs (65% of total) (Fig. 1A) and corresponding to some 220 miRNA genes (see below and Table 1). The ranges of all sequence counts for each miRNA gene are plotted in Figure 1B. Both libraries show a similar distribution of expression levels with count ranges for a given unique miRNA species, spanning two (to minimize consideration of reads deriving from sequencing errors, singletons were excluded, see Methods) to over 1.3 × 105 (ND13) and 1 × 106 (ND13+Meis1) sequence counts, respectively. Exemplified for ND13 cells, ∼8% of miRNAs and miRNA*s were detected at high sequence counts (>104), and ∼14% were detected in the intermediate 103–104 sequence count range; the remaining 77% were detected in the low range of two to 1000 sequence counts. Thus the sequence data reveal a wide range of expression levels for miRNAs spanning over five orders of magnitude (Table 1).
Overview about detected miRNA/miRNA* species, expression range and distribution
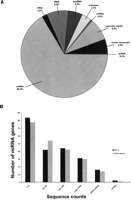
Overview of small RNA and miRNA gene expression in a preleukemic and leukemic cell model obtained by deep sequencing. (A) Breakdown of the proportions (in percent) of various classes of small RNAs detected by sequencing of the preleukemic ND13 library. The percentages are comparable to those found in the leukemic ND13+Meis1 library. Small RNAs belonging to the miRNA family constitute the majority (65.7% in the preleukemic and 66.2% in the leukemic cells). scRNA, small cytoplasmic RNA; snRNA, small nuclear RNA; snoRNA, small nucleolar RNA; rRNA, ribosomal RNA; tRNA, transferRNA; unknown, derived from unannotated/intergenic regions. (B) Distribution of miRNA genes expressed according to their sequence counts in the preleukemic (ND13) compared with leukemic (ND13+Meis1) cells. Shown are the numbers of unique miRNA genes plotted as a function of their expression levels as defined by a given range of sequence counts in the respective libraries of small RNAs. The total numbers of miRNA sequence counts were 1,240,570 and 1,030,414 for the preleukemic and leukemic libraries, respectively.
Sequence variations in miRNAs
In both libraries, miRNA sequences frequently exhibited variations from their “reference” sequences as currently described in miRBase, thus indicating multiple mature variants that we hereafter refer to as isomiRs. Evidence of isomiRs of a similar nature were also detected in a limited sequence analysis using a linker-based miRNA cloning approach from the same RNA pools (Fu et al. 2005), suggesting that isomiRs are not due to artifacts created from massively parallel sequencing (Fig. 2). Our analysis revealed two major classes of variants or isomiRs. Most isomiRs show variability at their 5′ and/or 3′ ends, likely resulting from variations in the pre-miRNA secondary structures that result in variable cleavage sites for RNASEN and DICER. In total, we found 3390 isomiRs for a total of 336 sequenced miRNAs and miRNA*s, corresponding to 225 miRNA genes from both libraries (Table 1; Supplemental Table 1). The number of different isomiRs for a given miRNA ranged from one to 74. Only 22 miRNAs or miRNA*s (all with very low sequence counts, <50) did not exhibit any isomiRs. However, the number of isomiRs showed only a moderate correlation with the absolute expression levels for each miRNA (R2 = 0.40, Pearson), suggesting that the number of observed isomiRs is not directly related to the abundance of a miRNA. The more prevalent type of modification noted among the miRNAs were single-nucleotide 3′ extensions (3390 isomiRs) (Supplemental Table 1), compared with 151 miRNAs/miRNA*s with 3′ variations not matching the genome (Supplemental Table 2; Fig. 2), possibly arising from novel mechanisms of miRNA processing.

Example of high frequency of miRNA sequence variation (isomiRs). Shown are the unique sequences and number of times this sequence was detected matching the pre-miRNA sequence of miR-181a. The most frequent occurring miR-181a sequence is not in accordance with the miRBase reference sequence. The three most common sequences were also detectable by linker-based cloning, as indicated in the figure. An example of a miR-181 isomiR not matching the genome is shown in the bottom part of the figure.
An example is given in Figure 2, demonstrating sequenced isomiRs for miR-181a. As seen for miR-181a, we frequently found that the miRBase reference sequence was not the dominant species. Therefore, the apparent relative expression levels could substantially depend on which isomiR is interrogated and challenge current real-time PCR approaches, which are based on the miRBase reference sequence. Our own experiments (data not shown) using stem-loop primers lacking only 1 nt at the 3′ end, which should theoretically amplify different IsomiRs, showed ΔCT differences between isomiRs of the same miRNA as large as 2. However, these CT differences were not predictable based on the ratio of read counts for the longer and shorter isomiR, implying that more than one isomiR was amplified by either primer.
miRNAs are differentially expressed between the myeloid progenitor ND13 and the leukemic ND13+Meis1 cell line
Measuring the abundance of a miRNA or miRNA* using the sum of all isomiR sequence counts correlated well with the expression level of the most abundant miRNA/miRNA* sequence (ND13: R2 = 0.98, Pearson; ND13+Meis1: R2 = 0.97, Pearson). As the most abundant miRNA/miRNA* sequence detected did not correspond to the current miRBase reference sequence (Table 2; for all sequence counts, refer to Supplemental Table 3), we focused on the most abundant miRNA/miRNA* sequence for differential abundance analyses as previously described (Morin et al. 2008). Applying this measure of miRNA expression, we identified 336 miRNAs and miRNA*s in both libraries, corresponding to 225 miRNA genes. Only 12 miRNAs were unique to ND13 and eight were unique to ND13+Meis1 (Table 1), although all of these corresponded to the very low (<10) sequence count range, and thus their observed differential expression may represent limits of detection rather than biological variability.
Most differentially expressed miRNA/miRNA* species (counts >150 and >1.5 fold change), including miRBase annotation
Of the miRNAs detected in both the preleukemic and leukemic cells, 65 were significantly differentially expressed (≥1.5 fold change, ≥150 sequence counts, P < 0.001) (Fig. 3A). Table 2 lists all up-regulated and the top 20 down-regulated miRNAs with a sequence count ≥150 and a ≥1.5 fold change, thresholds set to minimize inflated fold changes values from miRNAs with very low expression levels. Supplemental Table 3 summarizes all differentially expressed miRNAs for different metrics. In general, more miRNAs were down-regulated (49 miRNAs) than up-regulated (16 miRNAs) (Fig. 3A; Table 2; Supplemental Table 3), a phenomenon that is consistent with recent profiling reports in leukemias (Lu et al. 2005; Garzon et al. 2008). Considering fold change, the most significant miRNA was miR-196b, exhibiting a 4.4× increase in the ND13+Meis1 library. Few miRNAs showed predominant expression in only one of the two libraries, with miR-223* (2684 counts) sequestered to ND13 cells (Table 2). The fact that the 5′ arm of miR-223, miR-223* is the highest down-regulated sequence, implies that miR-223*, previously thought to be nonfunctional, might also be an important factor for leukemic transformation.
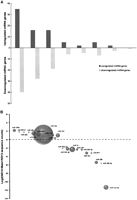
Analysis of differentially-expressed miRNA genes in leukemic cells compared with preleukemic cells. (A) Distribution of differentially-expressed miRNA genes according to their fold changes. Shown are the number of miRNA genes whose expression was up-regulated (positive values) or down-regulated (negative values) in the leukemic cells as a function of the fold change. Only changes >1.5 and achieving a P-value of <0.05 were included. (B) Bubble plot depicting the abundance of selected miRNA/miRNA* species and their relative expression levels. The bubbles represent the sum of the most common sequence counts from both libraries for a miRNA/miRNA* species plotted as a function of fold difference between the leukemic versus preleukemic cells.
Most differentially expressed miRNA/miRNA*s were detected with intermediate sequence count levels as depicted in Figure 3B, which shows the abundance of a miRNA and its relative expression. An exception is miR-10a, which is expressed at high levels (ND13: 14,700, ND13+Meis1: 32,064 counts) and displays a 2.18-fold up-regulation. Notably, all miRNAs located in the Hox cluster (miR-10a, miR-10b, and miR-196b) (Mansfield et al. 2004) and previously implicated in regulating Hox gene expression in AML (Debernardi et al. 2007; Isken et al. 2008) were up-regulated in the leukemic ND13+Meis1 cells. In contrast, almost all members of the let-7 family, some of them with very high expression levels (Supplemental Table 3), were found to be down-regulated in the ND13+Meis1 library (1.7- to 2.6-fold) and consistent with the proposed role of let-7 family members as tumor suppressors (Lee and Dutta 2007).
In order to compare our sequencing results with a secondary method, we used TaqMan probes to assay 23 of 27 miRNAs as presented in Table 2. Consistently, 17 out of 23 miRNAs (73.9%) correlated with the differential expression detected by Illumina sequencing (Table 2).
In summary, these results from comparing a preleukemic versus leukemic cell state document a large and overlapping repertoire of miRNA species, many of which are differentially expressed and span a large range of expression levels. These results suggest that processes involved in or reflecting leukemogenic states are dictated by a complex repertoire of shared, but differentially expressed miRNAs, rather than complete presence or absence of miRNAs between these cellular states.
Novel miRNA genes
We sought to identify novel miRNA genes among the unclassified sequences in our libraries. After annotation, 81,316 of the small RNA sequences in the ND13 library and 57,015 in the ND13+Meis1 library remained unclassified because they derived from unannotated regions of the mouse genome. To identify candidate novel miRNAs among these, we employed both in-house and publicly available algorithms as published (Morin et al. 2008). The total set of novel miRNA candidates comprises 94 unique miRNA sequences with mainly low expression levels, of which 55 have been accepted by miRBase (Supplemental Table 4) as novel miRNAs. Three novel miRNAs exhibited sequence counts >100 and a ≥1.5-fold change between the two libraries, and thus represent potentially interesting candidates as having functional relevance in leukemia (Table 3).
Top 12 most abundant differentially expressed novel miRNAs, including their genomic location
Targets of differentially expressed miRNAs
In an attempt to highlight the regulation of oncogenes through miRNAs in preleukemic ND13 and leukemic ND13+Meis1 cells, target sites of the 19 most abundant miRNAs from each cell line were predicted with the following three computational algorithms: TargetScan (http://www.targetscan.org), RNAhybrid (http://bibiserv.techfak.uni-bielefeld.de/rnahybrid/), and miRanda (http://www.microrna.org/microrna/home.do). Only genes with at least two predicted target sites and an accessible 3′ UTR were considered relevant (Zuker 2003; Long et al. 2007). Based on this, we identified 1461 and 937 target genes for miRNAs enriched in ND13 and ND13+Meis1 cells, respectively. In order to identify possible oncogenes from these lists, the predicted targets were compared with the Sanger Cancer Gene Census (http://www.sanger.ac.uk/genetics/CGP/Census/), a list comprising 367 genes documented to be involved in cancer development (Futreal et al. 2004). Remarkably, almost half of the 17 predicted cancer gene census targets of ND13-enriched miRNAs represented AML-specific oncogenes, including MLL genes and HOXA11 (Fig. 4). However, only three AML-specific oncogenes (DEK, CBFB and WT1) out of 15 predicted gene census targets were identified as targets for miRNAs of the ND13+Meis1 library (Fig. 4). Predicted miRNAs targeting DEK include miR-155 and miR-23a, which were confirmed as regulators in luciferase assays (Fig. 5A,B). Other predicted miRNAs, but not experimentally validated for CBFB and WT1, were miR-19b, miR-30e, and miR-669c (Supplemental Table 5). In addition, the down-regulation of the ND13+Meis1-specific tumor suppressor gene NR4A3 has been recently shown to cause AML (Mullican et al. 2007). Thus, the observation of an accentuated miRNA down-regulation and the miRNA-mediated release of oncogenes might facilitate leukemic progression from a preleukemic to leukemic state.
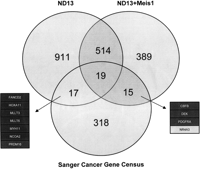
Venn diagram of the predicted miRNA targets for the 19 most abundant miRNAs from each library and their shared targets with the Sanger Cancer Gene Census. The dark boxes indicate AML-specific oncogenes, whereas the gray box highlights a tumor suppressor gene targeted by miRNAs enriched in ND13+Meis1 cells.
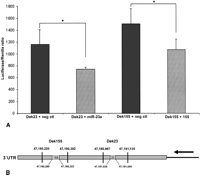
Dek-3′ UTR luciferase assays for miR-23a and miR-155. (A) Bar diagram demonstrating the binding of miR-23a and miR-155 to the 3′ UTR of the Dek oncogene. Dek23 comprises only binding sites for miR-23a, whereas Dek155 exhibits only predicted binding sites for miR-155. A nonbinding miRNA was used as negative control. *P < 0.05. (B) Schematic representation of the Dek 3′ UTR constructs and the predicted miRNA binding sites.
Discussion
To gain insight into the miRNA transcriptome in the development of leukemia, we have exploited a massively parallel sequencing strategy coupled to a novel leukemic progression cell line model. Our results reveal a large number and remarkable overlap in miRNAs detected in both libraries and also a broad range in miRNA expression levels. Furthermore, we support the recent finding in humans that the miRNA transcriptome includes a diversity of miRNA isoforms, and have identified over a hundred putative novel miRNAs (Morin et al. 2008). Though these two cellular states share many miRNAs, we documented numerous differences in miRNA expression levels between the preleukemic versus leukemic stages in the Hox-based progression model.
Among the dramatic findings from this analysis are the large numbers of 205 different miRNA genes and 55 novel miRNAs detected. This exceeds a previous estimate of only 43 miRNAs in various leukemia cell lines as detected by Northern blot analysis (Yu et al. 2006). Recent analysis of the miRNA transcriptome in ES cells using the Illumina high-throughput sequencing platform also revealed a remarkable number of miRNA species (Morin et al. 2008). Strikingly, over 60% of the miRNAs detected in ES cells were also found in our analysis of a leukemia progression model. Such overlaps of miRNA expression patterns suggest that many key functional roles of miRNAs depend more on relative levels rather than unique, tissue-specific expression (Morin et al. 2008). Indeed, we observed a high overlap (∼80%) in the miRNA sequences expressed in both preleukemic and leukemic bone marrow cells. All miRNAs unique to a single library were found at very low expression levels (<30 copies), and thus were likely at the limits of detection and of questionable functional significance. An exception, with relatively high expression levels, was miR-223*, which was highly detected in nonleukemic cells (2684 sequence counts) and only one copy was detected in the leukemic cells. The fact that a miRNA* species from a well-characterized miRNA such as miR-223 (Fazi et al. 2005, 2007) was overrepresented in ND13 bone marrow cells supports current speculations that both pre/miRNA arms can be tissue-dependently expressed and may have functional relevance (Ro et al. 2007; Okamura et al. 2008; Seitz et al. 2008). Recently published works (Okamura et al. 2008; Seitz et al. 2008) suggest that the miRNA* strand is not merely a carrier strand but can bind to the RISC complex and have inhibitory potential. Interestingly, based on the targetscan prediction algorithm (www.targetscan.com), the only target with two conserved target sites for the miR-223* seed region (GUGUAUU) is CUTL1, a gene essentially involved in the pathogenesis of leukemia. Another potential highly ranked target of miR-223* is MDS1 (also known as EVI1), a known oncogene involved in AML pathogenesis. Based on these facts, it is not unlikely that miR-223* might contribute to the myeloid differentiation potential of mir-223.
The complexity of the miRNA transcriptome dramatically increases when the miRNA miRBase reference sequences and their isoforms (isomiRs) (Morin et al. 2008) are taken into consideration. Our analysis revealed a large number of isomiRs, derived from almost all detected miRNAs; in total, we detected 3390 isomiRs in both libraries, greatly exceeding and extending previous reports (Ruby et al. 2006; Landgraf et al. 2007) but comparable to Illumina sequencing of human ES cells (Morin et al. 2008). This confirms recent suggestions that the miRNA transcriptome is more complex than previously assumed. Despite the large number of isomiRs detected, their role in post-transcriptional regulation remains to be experimentally determined. However, isomiRs resulting from variation at the 5′ end may be of particular interest as they have different seed sequences than the reference miRNA, with the ability to potentially target different transcripts. Besides changes of the seed region, end variations can putatively change the secondary structure of the miRNA and thus facilitate or prevent target UTR binding. These results suggest that a fuller description of the expression of isomiRs for each miRNA will be of interest to determine if there are tissue-specific isomiR distributions relevant to development and disease.
Another striking observation was the large range in miRNA expression levels for both libraries with count ranges for a given unique miRNA and miRNA* species, spanning from two to >1.3 × 107 sequence counts. This documented range in expression spanning over five orders of magnitude is some 100-fold greater than reported in previous studies (Berezikov et al. 2006a), likely reflecting the improved sampling depth possible with the Illumina sequencing method. MiRNAs with high expression levels in both libraries included members of the let-7 family, miR-21, and miR-25, suggesting a fundamental role in cell survival and/or proliferation. Indeed, some of these miRNAs like the let-7 family and miR-21 have been shown to be highly expressed in other tissue libraries (Ibarra et al. 2007; Morin et al. 2008) and in the context of cancer (Chan et al. 2005; Mayr et al. 2007).
Interestingly, all miRNAs located in the Hox cluster (miR-10a, miR-10b, and miR-196b) were up-regulated in the leukemic ND13+Meis1 cells. Although the function of these miRNAs in AML is unknown, recent AML profiling reports point toward distinct roles in leukemogenesis (Garzon et al. 2008; Isken et al. 2008; Jongen-Lavrencic et al. 2008). Furthermore, miR-10b seems to be a key factor modulating the ability of breast cancer cells to metastasize (Ma et al. 2007). In contrast, almost all members of the known tumor suppressor miRNA family let-7 (Johnson et al. 2005; Akao et al. 2006; Lee and Dutta 2006, 2007; Mayr et al. 2007; Yu et al. 2007a) were down-regulated in the leukemic state examined here, raising the intriguing possibility that this down-regulation is linked to the erosion of key self-renewal and differentiation programs in leukemic stem cells similar to breast cancer stem cells as shown recently (Johnson et al. 2005; Akao et al. 2006; Lee and Dutta 2006, 2007; Mayr et al. 2007; Yu et al. 2007a). Although it is difficult to compare our model system to recent miRNA profiling approaches in human AML, the depth of the presented work might complement these studies (Garzon et al. 2008; Isken et al. 2008; Jongen-Lavrencic et al. 2008). Similar as in the mentioned works, we also found an up-regulation of all miRNAs located in the Hox cluster, as well as a down-regulation of let-7 family members. However, all published works do not provide more than a quantitative approach, which does not cover questions about isomiRs, novel miRNAs, and disease specific mutations within the miRNA transcriptome. These questions will be more likely covered by future high-throughput sequencing approaches, providing the genomic resolution to understand the undergoing changes of the miRNA transcriptome within the development of AML.
In order to highlight potential oncogenes as targets for miRNAs highly expressed in both libraries, we took into account cooperative target selection and 3′ UTR accessibility. Comparing all predicted targets with the Sanger Cancer Gene Census (Futreal et al. 2004), we identified unique cancer related target genes for miRNAs of both libraries. Notably, our analysis suggested that in nonleukemic ND13, cell leukemia-specific oncogenes were more frequently targeted, whereas within ND13+Meis1 almost no leukemia-specific oncogenes were targeted. These predictions were experimentally validated for the Dek oncogene. Therefore, it can be speculated if targeting of specific oncogenes through miRNAs could tip the balance from the preleukemic to leukemic state.
With few exceptions, recent large-scale cloning efforts have provided minimal yields of new miRNA genes, mainly due to the dominance of the highly expressed miRNAs in small RNA libraries (Landgraf et al. 2007). We present 55 novel miRNA genes that have passed multiple levels of annotation criteria (Morin et al. 2008). Although the majority of these novel miRNAs were expressed at modest to low levels and only three showed differential expression in this model, further assessment of their expression and roles in other tissues and diseases will be of interest. Notably, the predicted novel miRNAs also exhibit 3′ variations, which match or do not match the genome, similar to the above-mentioned isomiRs (data not shown).
Applying the massively parallel Illumina sequencing platform has allowed us to generate an accurate and comprehensive picture of the miRNA transcriptome in a Hox/Meis1 leukemia progression model at great depth. Following a combination of novel miRNA annotation and discovery techniques, we have revealed a large list of expressed miRNA/miRNA* sequences. In addition to a large range of expressed miRNA genes with dramatic expression ranges, we detected massive 5′ and 3′ sequence variations within each miRNA/miRNA* species, called isomiRs, adding an additional layer of complexity to the known miRNA sequences.
Methods
Generation of the ND13 and ND13+Meis1 bone marrow cell lines
Mice were bred and maintained at the British Columbia Cancer Research Center Animal Facility (Vancouver, Canada). All experimental protocols were approved by the University of British Columbia Animal Care Committee. Establishment and characterization of the ND13 preleukemic BM cell lines was as previously described (Pineault et al. 2005). In brief, a polyclonal representative line was established from BM cells from C57Bl/6J mice freshly transduced with the ND13-PAC virus, selected with puromycin at a concentration of 3 mg/mL for 5 d and maintained at a concentration 2 mg/mL in liquid culture (Dulbecco’s modified Eagle's medium [DMEM]) supplemented with 15% fetal bovine serum (FBS), 10 ng/mL of human interleukin-6 (hIL-6), 6 ng/mL of murine interleukin-3 (mIL3), and 100 ng/mL of murine stem cell factor (mSCF). All culture media and growth factors were obtained from StemCell Technologies Inc. (Vancouver, Canada). Cells were counted with the Vi-Cell XR Cell Viability Analyzer (Beckman Coulter Inc.). To generate the ND13+Meis1 BM cell line, puromycin-selected ND13 BM cells as described above were transduced by co-cultivation on irradiated (4000 cGy) E86 producers for Meis1-YFP, respectively, for a period of 2 d in the presence of 5 μg/mL of protamine sulfate (Sigma) and sorted for YFP-positive cells with the FACSVantage SE (Becton Dickinson). Both, ND13 and ND13+Meis1 bone marrow cell lines were kept in culture for 40–50 d and tested for YFP by flow-cytometry analysis (FACS), immunophenotyped by FACS, and injected into C57Bl/6J mice to test their in vivo properties. The lines were frozen in multiple vials with between 1 × 106 and 3 × 106 cells from both lines in 1 mL of 90% newborn calf serum (Invitrogen) and 10% DMSO (Sigma).
Small RNA library preparation
Cultured ND13 and ND13+Meis1 cells were harvested and RNA extracted with TRIzol, as previously described (Argiropoulos et al. 2008). The extracted RNA was subjected to miRNA library construction (ND13, ND13+Meis1) according to the protocol published by Morin et al. (2008).
Differential expression detection
All unique small RNA sequences were compared between the two libraries (ND13 and ND13+Meis1) for differential expression using the Fisher exact test and Bonferroni correction. Sequences were deemed significantly differentially expressed if the P-value given by this method was <0.001, and there was at least a 1.5-fold change in sequence counts between the two libraries. In practice, all miRNAs with this combination of fold change and expression level were deemed statistically significant. Unless stated otherwise, comparison of miRNA expression between libraries regards the most frequently observed isomiR as the diagnostic sequence for evaluation of differential expression.
Cloning, annotation, and prediction of novel miRNAs
A limited small RNA sequence analysis was performed according to the protocol of Fu et al. (2005). The annotation procedure was performed as described but employed annotations from miRBase version 11 and the Mus musculus genome (NCBI build 37). Novel miRNAs were predicted as previously described (Morin et al. 2008).
Real-time quantitative TaqMan PCR assays
MiRNA real-time quantification was performed using the BioMark 48.48 Dynamic Array System (Fluidigm Corporation) and TaqMan MicroRNA Assays (Applied Biosystems) according to the manufacturer’s instructions. The reverse transcriptase reaction using TaqMan stem-loop primers was performed according to the protocol of Tang et al. (2006).
Cooperative miRNA target prediction
Predicted targets for the 19 highest expressed miRNAs from each library were downloaded from TargetScan (http://www. targetscan.org), RNAHybrid (http://bibiserv.techfak.uni-bielefeld. de/rnahybrid/), and miRanda (http://www.microrna.org/ miranda_new.html). Only miRNAs with counts of at least 100 in ND13 or ND13+Meis1 were included in the target analyses. Genes with target sites for at least two coexpressed miRNAs from one or both libraries were identified as potential cooperative targets. To compensate for potential bias, genes with numerous predicted miRNA target sites were given a lower rank than those with few predicted target sites. The rank score of a gene was calculated by dividing the number of target sites for coexpressed miRNAs by the total number of target sites for that gene. We used a cutoff of 0.15 (rank) to produce the two sets of high-ranked candidate cooperative targets of ND13-enriched and ND13+Meis1-enriched miRNAs. Predicted targets were only considered relevant if their 3′ UTR was accessible based by secondary structure folding predicted by Mfold 3.2 (Zuker 2003).
Luciferase assays
Two fragments of Dek-3′ UTR with binding sites for either miR-23a (Dek23; chr13:47180220–47180392) or miR-155 (Dek155; chr13:47180967–47181135) (Fig. 5B) were cloned into pMirReport (Ambion) and transfected with hsa-miR-155 (Ambion), hsa-miR-23a (Ambion) or a negative control miRNA (Ambion) into 293T cells. For the 3′ UTR-luciferase assays, 200 ng of pMirReport-3′ UTR, 10 pmol of miRNAs, and 0.17 ng of thymidine kinase-Renilla were cotransfected into 6 × 105 293T cells (24-well format) using the Lipofectamine 2000 transfection reagent (Invitrogen). The assays were read in the Lumat LB 9507 tube luminometer (EG&G Berthold) and the luciferase/Renilla ratio calculated. Student’s t-test was used for statistical analysis, and P < 0.05 was considered as significant.
Acknowledgments
We thank the staff at Illumina, Inc. for technical assistance. We also thank Patty Rosten for her invaluable knowledge, as well as Dr. Daniel Starzynowski, Dr. Aly Karsan, and Dr. Andrew Weng for inspiring discussions and helpful advice. This project was funded in part by The Terry Fox Foundation through the National Cancer Institute of Canada and by the Canadian Stem Cell Network. F.K. is supported by the Deutsche Forschungsgemeinschaft Germany (grant no. Ku 2288/1-1). M.H. is supported by the Deutsche Forschungsgemeinschaft Germany (grant no. He 5240/1-1). E.Y. is a recipient of postdoctoral fellowships from the Michael Smith Foundation for Health Research and the Canadian Institute of Health Research. M.A.M. is a Michael Smith Foundation for Health Research Senior Scholar. R.D.M. receives stipends from the Michael Smith Foundation for Health Research and the Canadian Institutes for Health Research. M.G. is supported by scholarships from the Michael Smith Foundation for Health Research and the Natural Sciences and Engineering Research Council.
Footnotes
-
↵6 Corresponding authors.
↵6 E-mail mmarra{at}bcgsc.ca; fax (604) 675-8178.
↵6 E-mail khumphri{at}bccrc.ca; fax (604) 877-0712.
-
[Supplemental material is available online at www.genome.org.]
-
Article published online before print. Article and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.077578.108.
-
- Received February 17, 2008.
- Accepted August 13, 2008.
- Copyright © 2008, Cold Spring Harbor Laboratory Press








