
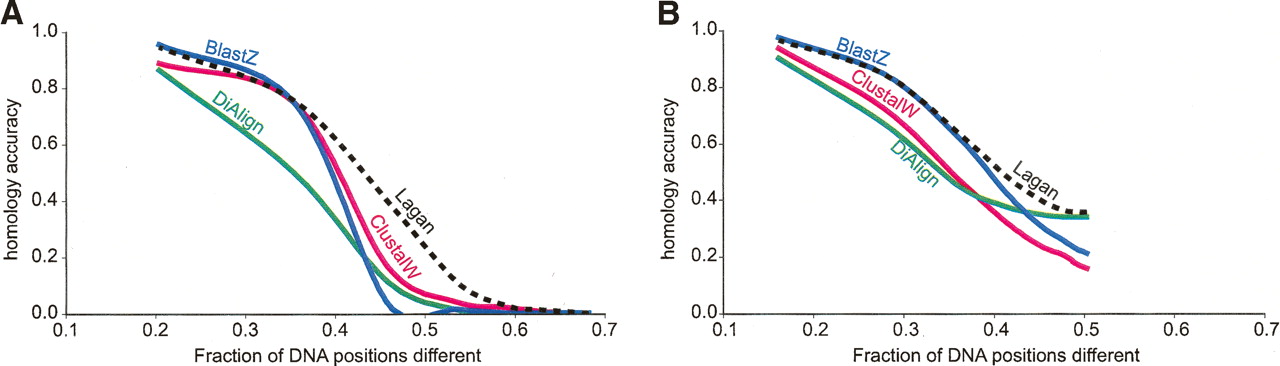
Graphs showing how the accuracy of pairwise alignment varies with evolutionary distance in computer simulations with only insertion–deletions (A) and insertion–deletions together with the constraint that 20% of all DNA positions are occupied by interspersed highly conserved blocks that evolve 10 times slower than other positions (Pollard et al. 2004) (B). The homology accuracy is calculated as a fraction of simulated positions that were aligned correctly, and it is plotted against the fraction of sites different, as reported in Figures 2 and 4 of Pollard et al. (2004). Even though the same set of evolutionary distances is simulated for both panels, the percent sequence difference in B is smaller for a given simulated distance because of the existence of highly conserved blocks. Programs compared for homology accuracy are ClustalW (Thompson et al. 1994), BLASTZ (Schwartz et al. 2003), DiAlign (Morgenstern 1999), and Lagan (Brudno et al. 2003a).








