
Nucleosome positioning in human HOX gene clusters
- Peter V. Kharchenko1,2,4,
- Caroline J. Woo3,4,
- Michael Y. Tolstorukov1,
- Robert E. Kingston3,5, and
- Peter J. Park1,2,5
- 1 Harvard Partners Center for Genetics and Genomics, Boston, Massachusetts 02115, USA;
- 2 Children’s Hospital Informatics Program, Boston, Massachusetts 02115, USA;
- 3 Department of Molecular Biology, Massachusetts General Hospital, Boston, Massachusetts 02114, USA
-
↵4 These authors contributed equally to this work.
Abstract
The distribution of nucleosomes along the genome is a significant aspect of chromatin structure and is thought to influence gene regulation through modulation of DNA accessibility. However, properties of nucleosome organization remain poorly understood, particularly in mammalian genomes. Toward this goal we used tiled microarrays to identify stable nucleosome positions along the HOX gene clusters in human cell lines. We show that nucleosome positions exhibit sequence properties and long-range organization that are different from those characterized in other organisms. Despite overall variability of internucleosome distances, specific loci contain regular nucleosomal arrays with 195-bp periodicity. Moreover, such arrays tend to occur preferentially toward the 3′ ends of genes. Through comparison of different cell lines, we find that active transcription is correlated with increased positioning of nucleosomes, suggesting an unexpected role for transcription in the establishment of well-positioned nucleosomes.
Epigenetic regulation of the primary sequence of a genome determines a distinctive cell fate by controlling transcription, replication, and DNA repair mechanisms. The regulation is achieved in part by modulating accessibility of the genomic loci. On a large scale, transcriptional activation has been observed to result in transition of compact domains containing multiple genes into decondensed loops that extend beyond chromosome territories (Chambeyron and Bickmore 2004; Fraser and Bickmore 2007). Perhaps equally important are the alterations of chromatin structure occurring at the most detailed scale—that of the individual nucleosomes (Workman 2006). The DNA directly in contact with the nucleosome core may be inaccessible to trans-acting proteins, and association of nucleosomes into larger complexes may reduce DNA accessibility even further (Workman and Kingston 1998; Li et al. 2007; Morse 2007). In the case of transcription, the accessibility of activators or repressors to their binding sites plays a significant role in determining the transcriptional state of the gene. The plasticity of chromatin architecture allows for regulatory sequences within promoter regions to gain or lose accessibility by mechanisms such as nucleosome remodeling, covalent modifications of histones, and histone replacement or loss.
DNA fragmentation patterns resulting from digestion by micrococcal nuclease (MNase) and other endonucleases have been used extensively to examine chromatin structure (Van Holde 1989). Only recent technological advances, however, allowed genome-scale examination of nucleosome patterns, in particular with respect to specific positions in the chromosome. A study of MNase protection along continuous chromosome regions in Saccharomyces cerevisiae has demonstrated that a substantial fraction of nucleosomes occupy fixed chromosomal positions, and that functionally important positions such as transcription start sites or transcription factor binding sites were selectively devoid of nucleosomes (Yuan et al. 2005; Lee et al. 2007). These findings were confirmed for the promoter regions of human genes using genome-tiling microarrays (Ozsolak et al. 2007), and more recently by the analysis of high-throughput sequencing data (Schones et al. 2008). However, our understanding of the spatial properties and functional significance of nucleosome organization beyond promoter regions is still limited. While the high-throughput sequencing has provided a genome-wide snapshot of the nucleosome positioning, detection of all stable nucleosome positions within a particular region requires greater sequencing depth (see Supplemental Section 2). In this study we have examined nucleosome organization throughout 0.5 Mb of the human genome that spans across HOX gene clusters. The HOX clusters were chosen because they have been well-characterized across a broad range of species and are amongst the most studied regions of the human genome.
Results
Stable nucleosome protection patterns are visible under detergent conditions
To examine chromatin architecture we have measured DNA protection patterns formed by digestion with MNase. Nuclei isolated from K562 and HeLa cells were pre-extracted and treated with MNase to obtain predominantly nucleosome-sized DNA fragments (Fig. 1B; Methods). The digested chromatin was hybridized against differentially labeled MNase-digested bare genomic DNA, using a custom genome tiling array that covers the four human HOX clusters at 5-bp resolution along both DNA strands (Fig. 1C).

(A) Schematic view of the chromatin measurements. Nucleosome positions were predicted based on the hybridization of MNase-digested pre-extracted chromatin against differentially labeled MNase-digested bare genomic DNA. (B) Agarose gel separation showing nucleosome-sized DNA fragments (arrow) in K562 and HeLa after MNase digestion. (C) Tiling of the DNA microarray used for chromatin measurements: 50-bp probes were tiled with 5-bp steps on both strands.
While MNase preferentially cleaves internucleosomal linker DNA, the cleavage pattern is also biased by the nucleotide sequence (Wingert and Von Hippel 1968; Dingwall et al. 1981). To minimize the contribution of the MNase bias, chromatin profiles for the K562 and HeLa cells were obtained by competitive hybridization against MNase-digested bare genomic DNA (Fig. 1A). We have separately assessed MNase digestion bias on bare genomic DNA from K562 cells by hybridizing it against sonicated bare genomic DNA on the same microarray (see Methods). The resulting MNase bias profile exhibits statistically significant negative correlation with the K562 pre-extracted chromatin profile (Spearman r = −0.12, P < 10−10). This indicates that MNase sequence specificity is indeed more pronounced on bare genomic DNA than on the nucleosomal DNA, so that competitive hybridization of mononucleosome material against MNase-digested bare genomic DNA overcompensates for the MNase sequence bias. To reduce this residual contribution of the MNase bias, pre-extracted chromatin profiles were renormalized based on a local regression model describing contribution of MNase bias to the pre-extracted profile measurements (see Methods). Such renormalization reduced the correlation with the MNase bias measurement to r = −0.028 (P < 10−10).
The resulting chromatin profiles exhibit regions with regularly spaced peaks of width in the range of 150–200 bp that are expected from positioned nucleosomes (Fig. 2). These patterns are highly reproducible between biological replicates, and are also observed in samples prepared without the formaldehyde crosslinking step (see Methods). Chromosome locations occupied by well-positioned (fixed) nucleosomes were identified from the pre-extracted chromatin profiles using a hidden Markov model segmentation (see Methods). A total of 1169 fixed nucleosome positions were identified for K562 cells, and 1086 positions for HeLa cells. This indicates that >60% of nucleosomes are well positioned (67% for K562, 62% for HeLa, assuming optimal nucleosome packing with 195 bp distance). Based on the agreement between the biological replicate measurements, we estimate the error rate for fixed nucleosome predictions to be ∼20%. In addition, the predictions based on the K562 and HeLa data agree over 70% of nucleosome positions (71% of K562 nucleosome predictions, 77% of HeLa).
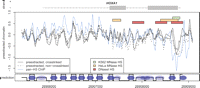
An example of measured chromatin profiles and nucleosome calls in the K562 cell line. (Top, gray) HOXA1 gene body (line) and exons (boxes). (Center main plot) Log intensity ratios for pre-extracted chromatin, with (solid lines) and without (dashed line) crosslinking. (Blue) Profile of pan-H3 ChIP measurement. (Colored bars) Positions of MNase hypersensitive sites identified in K562 cells (green), HeLa cells (orange, profiles not shown), and DNaseI hypersensitive site (red bar) identified by Sabo et al. (2006) in human lymphoblastoid cells. (Bottom track) Generated nucleosome predictions: (high values) fixed nucleosome predictions, (low values) linker regions, (intermediate values) likely positions of delocalized nucleosomes.
To validate that the observed peaks correspond to protection by nucleosome complexes, we performed ChIP–chip with a pan-histone H3 antibody, which allows for the enrichment of histone H3 regardless of covalent modifications present along the amino-terminal tail. The resulting profile strongly correlates with the pre-extracted chromatin measurements (Spearman r = 0.57, P < 10−16; see Figs. 2, 3). Overall, 94% of predicted nucleosome positions are associated with the peaks in the pan-H3 profile (50% expected by chance, 64% for control rabbit immunoglobulin antibody profile). The remaining 6% of predicted nucleosome peaks could include instances where non-histone proteins create nucleosome-sized protection or bind to nucleosomes in a manner that shields the pan-H3 epitope. To validate that the pre-extraction procedure does not have a notable effect on nucleosome positions, we have repeated the experiment for the K562 cells, omitting the pre-extraction step. The resulting profile is well-correlated with the pre-extracted measurements, and exhibits prominent peaks at the positions of the nearly all stable nucleosomes identified from the pre-extracted samples (Supplemental Fig. 13). However, the chromatin profile obtained without pre-extraction is notably noisier and exhibits additional, closely spaced peaks that most likely correspond to non-nucleosomal proteins. The identified nucleosome positions are also in general agreement with earlier predictions (Supplemental Sections 1, 2). These results strongly suggest that our methods of MNase-based nucleosome mapping accurately reflect long-range chromatin architecture.
In agreement with earlier studies (Kaye et al. 1984; Yuan et al. 2005; Ozsolak et al. 2007; Schones et al. 2008), we observe extended regions of decreased MNase protection near transcription start sites (TSS) (Supplemental Fig. 1). These MNase hypersensitive sites (MHS) are statistically overrepresented in the 500-bp region around TSS (P < 1.6 × 10−3 in K562, P < 2.8 × 10−4 in HeLa). Such MHS are observed in both transcriptionally active and silent genes. In the K562 cell line we also find a statistically significant overrepresentation of MHS 500 bp upstream of the 5′ ends of the exonic regions, even when regions within 1 kb of annotated TSS are excluded (P < 7.2 × 10−4). These MHS occurrences most likely indicate alternative TSS.
Energetically favorable sequence properties at fixed nucleosome positions
Nucleosome positioning may in part be determined by the physical properties of the underlying sequence (Trifonov 1980; Widom 2001). Nucleosomes in yeast, worm, and chicken display preferential affinity to sequences containing 10-bp AA/TT dinucleotide repeats, which is favorable for wrapping DNA around the histone core (Trifonov and Sussman 1980; Satchwell et al. 1986; Johnson et al. 2006; Segal et al. 2006). However, autocorrelation analysis of dinucleotide occurrences fails to detect any such periodicity at the fixed nucleosome positions predicted by our measurements (Supplemental Fig. 2).
It is possible that sequence patterns other than the 10-bp periodicity in dinucleotide placement may distinguish energetically favorable nucleosome positions. One approach to distinguish such positions is to estimate the deformation energy required for nucleosome formation at these sites (Sivolob and Khrapunov 1995; Anselmi et al. 2000; Lee et al. 2007; Miele et al. 2008). To evaluate if the nucleosome sequences identified in this study are favorable for nucleosome positioning, we employed a knowledge-based approach, assessing the energy cost of the DNA deformation required for a duplex of given sequence to follow the trajectory of the DNA in the experimentally determined nucleosome structures (Tolstorukov et al. 2007). This approach is based on the analysis of the anisotropic DNA deformability in a large set of protein–DNA complexes (Olson et al. 1998; see Methods).
The predicted nucleosome positions exhibit notably lower DNA deformation energy than the surrounding linker regions, indicating that such positions are favorable for nucleosome binding (Fig. 3B). The principal contribution toward the nucleosome binding energy difference comes from the increased G/C content around the predicted nucleosome dyad positions, and decreased G/C content in the linker regions (Fig. 3A; see Supplemental Fig. 3 for dinucleotide analysis).
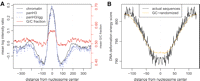
Average profiles and energy properties of nucleosome positions. (A) (Black line) Average pre-extracted chromatin log intensity ratio around fixed nucleosome positions. (Blue lines) Average pan-H3/input (solid) and pan-H3/IGG (dashed) log intensity ratio profiles, confirming that predicted fixed nucleosome positions correspond to areas of histone H3 enrichment. (Red line) Average G/C fraction smoothed with a window of 11 bp, with the G/C fraction scale shown on the left. Data from the K562 cell line are used. (B) Average nucleosome deformation energy profile around predicted fixed nucleosome positions. The score assesses the energy required to wrap a given DNA sequence around the histone core. A pronounced dip in the energy profile indicates that the predicted positions are energetically favorable for nucleosome integration. (Orange line) Expected energy profile based only on the dinucleotide properties of the predicted positions (see Methods). This illustrates that much of the energy dip can be attributed to increased occurrence of GC-rich dinucleotides around predicted nucleosome dyad positions. These dinucleotides are associated with reduced DNA deformation energy, which would facilitate histone core integration (Olson et al. 1998; Vinogradov 2003).
Fixed nucleosomes form localized groups with 195-bp periodicity
To examine long-range properties of the nucleosome organization, we first calculated the autocorrelation function of the average pre-extracted chromatin profile. A periodic pattern, if present, will manifest itself in higher autocorrelation values at distances that are multiples of the period. The autocorrelation of the chromatin profile shows prominent secondary peaks spaced 195 bp apart can be seen extending to 1200 bp (Fig. 4A). The 195-bp periodicity indicated by the average profiles is confirmed by the distribution of internucleosome distances based on the predicted nucleosome positions, which clearly shows preferred distances to be multiples of 195 bp (Fig. 4B). The statistical significance of the 195-bp period peak was established based on the randomizations of nucleosome positions (P < 10−4; Fig. 4C). Based on the width of the frequency peak, we estimate the true period to be 195 ± 7 bp. Such periodicity is apparent despite variable internucleosome distances in the bulk chromatin (Lohr et al. 1977).

Fixed nucleosomes are organized into regular periodic arrays. (A) Autocorrelation function of the average pre-extracted chromatin profiles in K562 cells. (Dashed vertical lines) Locations of secondary peaks, spaced 195 bp apart. This demonstrates presence of pronounced 195-bp periodicity in the chromatin profiles. (B) Distribution of distances between identified fixed nucleosome positions is shown for nucleosomes 10 or fewer nucleosomes away from each other. The distance distribution clearly shows preferred internucleosomal distances that are multiples of 195 bp. (C) Fourier frequency decomposition of the nucleosome distance distribution confirming periodicity peak of 195 ± 7 bp in nucleosome positions. (Gray lines) Statistical significance levels of spectral power based on random positioning of nucleosomes in the tiled regions. (D) Mean 195-bp periodicity scores for different genomic elements. The periodic nucleosome arrays are preferentially localized near the 3′ regions of annotated genes. The periodicity scores at 3′ UTR regions are on average significantly higher than scores at the 5′ proximal, 5′ UTR, and exonic regions (t-test P-values < 4.1 × 10−3, 6.2 × 10−4, 7.5 × 10−3, respectively, in K562; 0.013, 0.0013, 0.019 in HeLa). The proximal regions are defined as 2-kb segments upstream and downstream of the annotated gene boundaries. The periodicity score is calculated using local autocorrelation (see Methods).
The limited extent of the periodic peaks in the autocorrelation and distance distribution functions (Fig. 4A,B) may indicate that the periodically spaced arrangements are limited to groups of six or fewer nucleosomes. Alternatively, such periodical nucleosome spacing may be found throughout the tiled regions, but gradual loss of phase at larger distances precludes us from observing regular patterns beyond 1200 bp. To distinguish between the two possibilities we quantified the degree to which 195-bp periodicity is pronounced at different locations within the HOX clusters using local autocorrelation. We find that the periodic nucleosome placement does not occur evenly throughout the tiled regions, but is localized to specific positions (Supplemental Figs. 5, 6). Such periodic nucleosome groups tend to occur toward the 3′ end of annotated genes (Fig. 4D).
Density of fixed nucleosome positions in genic regions
Since chromatin properties are interlinked with gene regulation, we examined the relationships between fixed nucleosome density and gene expression. We find that gene body regions associated with different transcriptional states show differences in 31% of predicted nucleosomes, while only 15% are affected for regions whose transcriptional state remains the same (P < 4.8 × 10−4; Fig. 5A). We calculated pairwise differences of fixed nucleosome density for the 13 genes that are expressed only in HeLa cells, and for 23 genes that maintain the same transcriptional state in both cell lines (see Methods; Supplemental Fig. 12). On average, transcriptional activity is associated with an increase in the density of fixed nucleosome positions relative to genes where transcriptional state remained the same (P < 9.0 × 10−4). These changes appear to be evenly distributed among exonic, intronic, and untranslated regions (UTRs) (Supplemental Fig. 7). At the same time, the opposite trend is observed for predictions of delocalized nucleosomes (P < 1.5 × 10−5). Therefore, this observation likely reflects improved positioning of nucleosomes or improved ability to detect nucleosomes in transcribed regions, rather than a change in absolute nucleosomal density. It is also noteworthy that the differences between the chromatin profiles of the two cell lines do not always involve changes in nucleosome positions, but often appear to involve changes in relative occupancy within the same positioning pattern (Supplemental Section 3).
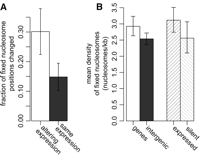
Fixed nucleosome density in gene regions. (A) Fraction of fixed nucleosome positions changing between HeLa and K562 cell lines is higher for protein-coding genes altering their transcription state (altering) and genes whose transcription state remains the same (same) between the two cell lines (t-test P-value < 0.0011). (B) Average mean nucleosome density is shown for gene body and intergenic regions. The average density for actively transcribed and silent genes is shown separately. Data from HeLa cells are used. Gene regions display higher density of fixed nucleosomes than the intergenic regions (P < 0.014); in HeLa cells, such a difference may be attributed to higher density at transcribed genes (P < 0.041). The error bars show 95% confidence intervals based on the exponential distribution models.
Overall, we observe that gene body regions possess a higher density of predicted fixed nucleosomes (Fig. 5B). The increase in fixed nucleosome density within gene bodies is statistically significant within a 95% confidence interval in both cell lines (P < 0.014 for HeLa, P < 0.029 for K562). In HeLa cells, this difference may be attributed to actively transcribed genes, whose average density is higher than at transcriptionally silent genes (P-value < 0.041); however, no significant difference can be observed in K562 cells, where fewer genes are expressed.
Discussion
Given the paramount role of chromatin in transcriptional regulation, high-throughput methods for assessing chromatin organization at the level of individual nucleosomes are of significant interest.
Using a combination of MNase digestion and high-resolution tiled microarrays we have characterized stable nucleosome positions throughout HOX gene clusters in two well-studied human cell lines. These positions exhibit a number of organizational and sequence properties that differ from those observed previously.
The energy of the nucleosome–DNA interaction is determined in part by the underlying nucleotide sequence. This has been clearly demonstrated by in vitro reconstitution of nucleosomes on bare DNA (Widom 2001). However, despite extensive investigations, the sequence determinants of nucleosome positioning and the degree of their importance in vivo remain a subject of continuing research. A 10-bp periodicity in the occurrence of AA/TT dinucleotides has been suggested to position nucleosomes in vivo (Trifonov and Sussman 1980; Satchwell et al. 1986; Ioshikhes et al. 2006; Johnson et al. 2006; Segal et al. 2006). Our analyses of fixed nucleosome positions in human HOX clusters, however, fail to detect such 10-bp periodic patterns. Instead, the identified sequences exhibit a simple trend in which the central region (100–130 bp) surrounding the nucleosome dyad is associated with increased G/C content and is flanked by linker regions with decreased G/C content. This pattern is consistent with energy models, as G/C-containing dimers have lower DNA deformation energy and would facilitate wrapping of DNA around the nucleosomes (Olson et al. 1998; Vinogradov 2003). Randomized sequence sampling indicates that while specific dimer organization of identified sequences contributes to the observed energetic minimum, the principal contribution comes from the variation in G/C-content.
The elevation of G/C content at the nucleosome positions compared with the linker regions has also been observed in the experimental measurement of nucleosome positions on continuous segments of S. cerevisiae chromosomes (Peckham et al. 2007). This is also the case for the recently derived set of nucleosome-positioning sequences in Caenorhabditis elegans (Johnson et al. 2006). Our finding is also in accord with the previous observation of increased fraction of poly(A) or poly(T) tracts in the linker regions in S. cerevisiae, characterized by lower affinity to histones due to intrinsic structural rigidity (Struhl 1985; Nelson et al. 1987; Anderson and Widom 2001; Yuan et al. 2005). Finally, such a nucleotide composition pattern is consistent with the sequence motif enrichment identified in a study of stable nucleosome positions within human promoter regions (Ozsolak et al. 2007). Our results provide a simple explanation in terms of DNA deformation energy for the observations discussed above and suggest a widespread role of this sequence pattern in nucleosome positioning in human chromatin.
While the observed G/C content trend can be potentially attributed to the MNase sequence bias, this is unlikely given very low correlation between measured MNase bias and chromatin profiles, high agreement with pan-H3 ChIP, and strong 195-bp periodicity in nucleosome placement. The lack of the expected 10-bp sequence periodicity may reflect differences in nucleosome positioning mechanisms in humans, but may also be a consequence of insufficient number or precision of the examined nucleosome positions. These questions will be addressed in detail by a subsequent manuscript (M.Y. Tolstorukov, P.V. Kharchenko, J. Goldman, R.E. Kingston, and P.J. Park, in prep.).
Studies of nucleosome positioning in yeast demonstrated variation in nucleosome density (Lee et al. 2004; Yuan et al. 2005) and coordination of nucleosome positioning around transcription start sites (Guillemette et al. 2005). In mammalian systems, nucleosome positions at promoter regions have been studied using similar mapping strategies (Ozsolak et al. 2007), and a decrease of density at the transcription start site was also observed. Our analysis extends beyond the promoters of genes to cover contiguous regions spanning a total of 0.5 Mb. We observed transcription-associated variations in nucleosome positioning, and, more unexpectedly, that transcribed genes had stronger positioning of nucleosomes than nontranscribed genes. This is consistent with previous data suggesting that transcription itself rearranges nucleosomes; however, it is surprising that this rearrangement leads to increased positioning as opposed to randomization. While a number of earlier studies have noted corresponding decreases in the nucleosome repeat lengths in actively transcribed genes (Gottschling et al. 1983; Villeponteau et al. 1992; Cavalli and Thoma 1993), transcriptional activity in yeast has been linked to an overall decrease in nucleosome density within highly transcribed genes (Lee et al. 2004). Our findings are also supported by the recent high-throughput sequencing study of human nucleosomes (Schones et al. 2008), which found that phased nucleosome arrays are observed around the TSS of expressed, but not silent, genes. We note that the observed differences are small in magnitude and potentially can be attributed to an improved ability to detect stable nucleosome positions within actively transcribed regions, for example as a result of increased MNase accessibility or chromatin extraction efficiency. It is also possible that the observed changes within the HOX genes may be gene-specific; therefore, an assessment of nucleosome density on a larger scale would be necessary to establish the predominant trend.
Higher-order organization of chromatin fibers is of a great interest due to its potential to illuminate the mechanisms involved in activation and repression of genomic loci. Such organization may be reflected to some degree in the linear arrangement of nucleosomes along the chromosome. We found periodic positioning of nucleosomes near the 3′ regions of HOX genes spanning up to 1200 bp, suggesting an importance for regularly spaced nucleosomes in these regions. While the presence of such repeats does not appear to be directly correlated with gene transcription, their localization near the 3′ ends may reflect requirements of transcriptional termination, as has been suggested for other organisms (Alen et al. 2002).
Together, these results describe an active organization of chromatin structure throughout the examined HOX gene clusters that does not conform to the nucleosome positioning observations from other eukaryotes. Our results indicate the significant potential for uncovering novel levels of regulation via mapping of nucleosome positions over large chromosome regions in mammals.
Methods
Cell culture
K562 and HeLa S3 cells were obtained from ATCC and maintained in culture at the National Cell Culture Center, NCCC (Bethesda, Maryland). K562 cells were grown in RPMI and 10% newborn calf serum. HeLa S3 cells were grown in Joklik’s modified MEM supplemented with 5% newborn calf serum.
Pre-extraction and crosslinking conditions
K562 and HeLa cells were washed with cold PBS (Ca/Mg-free) and the pellets were shipped on wet ice overnight from the NCCC. Under pre-extraction conditions, the K562 and HeLa cells were resuspended with 30 mL of PBS supplemented with 150 mM NaCl, 0.2% Tween-20, 0.2% Triton-X 100 for 5 min at room temperature. The cells were crosslinked with one-tenth volume of a freshly prepared 11% formaldehyde crosslinking solution (50 mM HEPES–KOH at pH 7.5, 100 mM NaCl, 1 mM EDTA, 0.5 mM EGTA, and 11% formaldehyde) for 10 min at room temperature. The formaldehyde crosslinking reactions were quenched with glycine to a final concentration of 125 mM by incubating for 2 min on ice. Cells were washed with 50 mL of cold PBS twice at 4°C.
Micrococcal nuclease digestion
The K562 cells were resuspended at ∼1.5 × 107 cells/mL in TM-2 buffer (10 mM Tris at pH 7.4, 2 mM MgCl2) and incubated for 5 min at on ice. Triton-X 100 was added to 0.5% (v/v). Cells were lysed by one passage through a 25-gauge needle fitted to a 10-mL plastic syringe. Nuclei were pelleted by centrifugation for 5 min at 500g at 4°C, washed once in TM-2 buffer, resuspended in STM-5 buffer (10 mM Tris at pH 7.4, 0.25 M sucrose, 5 mM MgCl2), and spun for 10 min as before. Nuclei were resuspended in buffer W (10 mM Tris at pH 7.4, 15 mM NaCl, 60 mM KCl) and passed through a 0.45-μM cellulose filter. Leupeptin, aprotinin, PMSF, and pepstatin (Roche) were added freshly just before the buffers were used.
The HeLa cells were lysed by gently resuspending the pellet at ∼1.5 × 107 cells/mL in buffer H (10 mM Tris at pH 7.4, 15 mM NaCl, 60 mM KCl, 1 mM EDTA, 5% [w/v] sucrose, 0.2% Nonidet P-40), incubated for 5 min on ice, and dounce homogenized with 10 strokes with a B pestle.
The nuclear suspension was carefully layered on a sucrose cushion (10 mM Tris at pH 7.4, 15 mM NaCl, 60 mM KCl, 1 mM EDTA, 10% [w/v] sucrose) and spun for 5 min at 500g. Nuclei were resuspended in buffer W (10 mM Tris at pH 7.4, 15 mM NaCl, 60 mM KCl) and passed through a 0.45-μM cellulose filter. Leupeptin, aprotinin, PMSF, and pepstatin (Roche) were added freshly just before the buffers were used.
CaCl2 (0.1 M) was added to each nuclei cutting reaction at a final concentration of 2.2 mM, and the appropriate concentrations of MNase (0–300 units/mL; Worthington) were added. Reactions were mixed well, incubated for 15 min at 25°C, and then stopped by adding 50 μL of Proteinase K (20 mg/mL, Roche) and a stop solution (50 mM Tris-HCl, 20 mM EDTA, 1% SDS). Reactions were incubated for 2 h at 55°C, and crosslinks were reversed overnight at 65°C.
The undigested DNA was purified with phenol:chloroform:isoamyl alcohol (Sigma). The purifications were repeated until the interface was clear. Water was extracted with isobutanol, and the DNA was precipitated with ethanol and resuspended in ddH2O. DNA was reprecipitated with 3 M NaOAc and ethanol. Pellets were washed in 70% EtOH, and DNA was resuspended in ddH2O and run on a 3% metaphor agarose gel (Cambrex) to isolate the mononucleosome-sized band. The mononucleosome-sized DNA was eluted from the gel and purified with phenol:chloroform:isoamyl alcohol. The DNA was precipitated and washed as stated above. Samples were resuspended and sent to Nimblegen for labeling and hybridization.
Bare genomic DNA was isolated from nuclei and treated with proteinase K. Formaldehyde crosslinks were reversed overnight at 65°C. DNA was purified and precipitated, and bare genomic DNA was digested with a range of micrococcal nuclease concentrations to obtain a smear ranging from 1 kb to 100 bp. MNase-digested bare genomic DNA was sent to Nimblegen for labeling and hybridization as the control.
Approximately 107 cells were used for each biological experiment. Half of the nuclei were used to obtain genomic DNA. The other half was used in an MNase digestion reaction. After RNaseA treatment and proteinase K digestion, the DNA was purified and precipitated. The DNA was run on a gel and mononucleosomes were isolated. This would be ∼70%–80% of the total DNA. Assuming that a loss of DNA occurs with every step of the processing, we estimate that the equivalent of 2 × 106 human genomes were sent for hybridization.
ChIP–chip
After chromatin samples were treated with MNase, reactions were stopped with the addition of EDTA to 5 mM and Na3EDTA to 5 mM. One microgram of anti-H3, CT, pan (Upstate), or rabbit IgG (Santa Cruz) antibodies were coupled to Protein A Dynabeads (Dynal). Digested chromatin samples were directly added to the antibody–Dynabead complex for incubation overnight at 4°C. Dynabead–antibody–chromatin complexes were washed with RIPA buffer, eluted, and treated with RNase A (Roche) and proteinase K (Roche). DNA was purified and sent to Nimblegen for labeling and hybridization.
Overall, two biological replicate measurements of pre-extracted chromatin profiles were obtained for HeLa and K562 cells, with one additional measurement without crosslinking for K562 cells. To evaluate MNase sequence bias, MNase-digested bare genomic DNA was competitively hybridized against sonicated bare genomic DNA. The bare genomic DNA was sonicated with a Misonix 3000 sonicator on wet ice for seven cycles at 30 sec “on” and 60 sec “off.” After sonication, the DNA was centrifuged at 20,000g for 10 min at 4°C to pellet debris. The supernatant containing the sonicated DNA was precipitated, resuspended, and used as the sonicated bare genomic DNA sample for hybridization to the microarray. Two such measurements were conducted using different dye assignments, with both measurements showing consistent results.
Microarray designs
385,000-feature microarrays from Nimblegen were designed across the HOXA, HOXB, HOXC, and HOXD clusters at 5-bp resolution with 60-mer oligonucleotides, based on NCBI build 35.1 of the human genome. Both strands of the DNA sequence were tiled and in duplicate on the array. All experimental DNA samples were labeled with Cy5 and all digested bare genomic control DNA samples were labeled with Cy3.
Gene expression
Total RNA was isolated with TRIzol (Invitrogen) from cells. RNA was treated with RNase-free DNaseI (Roche) for 30 min at room temperature. The reaction was stopped with 1 mM EDTA and heated for 15 min at 65°C. The DNaseI-treated RNA was used in a first-strand cDNA synthesis reaction using Superscript III reverse transcriptase (Invitrogen) with random hexamer primers. TaqMan Universal PCR master mix (Applied Biosystems) was used with validated TaqMan probe and primer sets for HOXA5, HOXA9, HOXA11, HOXB2, HOXC10, HOXC11, HOXD10, HOXD11, and the TaqMan endogenous controls HPRT1 and PGK1 with cDNA from HeLa or K562 cells. Reactions were read using the Applied Biosystems 7500 Real Time PCR system.
The mRNA levels across all tiled regions were evaluated by hybridizing the resulting cDNA to the same tiling microarrays. Total RNA was isolated using TRIzol (Invitrogen). The RNA was treated with DNaseI (Roche) before 2 μg of each was used to generate cDNA with the Superscript III kit (Invitrogen). Random hexamers were used to prime the cDNA synthesis. The cDNA was purified with phenol:chloroform:isoamyl alcohol and precipitated before being sent to Nimblegen to be analyzed for gene expression. The resulting profiles are showing in Supplemental Figure 12. We find microarray and real-time PCR results in perfect agreement with each other.
Normalization of microarray data
The log intensity ratio values (M) for all measurements were corrected using M-A loess normalization, where A is the total log intensity. Briefly, a smooth curve was fit (using R loess function with smoothing span of 0.05) and subtracted from the initial M values. To correct for MNase sequence bias, each pre-extracted chromatin measurement was normalized based on a generalized linear model (Wood 2006): M = s(A) + s(N), where M and A are the log intensity ratio and total log intensity of the chromatin measurement, N is the log intensity ratio of the MNase bias measurement on bare genomic DNA (comparing MNase-digested with sonicated bare genomic DNA, measurement #1), and s() is a spatial smoothing function (penalized regression splines were used with implementation provided by R the mgcv package). The probes were then binned into groups of 3000 probes according to the values of M predicted by the model. The original M values within each bin were normalized so that variance and mean are equal across all bins.
Detection and analysis of fixed nucleosome positions
Positions of fixed nucleosomes were identified using an HMM method similar to that used by Yuan et al. (2005). Large-scale trends were removed by subtracting a loess curve fit with 400-bp smoothing span. The HMM contained 18 hidden states: 16 states describing fixed nucleosome peaks, one state describing linker regions, and one for the intermediate state. To minimize the number of states and associated parameters, each observed state included information from two adjacent probes (treated as replicate measurements). The HMM parameters were estimated based on the data from all tiled regions simultaneously, with separate parameters for each measurement replicate. Positive and negative strand probes were treated as separate replicates. The nucleosome calls were determined based on the fixed nucleosome probability peaks (P > 0.1) as determined by the Viterbi algorithm.
To estimate the error rate in the identification of stable nucleosome positions we have compared predictions from different biological replicates. Specifically, predictions based on any two K562 biological replicates were compared with the predictions based on the third replicate, and the average of all comparisons was used. The nucleosomes were considered present in both predictions if they overlapped by at least 50%.
MNase hypersensitive sites
Hypersensitive sites were identified as regions significantly below the mean (threshold determined based on lowest 10% of
the signal after de-trending described in the previous section) that extend over 60 bp. To assess statistical significance
of MHS occurrences near TSS (or near exon starts), positions of the MHS segments were randomized within the HOX clusters. A total of 4 × 104 randomizations were performed. The P-values corresponding to the actual number of MHS observed near 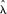 (χ
(χ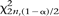 /2n),
/2n), 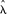 (χ
(χ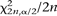 /2n) TSS were then calculated from this empirical distribution.
/2n) TSS were then calculated from this empirical distribution.
Nucleosome sequence analysis
The DNA deformation energy profiles were calculated as described by Tolstorukov et al. (2007). Briefly, we calculated threading scores for the 147-bp tiling DNA fragments (1-bp shift) around each fixed nucleosomal position using the DNA structure from the nucleosome core particle best resolved to date (Davey et al. 2002) as a template. The threading template was symmetrized relative to the dyad position. To obtain the average deformation profile, the individual profiles were averaged position-wise. The empirical force field used for the threading calculations is based on the analysis of a large data set of crystal structures of protein–DNA complexes and accounts for all six principal conformational parameters of a dinucleotide step (Twist, Tilt, Roll, Slide, Shift, and Rise) (Olson et al. 1998). Randomized energy profiles were generated by taking a 147-bp window around each position, generating 100 random sequences with the same average dinucleotide composition, and taking an average energy score of these sequences as the randomized energy profile value at this position.
Periodic nucleosome placement
The autocorrelation function of the pre-extracted chromatin profiles was determined as a sum of autocorrelation functions of individual HOX clusters. Internucleosome distance distribution was determined by calculating the density of all distances between nucleosomes that are 10 or fewer nucleosomes apart. The density shown in Figure 5B was determined based on a Gaussian kernel with a standard deviation of 30 bp. The power spectrum of the distance distribution was calculated based on the 3-bp Gaussian smoothing.
Local autocorrelation was calculated as a sum of Spearman rank correlation coefficients between the pre-extracted chromatin profile at a given position and the profile at distances np, where p is the desired period (195 bp) and n = (−3, −2, −1, 1, 2, 3).
Nucleosome density
To assess the significance of the differences in density of fixed nucleosome positions, the occurrence of fixed nucleosomes
along the chromosome was modeled using exponential distribution. The 95% confidence intervals on the rates (Fig. 5B) were calculated as,
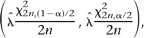 where
where 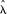 is the maximum likelihood estimate of fixed nucleosome rate (nucleosomes/Kbp), n is the number of fixed nucleosomes, and χ
is the maximum likelihood estimate of fixed nucleosome rate (nucleosomes/Kbp), n is the number of fixed nucleosomes, and χ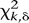 is the inverse CDF value of χ2 distribution with k degrees of freedom and probability δ. The 95% confidence interval plotted corresponds to α = 0.05. The statistical significance
of the nucleosome rate difference was calculated based on the rate ratio, from F distribution.
is the inverse CDF value of χ2 distribution with k degrees of freedom and probability δ. The 95% confidence interval plotted corresponds to α = 0.05. The statistical significance
of the nucleosome rate difference was calculated based on the rate ratio, from F distribution.
Acknowledgments
We thank John Rinn for sharing the design of the tiling microarray. This research was supported by NLM training fellowship T15LM007092 (P.V.K.), NSRA fellowship F32 GM062265 (C.J.W.), NIH R01 GM082798 (P.J.P.), and NIH grant GM49301 (R.E.K.).
Footnotes
-
↵5 Corresponding authors.
↵5 E-mail peter_park{at}harvard.edu; fax (617) 525-4488.
↵5 E-mail kingston{at}molbio.mgh.harvard.edu; fax (617) 643-2119.
-
[Supplemental material is available online at www.genome.org. The sequence data from this study have been submitted to GEO under accession no. GSE10042.]
-
Article published online before print. Article and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.075952.107.
-
- Received December 29, 2007.
- Accepted June 24, 2008.
- Copyright © 2008, Cold Spring Harbor Laboratory Press








