
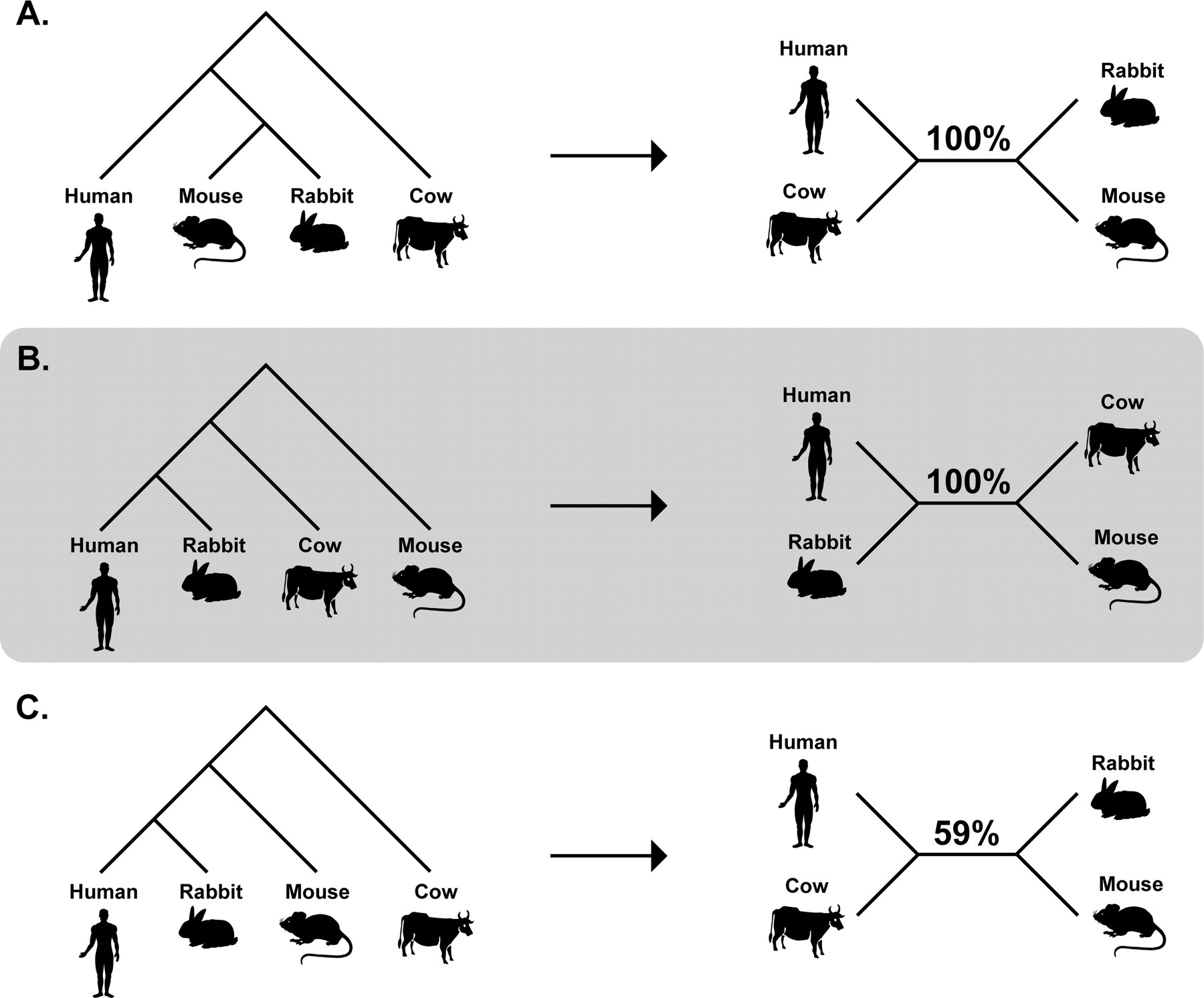
Diagrams show different guide trees used in a progressive alignment (left) and the phylogenies inferred from the resulting alignments (right). The bootstrap support for the inferred interior branch is shown (1000 replicates). Three different rooted guide trees (A, B, and C) were used to align 20,000 base pairs from human, mouse, rabbit, and cow sequences in the ENCODE data set ENm001 (ENCODE Project Consortium 2004; http://www.genome.gov/10005). Default options in ClustalW (Thompson et al. 1994) were used and the phylogenetic trees were inferred from the alignments using the Neighbor-Joining (NJ) method as implemented in MEGA3 (Kumar et al. 2004). (Maximum Likelihood (ML) analyses also support the same phylogenies.) All sites containing alignment gaps were removed before phylogenetic analysis, resulting in a data set of more than 8000 positions in each alignment. Because the NJ and ML methods both produce unrooted phylogenies, results are shown as such. In A and B, the contradictory inferred phylogenetic trees are identical to the guide trees, and each obtains 100% NJ bootstrap support. The influence of the root of the guide tree can be seen by comparing results in B and C: The guide trees are identical except for the location of the root, yet the alignments they produce result in incompatible inferences about the relationships of species.








