
Multiple sequence alignment: In pursuit of homologous DNA positions
Abstract
DNA sequence alignment is a prerequisite to virtually all comparative genomic analyses, including the identification of conserved sequence motifs, estimation of evolutionary divergence between sequences, and inference of historical relationships among genes and species. While it is mere common sense that inaccuracies in multiple sequence alignments can have detrimental effects on downstream analyses, it is important to know the extent to which the inferences drawn from these alignments are robust to errors and biases inherent in all sequence alignments. A survey of investigations into strengths and weaknesses of sequence alignments reveals, as expected, that alignment quality is generally poor for two distantly related sequences and can often be improved by adding additional sequences as stepping stones between distantly related species. Errors in sequence alignment are also found to have a significant negative effect on subsequent inference of sequence divergence, phylogenetic trees, and conserved motifs. However, our understanding of alignment biases remains rudimentary, and sequence alignment procedures continue to be used somewhat like benign formatting operations to make sequences equal in length. Because of the central role these alignments now play in our endeavors to establish the tree of life and to identify important parts of genomes through evolutionary functional genomics, we see a need for increased community effort to investigate influences of alignment bias on the accuracy of large-scale comparative genomics.
The relative positions of nucleotides within the same gene in different species and in duplicated genomic regions are disturbed by insertion and deletion of stretches of DNA over evolutionary time. This leads to differences in the length of the homologous regions in the genome, with more distant relatives having a higher likelihood of sequence length difference. A comparison of lengths of genome segments spanning protein-coding genes in human and mouse shows the extent of the effect of evolution by insertions and deletions (Fig. 1). The lengths of noncoding orthologous sequences have also evolved substantially after divergence over 90 million years ago. A grand challenge in comparative genomics is to line up these bases by inserting gaps in sequences, because genomic analyses must be based on comparisons between bases at positions (sites) that coincided in a common ancestor. The task is to re-establish (estimate) the ancestral site-wise homology obfuscated by the insertion–deletion and substitution processes. Naturally, this operation has come to be known as “alignment,” and the resulting set of sequences, all of which are the same length (taking gaps in to account), is also called an alignment (Fig. 2). We can distinguish between “pairwise” alignments, in which sequences, even if they are part of a larger set, are aligned only in pairs, and “multiple” alignments, in which more than two sequences are aligned simultaneously.
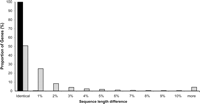
Frequency distributions of percent difference in total coding sequence length (exons only) between human and mouse orthologs (light gray) and between human and chimpanzee orthologs (black) for 7645 protein-coding genes. Forty-nine percent of the genes exhibit some length difference between human and mouse; human and chimpanzee orthologs exhibit length difference in only 17 of these 7645 genes. Data from Clark et al. (2003).

Example of an alignment. A phylogenetic tree is shown for the taxa (rows), and a G+C content profile is shown for the sites (columns).
Alignment procedures may also be classified as either “global” or “local.” In the simplest form, sequences are aligned beginning to end to produce global alignments. This is appropriate for sequences of protein-coding genes and for short stretches of the genomic sequence. For longer genomic DNA, it is necessary to account for medium and large-scale rearrangements in addition to large sequence insertions and deletions, which necessitates the building of local alignments. Local alignments differ from global alignments in that the former focus on shared regions of high similarity while ignoring regions that do not show high sequence homology between sequences. Unlike traditional global and local alignment methods that assume colinearity of homologous segments among sequences, “glocal” alignment methods model the rearrangement process explicitly during the alignment procedure itself and result in a nonlinear mapping between homologous regions of different sequences (Brudno et al. 2003b).
For most applications in the areas of molecular phylogenetics and evolution, we are interested in properties and relationships of “rows” of the alignment, which represent species, genes, or genomic regions (Fig. 2). Examples of these include inference of multigene family and species phylogenies, determination of evolutionary rates in different lineages, and identification of bouts of selection and patterns of DNA sequence change over time (Nei and Kumar 2000; Felsenstein 2002). The second use of sequence alignments (local as well as global) stems from the need to understand properties of individual or groups of contiguous “columns.” A common example is the building of genomic profiles of conservation and divergence in different DNA positions or sets of contiguous positions (genomic regions). These applications are considered to fall in the area of functional genomics (Hardison 2000; Gaucher et al. 2002; Town 2002; Koonin and Galperin 2003; Pevsner 2003). Of course, many studies involve joint analyses of rows and columns, but from the perspective of accuracy, the requirements of these two alignment usages are quite different. In the following sections, we assess the existing knowledge of the fidelity of the alignment process, focusing primarily on finding motifs, estimating sequence divergence, and inferring phylogenetic relationships.
Practical sequence alignment
The amount of nucleotide sequence data in GenBank and other public databases has expanded exponentially since the inception of these electronic warehouses; the data now consist of over 125 billion base pairs of sequence data from over 200,000 organisms. Understanding the functional significance of these data has become the central problem in comparative genomics. Naturally, because of this great volume of data, scientists would like to establish DNA homologies by applying one or more of the highly innovative alignment methods available today in an automated high-throughput fashion (Thompson et al. 1994; Eddy 1995; Morgenstern et al. 1998; Notredame et al. 2000; Brudno et al. 2003a; Covert et al. 2004).
The process of alignment involves insertion of gaps into sequences to make them the same length. These gaps are hypotheses about the site homologies resulting from historical insertion–deletion events. Since mutations can cause two homologous sites to differ from each other (substitutions), the complexities of the alignment process transcend traditional string-matching problems in computer science. The interplay of insertion–deletion events and substitutions over thousands and millions of years produces sequences that may lead to many different alignments, with some containing more gaps than others. One may prefer a specific alignment over another if some “optimality” score is better (Needleman and Wunsch 1970). Such a score typically represents some function of the numbers and positions of columns that contain identical bases, different bases, or gaps. Each difference is penalized and there are penalties for inserting and extending gaps.
The scoring functions incorporate differences in the likelihood of change from one base to another and of inserting of gaps of various lengths. It is well-established, for example, that transitional mutations are much more common than transversional mutations (Vogel and Kopun 1977; Rosenberg and Kumar 2003). Hence, transitional differences are penalized less (i.e., they are allowed in the alignments more frequently). Longer gaps are also known to be rare as compared with shorter ones, so alignments containing long gaps are favored less (Waterman et al. 1976; Gu and Li 1995; Qian and Goldstein 2001; Miklos et al. 2004). However, the exact value to be assigned to each parameter is difficult to determine, as it is expected to differ between closely and distantly related species and vary from species to species (Vingron and Waterman 1994; Wheeler 1995; Zhu et al. 1998; Yuan et al. 1999). Although progress has been made in establishing biological justification for the use of some scoring schemes, a comprehensive theoretical framework for gap and substitution penalties still eludes us (Gu and Li 1995; Miklos et al. 2004). Thus, the optimal alignment under a specific scoring scheme may not be the true one (Landan 2005; Morgenstern et al. 2006). In fact, many different alignments for the same sequence set may be equally optimal under the scoring scheme chosen, and it is often difficult to choose among them (Fitch and Smith 1983; Wheeler and Gladstein 1994; Vingron 1996; Cerchio and Tucker 1998; Landan 2005). Furthermore, the accuracy of the alignments obtained using common default values of alignment parameters is only slightly worse than those obtained were we to know the true penalties for inserting gaps and allowing base substitutions (Landan 2005). This means that the absence of knowledge of true parameter value does not substantially affect the alignment accuracy, although it also points to the need for developing methods wherein the true parameters can be used more effectively.
Insurmountable computational demands limit our ability to generate optimal alignment for more than a few sequences. Finding an optimal alignment for a single pair of sequences, even very long genomic ones, has become practical using dynamic programming methods that now require running times and memory requirement proportional to the total lengths of the two sequences (Needleman and Wunsch 1970; Myers and Miller 1988; Delcher et al. 2002; Kurtz et al. 2004). However, the time requirements go up exponentially with increasing numbers of sequences and quickly exceed the capabilities of available computers, even when cleverly optimized (Lipman et al. 1989). Therefore, all practical applications today resort to the use of heuristics when aligning more than a few sequences. Progressive alignment is the most commonly employed heuristic procedure. In this method, a multiple sequence alignment is generated in a step-wise fashion by iteratively aligning pairs of individual sequences or aligned groups of sequences (Barton and Sternberg 1987; Feng and Doolittle 1987; Taylor 1988; Thompson et al. 1994; Brudno et al. 2003a; Bray and Pachter 2004). Because the algorithm needs to start somewhere, a hierarchical alignment order is first created by determining which pairs of sequences and pairs of groups of sequences are the most similar. This order is often referred to as the “guide tree.” Using progressive alignment, it was possible to align one thousand short (<500 base pairs each) sequences in a little over an hour using an ordinary PC workstation, and this time was reduced to less than seven minutes using special-purpose hardware (Oliver et al. 1992).
In heuristic approaches, no overall optimal alignment is sought. Instead, it is hoped that optimizing pairwise alignments will lead to a “good” solution. The time efficiency of this approach has led to its becoming the standard, as reflected in its implementation in a variety of well-used software packages (Thompson et al. 1994; Notredame et al. 2000; Brudno et al. 2003a). While some other methods are also available, they have not yet become mainstream because of the lack of unequivocal superiority over classical methods (for review, see Eddy 1995; Notredame and Higgins 1996; Morrison and Ellis 1997; Morgenstern et al. 1998; Thompson et al. 1999; Pollard et al. 2004). Finally, in light of the fact that over 75 approaches to the multiple alignment problem are available (M.S. Rosenberg, pers. comm.), methods have been developed to produce a single consensus alignment out of many (Wallace et al. 2006). This consensus approach remains to be thoroughly evaluated.
Up to this point, we have primarily focused on DNA sequence alignments, but the discussion applies in principle to the alignment of amino acid sequences. In the latter, the probability of substitution from one amino acid to another is incorporated by using 20 × 20 scoring matrices (e.g., PAM and BLOSUM) to accommodate differences in different types of amino acid substitutions (Dayhoff et al. 1978; Altschul 1991; Henikoff and Henikoff 1992). Furthermore, the alignment of coding DNA sequences of exons is regularly carried out by first aligning the translated amino acid sequences and then adjusting the DNA sequence to reflect the protein alignment. In addition to avoiding the disruption of coding frames, this approach offers the added benefit of achieving better alignment accuracy, because amino acid changes accumulate more slowly than DNA base changes (Zhang et al. 1997; Nei and Kumar 2000).
Finding functionally important motifs via sequence alignment
A major current focus of study in comparative genomics is the identification of short motifs important for gene regulation. Many motif discovery tools have been developed with the common approach to construct a multiple alignment of homologous sequences and identify short stretches of DNA positions that are more conserved over disparate genomes than would be expected by chance (see Stojanovic et al. 1999; Stormo 2000; Keich and Pevzner 2002; McCue et al. 2002; Bulyk 2003; Sinha et al. 2004; Tompa et al. 2005). Accurate motif discovery is known to pose challenging requirements for a multiple alignment program. The data used need to contain sufficiently numerous and diverged species to allow distinction to be made between short significantly conserved (potentially functional) regions and regions that are conserved by chance alone. ENCODE and other projects have addressed this situation for at least some groups of species by providing long homologous DNA segments for many closely related species (particularly mammals) along with some distant relatives of humans (ENCODE Project Consortium 2004; http://www.genome.gov/10005). The evaluation of the success in finding functionally important segments of DNA (motifs) requires knowledge of the accuracy of current procedures for aligning homologous sites correctly in all the sequences in the data set (perfect columns) as well as the accuracy of aligning successive DNA positions without gaps. Surprisingly, no investigations appear to have tackled both of these questions together in a comprehensive fashion; however, results from some specific studies may be tied together to obtain useful insights.
The accuracy of alignment algorithms has been assessed directly by measuring the fraction of positions that are aligned correctly between sequence pairs in multiple sequence alignments in computer-simulated data (Altschul and Gish 1996; Thompson et al. 1999; Pollard et al. 2004; Rosenberg 2005a). This metric is referred to as “homology accuracy” here. As expected, homology accuracy is found to be lower for distantly related sequences irrespective of the algorithm used (Fig. 3). The usual pattern is that the fraction of positions aligned correctly declines with increasing sequence divergence, with a rapid decline apparent once the difference between DNA sequences is greater than 35% (Fig. 3A). This point corresponds to one base substitution for every two positions, which is close to an evolutionary distance of 0.5 substitutions per site (Pollard et al. 2004; Rosenberg 2005a). This trend holds for several well-known alignment tools; all of them become ineffective by the time the sequence divergence reaches one substitution per site (even though their accuracy diminishes at different rates). To put these divergences in an evolutionary perspective, sequence divergences close to 0.5 substitutions per site are often reported for the comparison of neutrally evolving positions in human and mouse, whereas mouse–chicken and Drosophila melanogaster–D. pseudoobscura divergences are expected to exceed one substitution per site (Kumar and Subramanian 2002; Tamura et al. 2004).

Graphs showing how the accuracy of pairwise alignment varies with evolutionary distance in computer simulations with only insertion–deletions (A) and insertion–deletions together with the constraint that 20% of all DNA positions are occupied by interspersed highly conserved blocks that evolve 10 times slower than other positions (Pollard et al. 2004) (B). The homology accuracy is calculated as a fraction of simulated positions that were aligned correctly, and it is plotted against the fraction of sites different, as reported in Figures 2 and 4 of Pollard et al. (2004). Even though the same set of evolutionary distances is simulated for both panels, the percent sequence difference in B is smaller for a given simulated distance because of the existence of highly conserved blocks. Programs compared for homology accuracy are ClustalW (Thompson et al. 1994), BLASTZ (Schwartz et al. 2003), DiAlign (Morgenstern 1999), and Lagan (Brudno et al. 2003a).
Limits on the homology accuracy in Figure 3A are for DNA segments in which all positions are evolving strictly neutrally (i.e., without any natural selection). However, a more realistic scenario is to consider situations in which genomic segments contain highly conserved blocks, because natural selection acts to keep important motifs intact to maintain function. As expected, the existence of conserved blocks enhances the homology accuracy and makes the performance of different methods more similar. Differences do exist, however. Global alignment programs (e.g., ClustalW) perform worse than the procedures that are essentially local in nature (e.g., Lagan, DiAlign), especially for highly divergent sequences (Fig. 3B). The local alignment methods work better because they look for regions of very high sequence similarity and, thus, evolutionary conservation (Smith and Waterman 1981). The presence of large- and small-scale rearrangements will also severely affect the quality of the global alignments (Morgenstern et al. 1998; Siddharthan 2006).
The probability of aligning multiple adjacent sites correctly in a set of sequences is a direct function of the homology accuracy at each position, so it is evident that identifying short genomic segments involved in gene regulation via comparative sequence analysis is likely to produce many false-negatives if the sequences involved are highly divergent. This is clearly seen in a dramatic decline in finding short motifs after the addition of a nonmammalian species (chicken) to a data set containing placental mammals (human, chimp, mouse, and rat) (Prakash and Tompa 2005) (Fig. 4). Because the bird–mammal sequence divergence is expected to be about three times the divergence between human and mouse based on the timing of their divergence (Hedges and Kumar 2003), the inclusion of an avian sequence demands that the alignment methods correctly align DNA bases that have experienced up to 1.5 mutations per site. The homology accuracy is very low at such high divergences, as mentioned above, and the chances of correctly aligning conserved motifs are small (Fig. 3). Of course, it is possible, and would not be surprising, that fewer conserved motifs are found between birds and mammals simply because fewer such motifs exist (Cooper et al. 2005).

Graph showing the performance of different multiple sequence alignment programs in detecting conserved motifs of length 10 in sets of orthologous 1000 base-pair putative promoter regions of vertebrate genomes (Prakash and Tompa 2005). The axes represent the fraction of alignments (using the given program) of orthologous sequences in which at least one perfectly conserved motif of length 10 was detected. The x-axis represents 5073 alignments containing human, chimp, mouse, and rat sequences, while the y-axis represents 945 alignments of human, chimp, mouse, rat, and chicken. The smaller number of hits in the latter set is partly attributable to fewer conserved motifs and partly to increased difficulty of detection. The programs used are ClustalW (Thompson et al. 1994), MAVID (Bray and Pachter 2004), DiAlign (Morgenstern 1999), MLagan (Brudno et al. 2003a), TBA (Blanchette et al. 2004), and FootPrinter (Blanchette and Tompa 2003). Data are from Figure 2 of Prakash and Tompa (2005).
The use of multiple sequences and the choice of species critically impacts motif discovery. Certainly, pairwise sequence analysis can be carried out in the absence of a robust phylogeny (Elnitski et al. 2003), but this is less informative because of the difficulty in detecting multiple substitutions at the same site, and because of an inability to distinguish the direction of sequence change (Bergman and Kreitman 2001). Also, the use of multiple sequences improves statistical power to detect short conserved elements by bringing more data to bear on the problem (Sumiyama et al. 2001; Gottgens et al. 2002; Brudno et al. 2003a; Thomas et al. 2003). The term “data” here refers not only to the sequence length and the total number of differences among sequences but also to the distribution of sequence differences among the species represented. For example, a set of several closely related (e.g., primate) sequences tends to be more informative than a single pair of sequences (e.g., mouse and human), even though the total number of substitutions in the evolutionary history of the sequences considered is identical. We expect better homology accuracy in primate sequence alignments than that between human and mouse sequences, so we expect to find more highly conserved motifs when using the primate data set. This was indeed the case when a group of primate species was used to provide essentially the same amount of collective additive phylogenetic divergence as exists between mouse and human (Boffelli et al. 2003).
Many DNA segments will be found to be conserved in closely related species, but such false positives can be detected by using many sequences and by estimating false-positive rates (Cooper et al. 2005; Prakash and Tompa 2005). Still, all conserved motifs found may not be functional, and the pursuit of functionally important motifs via evolutionary conservation profiles implicitly assumes that the same motif(s) are functional in different species. The latter assumption is expected to be satisfied in analyses involving closely related species as compared with distantly related species (Cooper et al. 2005). Therefore, the species chosen are critical in multispecies analysis to find candidate, functional motifs.
Knowledge of correct evolutionary relationships is important in motif discovery. Efforts have been made to determine how the use of shared ancestry (specified using phylogenetic relationships) enhances the accuracy of motif detection over simple treatment of sequences as an aligned set (Dubchak and Frazer 2003; Sinha et al. 2004; Siddharthan et al. 2005; Gertz et al. 2006). However, the effect of using incorrect phylogeny on the accuracy of motif-discovery remains to be measured. Intuitively, it is clear that if two sister species are placed distantly in a phylogeny, then statistical methods will spuriously identify conserved regions as candidate motifs, even though they could be explained by chance alone if the correct phylogeny were to be used. Likewise, placing species nearer than they ought to be would discount the significance of similarities and lead to false-negatives. There is an urgent need for quantification of the false-positive and -negative rates in motif discovery caused by the use of incorrect phylogenetic trees.
Estimating sequence divergence
Evolutionary distances are routinely estimated from pairs of aligned sequences and are used for inferring phylogenies, divergence times, and rates of evolution (Nei and Kumar 2000). What effect does the alignment process have on distances estimated in this way? It appears that as long as the evolutionary distance is less than about one substitution per site between sequences, evolutionary distances estimated from pairwise alignments of DNA sequences are relatively insensitive to even large amounts (50%) of alignment error (Rosenberg 2005a). In contrast, and as expected, computer simulations exploring the effect of the use of a wide range of alignment parameter values leads to distances that often fall outside the 95% confidence interval around the optimal estimates (Fleissner et al. 2000, 2005) (Fig. 5), because a large fraction of alignment parameter values are likely to be biologically unrealistic. There is no single default set that works over a wide range of true distances and insertion–deletion models, even though methods are available to help select good values for alignment parameters (e.g., Holmes and Durbin 1998).
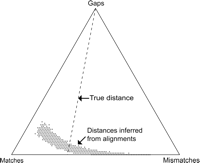
A barycentric representation of the distribution of matches, mismatches, and gaps for optimally aligned pairs of sequences having exactly 60 matches, 30 mismatches, and 10 gaps each over the entire range of alignment parameters (match, mismatch, and gap penalties) for which mismatches are penalized more than matches. Each point in the triangle represents a number of matches, mismatches, and gaps summing to 100. The lengths of the perpendiculars from a point in the diagram to the right, left, and bottom side of the triangle are proportional to the corresponding match, mismatch, and gap fractions, respectively. Simulation outcomes are represented by the dark gray points near the bottom of the triangle. In 1000 trials, the true sequence-pair description (60 matches, 30 mismatches, 10 gaps) was never recovered, and only those 1.1% of the alignments lying precisely on the line labeled “true distance” yielded estimated distances equal to the true distance (1/3) between the original pair of sequences. Adapted, with permission, from Figure 2.3 of Fleissner 2003.
Multiple sequence alignments are considered better than pairwise alignments because more similar sequences will act as intermediates between highly dissimilar ones (Lesk 2005). How does adding a third sequence to an alignment problem affect the accuracy of subsequently estimated distances? In a direct two-sequence comparison, a branch to a third sequence was added at varying intermediate points between the two. This improved the accuracy of estimated distance between the original two, but the highest accuracy for the estimates of evolutionary distance was not achieved when the maximal homology accuracy was found. Generally, the estimated distance between the two original sequences increased fairly linearly as the origin of the added branch was moved closer to the common ancestor of the original pair and the true evolutionary divergence was less than one substitution per site (Rosenberg 2005b).
Another measure of homology accuracy useful for motif detection is the fraction of sites successfully aligned when sequences are added one-by-one to the data set. This is used when the true sequence alignment is not known, as in empirical data analysis. Indeed, the number of sites aligned between two distantly related sequences increases as the data set is expanded by adding more sequences intermediate to two distantly related sequences in question (Margulies et al. 2006). This is similar to the effect observed for homology accuracy in the simulated data (Rosenberg 2005b), with the caveat that the relative divergence of the additional sequence is critically important.
Effect of alignment homology inaccuracies on phylogenetic inference
The common modus operandi for building phylogenetic trees is to align a data set using some program, inspect the result for obvious error, perhaps realign, and then infer a phylogeny from the result. Current computer simulations aimed at deciphering the effect of sequence homology accuracy on phylogenetic reconstruction for various shapes of trees and phylogenetic methods show that, on average, homology accuracy correlates with the accuracy of the inferred phylogeny (Ogden and Rosenberg 2006). However, the relationship is far from monotonic and phylogenetic accuracy may increase or decrease dramatically even with small changes in homology accuracy (Fig. 6). In fact, the phylogenies inferred appear to be more sensitive to alignment method and parameters than to the choice of the tree-building method (Morrison and Ellis 1997; Cammarano et al. 1999; Ogden and Whiting 2003; Hertwig et al. 2004; Lebrun et al. 2006).
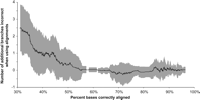
Graph illustrating the relationship between the accuracy of an alignment and the topological error of a phylogenetic tree reconstructed from that alignment by the Maximum Likelihood method in a large-scale simulation study. The x-axis is the average homology accuracy of all pairwise alignments in a multiple alignment. The y-axis is the number of incorrect branches in the tree obtained from the reconstructed alignment minus the number of incorrect branches in the tree reconstructed from the true alignment. Each point plotted is a moving average of 50 simulated results. Trees are based on 16 taxa and have a variety of topologies, relative branch lengths, and maximum distances. The shaded area shows plus and minus one standard deviation of points within the moving-average window. The results indicate that very poor alignments produce bad trees, but as long as 60% or more of the sites are accurately aligned, further improvements in alignment homology accuracy make little difference; even highly accurate alignments produce trees with substantial variation in quality. Results for Bayesian and Maximum Parsimony phylogenetic analyses are similar; Neighbor-Joining shows higher levels (an average of around 1 additional branch incorrect) of phylogenetic error. Based on data from Figure 4 of Ogden and Rosenberg (2006).
The inevitable need for the use of heuristic procedures in sequence alignment contributes significantly toward phylogenetic error, when the sequences being aligned have undergone a large number of substitutions and many insertion–deletion events. A key component of any progressive (heuristic) alignment procedure is the guide tree that sets up the order in which sequences (and sequence profiles) are aligned. Guide trees can be created in different ways. ClustalW, for example, creates a matrix of pairwise distances by aligning each sequence pair separately and computing a dissimilarity score from each pairwise alignment. This dissimilarity matrix is then subjected to the neighbor-joining algorithm to generate a directional hierarchy of sequence relationships (Thompson et al. 1994). Obviously, the guide tree is not guaranteed to reflect the true evolutionary history of sequences; because the dissimilarity scores are not evolutionary distances, the sequence alignment produced will most likely be based on incorrect inferences.
Guide-tree errors are known to have serious effects on downstream phylogenetic inference, as the increase in the phylogenetic error rate is found to be associated with errors in the guide trees (Lake 1991; Thorne and Kishino 1992; Landan 2005; Redelings and Suchard 2005). However, the error introduced by incorrect guide trees is generally considered a nuisance and thought to decrease the power of statistical inference by reducing our ability to distinguish among alternative phylogenetic hypotheses. Because the use of a guide tree is required to make sequence alignment feasible computationally, the implicit consolation has been that at least incorrect phylogenetic clusters will not garner high statistical support. This intuition is reasonable for short sequences, as the statistical biases introduced by guide trees in sequence alignments are expected to be smaller than the variances associated with estimation of evolutionary distances and branch lengths when conducting Maximum Likelihood and Least Squares analyses using sophisticated models of nucleotide substitution.
Disregarding the effect of guide-tree errors on evolutionary and phylogenetic inference is no longer tenable, because these errors are amplified in today’s large data sets. The genomic revolution in building the Tree of Life has now taken root and very long sequences are being used to establish key species relationships (Hedges 2002; Bashir et al. 2005; Margulies et al. 2005; Elango et al. 2006). First, these sequences need to be aligned, which is done invariably by using guide trees either explicitly or implicitly. In such data sets, the variance of estimates of evolutionary parameters is expected to become vanishingly small, as it decreases with sequence length. Alignment bias caused by errors in the guide tree will affect each position aligned, and it will not diminish with increasing sequence length. These two factors may combine to mislead phylogenomic inferences and incorrect phylogenetic clusters may be supported by spuriously high confidence. For example, alignments of homologous 20,000 base-pair regions of human, mouse, rabbit, and cow produce high bootstrap support for the species relationship implied by the guide tree itself when ClustalW is used (Fig. 7). A similar trend is observed when using genome sequence alignment approaches, such as the TBA (Blanchette et al. 2004) (V. Swarna, pers. comm.). We do not know which phylogeny is the true one, but it is clear that error in guide trees can produce incorrect inferences supported by high confidence. On a positive note, guide trees inferred using very long sequences may contain few or no errors, so the situation may not be as grim as it appears. However, this question has yet to be examined and is being investigated currently by our group.
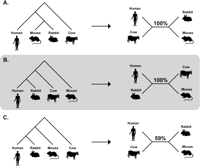
Diagrams show different guide trees used in a progressive alignment (left) and the phylogenies inferred from the resulting alignments (right). The bootstrap support for the inferred interior branch is shown (1000 replicates). Three different rooted guide trees (A, B, and C) were used to align 20,000 base pairs from human, mouse, rabbit, and cow sequences in the ENCODE data set ENm001 (ENCODE Project Consortium 2004; http://www.genome.gov/10005). Default options in ClustalW (Thompson et al. 1994) were used and the phylogenetic trees were inferred from the alignments using the Neighbor-Joining (NJ) method as implemented in MEGA3 (Kumar et al. 2004). (Maximum Likelihood (ML) analyses also support the same phylogenies.) All sites containing alignment gaps were removed before phylogenetic analysis, resulting in a data set of more than 8000 positions in each alignment. Because the NJ and ML methods both produce unrooted phylogenies, results are shown as such. In A and B, the contradictory inferred phylogenetic trees are identical to the guide trees, and each obtains 100% NJ bootstrap support. The influence of the root of the guide tree can be seen by comparing results in B and C: The guide trees are identical except for the location of the root, yet the alignments they produce result in incompatible inferences about the relationships of species.
When is it appropriate to use genomic multiple sequence alignments available in various database resources, which use a specific phylogeny as a guide tree? The answer depends on the purpose of the analysis. To begin with, the use of such multiple sequence alignments for inferring species phylogenies will be circular and will often (but not always) produce outcomes that merely reflect bias introduced by the alignment procedure (Fig. 7). This will be particularly problematic for traditionally hard-to-resolve phylogenetic relationships, because they are often associated with short internal branches (i.e., small amount of evolutionary change) in the phylogenetic trees. Errors introduced by alignment bias are expected to affect these parts of the phylogeny most severely, as the phylogenetic signal may be overwhelmed by the bias in the sequence alignment. For example, resolving the phylogenetic relationship of major groups of mammals using the sequence alignment of ENCODE data is not desirable, because these alignments are constructed using a guide tree that best reflects our current understanding of mammalian and vertebrate species relationships (http://www.genome.gov/10005). On the other hand, these alignments are appropriate for inferring ancestral genomes and times of species divergence events, because those procedures and all the results obtained are explicitly conditional on the evolutionary tree used. Obviously, one would generally use the same evolutionary tree for aligning sequences and for conducting evolutionary analyses, as long as it is substantiated. Otherwise, the use of an incorrect phylogeny will not only produce ancestral sequences and speciation times for ancestors that never existed, but it will also bias results for those that indeed existed. On the other hand, errors in the guide tree are expected to have a significantly lesser impact on the estimation of evolutionary rates at individual positions (or in sliding windows), and thus on motif finding, as such summary statistics are derived using a large amount of data for each position when many species are used (see, e.g., Yang and Kumar 1996; Siepel et al. 2005).
One unexplored wrinkle in the guide-tree conundrum is the observation that closely related species can show very different base compositions in homologous genomic segments. For instance, equality of substitution pattern can be rejected in ∼40% of human–mouse protein-coding gene orthologs, associated with differences in G+C content (Kumar and Gadagkar 2001; Jermiin et al. 2004). Among eubacteria, genome-wide G+C content can vary by a factor of two (Nakashima et al. 2003). This variation would serve to distort apparent distances among taxa and lead to biased guide trees. Either one may bias phylogenetic analyses (Steel et al. 1993; Kolaczkowski and Thornton 2004; Gadagkar and Kumar 2005). These deviations are seldom incorporated in simulated studies or models, but they are known to run rampant in real sequence data. One way of avoiding guide-tree bias on phylogenetic inference is to simultaneously infer multiple sequence alignment and phylogeny, so that one does not depend on the completion of the other (Hein 1990). Maximum Likelihood and Bayesian methods developed for this purpose seem promising for small-to-medium-sized problems (Fleissner et al. 2005; Lunter et al. 2005; Redelings and Suchard 2005), but their application to data sets with a large number of sequences and to long sequences is still computationally prohibitive.
Conclusions
The multiple sequence alignment procedure forms the backbone of comparative and evolutionary genomics. Results from some recent studies involving computer simulations and large-scale genomic data have begun to clarify and quantify sources of bias and the effects of alignment on subsequent downstream processing. Because of rapid growth of large scale data sets and increasing applications of multiple sequence alignments to understand patterns and processes that govern gene, genome, and species evolution, it would be prudent to further intensify these investigations and make their conclusions more accessible to practicing biologists.
Acknowledgments
We thank Vinod Swarna for his assistance with data analysis (Fig. 7), Drs. Sonja Prohaska and Michael Rosenberg for comments on an earlier version of this manuscript, and Ms. Kristi Garboushian for editorial support. We also thank three anonymous referees for many insightful suggestions. This work was supported in part by a research grant from National Institutes of Health to S.K.
Footnotes
-
↵1 Corresponding author.
↵1 E-mail s.kumar{at}asu.edu; fax (480) 727-6947.
-
Article is online at http://www.genome.org/cgi/doi/10.1101/gr.5232407
- Copyright © 2007, Cold Spring Harbor Laboratory Press








