
Traffic of genetic information between segmental duplications flanking the typical 22q11.2 deletion in velo-cardio-facial syndrome/DiGeorge syndrome
Abstract
Velo-cardio-facial syndrome/DiGeorge syndrome results from unequal crossing-over events between two 240-kb low-copy repeats termed LCR22 (LCR22-2 and LCR22-4) on Chromosome 22q11.2, comprised of modules, each of which are >99% identical in sequence. To delineate regions in the LCR22s that might contain hotspots for 22q11.2 rearrangements, we scanned the interval for increased rates of recombination with the hypothesis that these regions might be more prone to breakage. We generated an algorithm to detect sites of altered recombination by searching for single nucleotide polymorphic positions in BAC clones from different libraries mapped to LCR22-2 and LCR22-4. This method distinguishes single nucleotide polymorphisms from paralogous sequence variants and complex polymorphic positions. Sites of shared polymorphism are considered potential sites of gene conversion or double cross-over between the two LCR22s. We found an inverse correlation between regions of paralogous sequence variants that are unique to a given position within one LCR22 and clusters of shared polymorphic sites, suggesting that these clusters depict altered recombination and not remnants of ancestral single nucleotide polymorphisms. We postulate that most shared polymorphic sites are products of past transfers of DNA information between the LCR22s, suggesting that frequent traffic of genetic material may induce genomic instability in the two LCR22s. We also found that gaps up to 1.5 kb long can be transferred between LCR22s.
Diseases involving chromosome rearrangements of >1 Mb are referred to as genomic disorders, and most are mediated by region-specific low-copy repeats (LCRs) (Lupski 1998, 2003; Stankiewicz and Lupski 2002a,b). Both deletions and duplications can occur during meiosis, resulting in altered gene dosage associated with mental retardation and congenital malformation syndromes. The most well-recognized include Williams-Beuren syndrome on Chromosome 7q11.2, Prader-Willi/Angelman syndromes on Chromsome 15q11–13, Charcot-Marie-Tooth disease type 1A (CMT1A)/hereditary neuropathy with liability to pressure palsies (HNPP) on Chromosome 17p11.2, Smith-Magenis syndrome (SMS) on Chromosome 17p11.2–13, and neurofibromatosis (NF1) on Chromosome 17q11.2. Although each occurs rarely, when taken together they have a significant health impact. Understanding the mechanisms responsible for LCR-mediated chromosome rearrangements may identify sequence features responsible for genome instability leading to chromosome evolution or disease.
The 22q11.2 region is particularly susceptible to meiotic chromosome rearrangements associated with genomic disorders including velo-cardio-facial syndrome/DiGeorge syndrome (VCFS/DGS MIM192430/MIM188400) (DiGeorge 1965; Shprintzen et al. 1978); the reciprocal duplication, dup(22)(q11.2;q11.2) (Edelmann et al. 1999a; Bergman and Blenow 2000; Ensenauer et al. 2003); and cat-eye syndrome (CES; MIM 115470) (Guanti 1981). Unequal crossing-over events between LCRs on Chromosome 22q11.2 are responsible for these genomic disorders. Of the three, VCFS/DGS is one of the more common congenital malformation syndromes, occurring with a frequency of 1/4000 live births (Burn and Goodship 1996). Most affected individuals have a similar 3-Mb deletion (Lindsay et al. 1995; Morrow et al. 1995; Shaikh et al. 2000), flanked by two LCR22s (Edelmann et al. 1999b; Babcock et al. 2003). Both intrachromosomal and interchromosomal unequal crossing-over events between the two LCR22s, LCR22-2 and LCR22-4, are responsible for the typical 22q11.2 deletion in VCFS/DGS (Baumer et al. 1998; Edelmann et al. 1999a,b; Saitta et al. 2004). Both LCR22s are composed of blocks or modules consisting of genes and pseudogenes (Bailey et al. 2002; Babcock et al. 2003). Homologous blocks are >99% identical in sequence (Shaikh et al. 2000).
Recently, it has been found that there are positional recombination hotspots responsible for the CMT1A/HNPP rearrangements on Chromosome 17p12 (Reiter et al. 1998), NF1 deletion on Chromosome 17q11.2 (Lopez-Correa et al. 2000), SMS on Chromosome 17p11.2 (Bi et al. 2003), and Sotos syndrome deletion (Visser et al. 2005). Bi et al. (2003) found that the majority of breakpoints for both the SMS deletion and the reciprocal duplication occurred in a small interval of <12 kb, flanked by inverted AT-rich repeats in >200-kb LCRs. Interestingly, they narrowed the interval to <2 kb and found that this interval showed sequence evidence for frequent historical gene conversion (Bi et al. 2003). This suggests that there may be a correlation between regions of gene conversion or recombination and susceptibility to rearrangements.
To determine whether there are variations between gene conversion or recombination levels spanning the two LCR22s on Chromosome 22q11, we examined the sequence between clones spanning each. Using single nucleotide variants from multiple different BAC clone alignments from different libraries, we detected signatures or clusters of frequent gene conversion or recombination between LCR22-2 and LCR22-4 computationally and experimentally.
Results
Polymorphisms in LCR22-2 and LCR22-4
The LCR22s comprise 11% of the 22q11.2 region and contain genes and unprocessed pseudogene copies (Bailey et al. 2002; Babcock et al. 2003). The two largest are LCR22-2 and LCR22-4, each comprising 240 kb of genomic sequence. They flank the intervals deleted and duplicated in VCFS/DGS and dup(22)(q11.2;q11.2), respectively. The proximal end of LCR22-2 begins in the last exon of one of the LCR22 genes, USP18 (Fig. 1), and ends just centromeric to DGCR6, a gene present in two copies on 22q11.2 (Supplemental Fig. 1S). Two functional copies of the DGCR6 gene are present on human Chromosome 22q11 and are due to a duplication of an ancestral locus (Edelmann et al. 2001). LCR22-2 contains three large intra-LCR duplications denoted dupA-C (Fig. 1). DupA starts within the 3′-end of USP18 harboring the last exon. The last exon has been duplicated and is also found in dupB-C. The region defined by dupB-C is also present in LCR22-4, mapping 3 Mb telomeric to LCR22-2 (Babcock et al. 2003). The GGT, GGTLA, and BCR pseudogene copies are also present in LCR22-2 and LCR22-4 (Fig. 1). The BCR pseudogene is present once in LCR22-2 but twice in LCR22-4 (Babcock et al. 2003).
To detect potential recombination/gene conversion events between LCR22-2 and LCR22-4, we searched for polymorphic positions between the two. We first created a global alignment of all BAC clones that harbor the LCR22 segments but are anchored because of the asymmetric pattern of blocks in the two LCR22s (Supplemental Fig. 1S) and/or by the presence of flanking unique sequences (except for AP000551) (Edelmann et al. 1999a,b) as shown in Figure 2B. BAC genomic sequence was available for analysis, from at least two alleles, through the entire length of each LCR22.
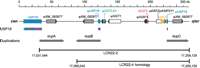
Organization of the LCR22-2 locus. We show a 128-kb-long segment homologous to LCR22-4. The locus contains one functional gene, USP18. There is a predicted gene, XM_092877, but it is not expressed (M. Babcock and B.E. Morrow, unpubl.), and many pseudogenes. The region contains three ∼35-kb-long duplications denoted dupA, dupB, and dupC (these correspond to the red blocks in Babcock et al. 2003). dupA starts in the 3′ part of the USP18 gene. USP18 introns are marked in blue, coding exons in red; the first noncoding exon is highlighted in yellow. dupB and dupC contain the 3′ part of the last internal intron, last exon, and 3′-UTR of USP18. The bottom part shows the exact localization of LCR22-2 and the region homologous to LCR22-4. The LCR22-4 homology corresponds to a segment defined by dupB and dupC.
The clone alignment revealed many polymorphic positions (Fig. 2C). Each type of single nucleotide variant was defined as shown in Figure 3. Paralogous sequence variants (PSVs) are positions that are conserved in each LCR22, but different between them; such as an A in LCR22-2 and a T in LCR22-4. LCR-specific single nucleotide polymorphisms (SNPs) correspond to positions that vary in one LCR22, but not in the other. If both LCR22 positions are variable, they are classed as either shared or nonshared. If a nucleotide variant in one LCR22 is equal, or included within the variation of the second LCR22, then the position is termed a shared polymorphism site (SPS); the other positions are unshared polymorphic sites (NPSs). SPSs are sites of potential recombination/gene conversion.
Positions from all categories were further divided into nonrepetitive (unique DNA); those found in interspersed repeats (copies of transposable elements); and polymorphic sites located in simple repeats such as micro- and minisatellites, satellites, or low complexity regions (Fig. 2C). We detected a total of 2492 non-gap (gap-free), polymorphic positions in the 176,245-bp-long alignment. Next, we excluded all positions that mapped to simple repeats. From the 2308 remaining positions, there were 1058 SNPs in LCR22-2, 443 SNPs in LCR22-4, 688 PSVs, 114 SPSs, and five NPSs.
The density of single nucleotide variants was quite high compared to the genome average of 1 SNP/kb (Li and Sadler 1991; Wang et al. 1998; Cargill et al. 1999). After removing simple repeats and the first 20 kb from both LCRs (because of the lack of polymorphism in this regions LCR22-4, we cannot separate potential SNPs from SPSs/NPSs), we detected 6.4 SNPs/kb in LCR22-2 and 3.0 SNPs/kb in LCR22-4. The presence of many polymorphic positions together with the potential transfer of genetic information between the LCRs (see below) significantly contributed to the relatively high divergence between individual BAC clones (Table 1). Pairwise identity between LCR22-2 clones is just 99.02%–99.53%. LCR22-4 clones are on average more similar, with identity ranges from 99.47% to 99.83%. Inter-LCR comparison revealed 99.13%–99.66% identity between LCR22-2 and LCR22-4 BAC clones.
Pairwise identities between BAC clones
We found that the distribution of individual groups of polymorphic sites is highly nonrandom (Fig. 2C,D). Sequences comprising the most centromeric ∼20 kb of LCR22-4 are almost identical between the LCR22-4 clones AC008018 and AC000550, and, as a consequence, PSVs, SPSs, and LCR22-4 SNPs are absent from the first 20 kb. The SPSs form several clusters, implicating high levels of recombination/gene conversion (see below). Using a probabilistic model (see Methods), we defined clusters of highly nonrandom concentration of SPSs (Fig. 2E). One such cluster is located at positions 35–40 kb corresponding to the pseudogene ψGGTLA. A large region of high SPS density is located at positions 65–165 kb. There are particularly SPS-rich regions at positions 75–83 kb and within pseudogenes ψDKFZp434P211 and ψBCR. No obvious correlation between SPS hotspots and unstable motifs such as palindromes (Fig. 2G) or repetitive DNA (Fig. 2H) was detected. Furthermore, analysis of various recognition motifs of endonucleases and recombinases failed to reveal any association (data not shown). Similar negative results have been reported for gene conversion in the AZFa region on Yq (Bosch et al. 2004). Finally, we should note that we have excluded all simple repeats including AT-rich repeats from our analysis because of possible alignment artifacts. However, positions close to simple repeats exhibit an increased rate of gene conversion in the human genome (Vowles and Amos 2004), and it is thus possible that simple repeats including those in LCR22-2/LCR22-4 can potentially also undergo concerted evolution.

Polymorphism between LCR22-2 and LCR22-4. (A) Schematic organization of LCR22-2/LCR22-4 (see Fig. 1). (B) Clone coverage. The scheme shows positions of clones (gray) on the genomic sequences (black). (C) Distribution of polymorphic positions. Individual categories of polymorphic positions (see the text and Fig. 3) are shown as total (black, top), nonrepetitive (unique, red), those in interspersed repeats (blue), and simple repeats (black, bottom). (D) Unequal distribution of PSVs and SPSs. Positions located in simple repeats were removed. (E) Probability of SPS clusters. Nonrandom clusters of SPSs were defined by a binomial model (see Methods). Dark blue segments are clusters of SPSs with probability ≤0.05, light blue marks ≤0.01, yellow ≤1e-03, and red highlight clusters with probability ≤1e-05. (F) Nucleotide identity between genomic sequences. The plot was obtained using 5-kb sliding windows and step 500 bp. (G) Positions of long palindromes. (H) Distribution of groups of repetitive elements.
Signature of DNA transfer between LCR22-2 and LCR22-4
Shared polymorphic positions can be considered as potential sites of information transfer by recombination (gene conversion or double cross-over) between LCR22-2 and LCR22-4. Nevertheless, shared polymorphic sites could have been created by independent mutations in both LCR22-2 and LCR22-4. The probability of such events can be estimated from nonshared polymorphic sites (NPSs); since random events should create both shared and nonshared polymorphisms. For simplicity, we consider only shared and nonshared sites with the most common dinucleotide (not tri- or tetranucleotide) polymorphism in both LCR22s, after excluding all simple repeat positions because of possible alignment artifacts. The expected ratio of shared polymorphism/nonshared polymorphism is 0.4 (six shared, 15 nonshared dinucleotide combinations in 21 possible). Having found five different NPSs in the entire clone alignment, we expect to find two shared polymorphic dinucleotide sites, compared to 114 observed. This discrepancy is highly statistically significant (p <10–8, Binomial test). As a consequence, most if not all SPSs are not independent, that is, they were not created by independent mutations between LCR22-2 and LCR22-4.
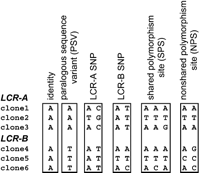
Classification of the sequence variants. Identical positions are invariant (fixed) in all clones from both LCRs. Paralogous sequence variants (PSVs) are positions invariant within each of the LCRs, but different between the LCRs. LCR-A and LCR-B SNPs correspond to positions where one LCR is polymorphic, but the second LCR is not. Shared polymorphism sites (SPSs) correspond to positions where both LCRs are polymorphic and the set of possible variants in one LCR is equal to, or contained within, the set of possible variants at the same positions in the other LCR. If both LCRs are polymorphic at a given site, but the clone variation does not overlap between the LCRs, this position is described as a nonshared polymorphism site (NPS).
The fact that SPSs seem to be interdependent between the LCRs can be explained by two different mechanisms: (1) by the preservation of ancestral, pre-duplication polymorphism, or (2) by transfer of genetic information between the LCRs by recombination/gene conversion (concerted evolution). If the second scenario is correct, the prediction is that in places of high concentration of shared polymorphism sites, we should find nearly no PSVs. PSVs should be homogenized between the LCRs by recombination/gene conversion. On the other hand, if shared polymorphic sites are just remnants of ancient, pre-duplication polymorphism, no correlation between PSVs and SPSs is expected (Fig. 4). Figure 2D shows that the PSVs are underrepresented in regions with frequent SPSs. This was confirmed by a statistical analysis of 10-kb-long, nonoverlapping segments after removal of the first 20 kb. The correlation between the number of PSVs and number of shared polymorphism sites was negative, –0.55 (p < 0.05; Spearman's correlation coefficient). This strongly indicates that many shared polymorphic sites are a result of true recombination and not remnants of ancestral polymorphisms. In conclusion, we can postulate that LCR22-2 and LCR22-4 SPSs are not independent and most of them are products of past transfers of DNA information between the LCRs.
Representative PSVs and SPSs in LCR22-2 and LCR22-4
To validate the PSVs and SPSs experimentally, we obtained large insert genomic clones from different human libraries and screened the DNA with PCR primers flanking seven PSVs and seven SPSs. We provide representative results from one set of PSVs and one set of SPSs (Fig. 5). For the PSVs, we amplified a 383-bp interval containing six PSVs (Chr22: 17,132,218–17,132,600) (Fig. 5A; Supplemental Fig. 2AS). In LCR22-2, all clones had the same C-T-C-G-T-C sequence, and all the LCR22-4 clones had a T-C-T-A-A-T sequence at the same positions. The interval is within the GGT locus, roughly 4 kb downstream from the 3′-UTR of the gene. The full-length functional GGT gene lies in LCR22-8. The sequence for this locus is T-C-C-T-A-C. Thus, there was significant alteration of sequences after the GGT locus had duplicated. In contrast, the 551-bp PCR product for the GGT locus (Chr22: 17,149,097–17,149,647), 17 kb downstream from the PSV PCR product, contains four shared polymorphic sites. There is a T-T-A-T/C and a C-C-G-C sequence in LCR22-2 clones; similarly, both occur in alleles in LCR22-4 clones (Fig. 5B; Supplemental Fig. 2BS).
Indel shuffling by inter-LCR recombination
LCR22-2/LCR22-4 BAC clone alignment contains several large gaps (Fig. 2B). Two of the indels can be characterized as shared polymorphism regions, because the gap and the full-length variant are found in both the LCR22-2 and LCR22-4 clones. We examined the flanking regions, and it appears that the two different deletions were stimulated by Alu-mediated rearrangements (Fig. 6; Supplemental Fig. 3S). Alu-mediated recombination typically occurs within a region of identity between two elements in the same orientation (Kapitonov et al. 2004) as is the case for both here (Fig. 6; Supplemental Fig. 3S).
Notably, both the indels are found in the large region of high SPS concentration at positions 65–165 kb. The short duplication is located around positions 77,473–77,635, within a particularly SPS-rich region, 75–83 kb. Given the high concentration of shared polymorphic sites and low concentration of PSVs, both the indel regions seem to be products of concerted evolution, rather than remnants of ancient pre-duplication polymorphism.
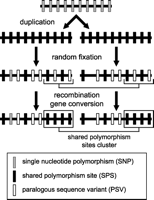
Simplified model of evolution of polymorphic sites after duplication. After a duplication, SNP sites are randomly fixed in the duplicated segments. In some cases, the process can fix the same nucleotide in both LCRs, the result being identity. In other cases, only one position is fixed and the second remains polymorphic, the result being an LCR-specific SNP (gray box). If both duplicated positions are fixed, but a different nucleotide in each case, the result is a PSV (white box). If both positions remain polymorphic, they will be detected as a shared polymorphism (black box). If the process of fixation was more or less random, we would expect to find PSVs interspersed with SPSs. This corresponds to the pattern expected from ancestral polymorphism. Recombination, on the other hand, produces separated clusters of SPSs depleted in PSVs. If the two LCRs occasionally exchange information in some individuals, all PSV positions become shared polymorphic positions (bottom), because some individuals in the population will have the original variant, and some will have a new variant transferred from the second LCR. In our model, we considered that polymorphism is transferred to both LCRs (possibly via initial gene conversion between identical segments). If only one copy remains polymorphic and the second LCR has no initial polymorphism (simple duplication event), no ancestral shared polymorphic sites are created and SPSs are solely results of other post-duplication processes (recombination). The model is simplified because we do not take into account de novo polymorphism by mutations in individual LCRs. Again, this random polymorphism would produce an interspersed pattern of PSVs and SPSs, not separate clusters.
Discussion
Sequence comparison of BAC clones covering 240-kb repeats on Chromosome 22q11.2, frequently deleted in patients with velo-cardio-facial syndrome/DiGeorge syndrome, revealed a complex pattern of polymorphic sites. Apart from paralogous sequence variants (PSVs) and LCR-specific SNPs, we have detected positions that are polymorphic in both LCRs. Based on equality/inclusion of the variations, these were classified as shared and nonshared polymorphic sites (SPSs and NPSs, respectively). The SPSs are equivalent to previously reported multisite variation type 2 (MSV2) (Fredman et al. 2004). The proportions after removal of simple repeats were 65% of LCR-specific SNPs, 29.8% PSVs, 4.9% SPSs, and 0.2% NPSs. The basic proportions are roughly similar to those obtained previously for segmental duplications (Fredman et al. 2004). We should mention, however, that in our analysis we concentrated only on LCR22-2/LCR22-4 comparisons. There are additional intra-LCR duplications (dupA-B) and other related but less similar LCRs on 22q11.2 and Chromosome 20 (Babcock et al. 2003). As a consequence, some of the detected polymorphic sites may belong in a different category of polymorphic sites, if the complete genome is considered.
The SNP density along LCR22-2/LCR22-4 is relatively high (6.4 and 3.0 SNPs/kb for LCRs 22-2 and 22-4, respectively), despite the fact that our method precisely maps polymorphic sites to the LCRs and avoids frequent identification of PSVs as ambiguous SNPs in segmental duplications (Estivill et al. 2002). In addition, it is possible that, owing to the low number of compared BAC clones, many SNPs may not have been discovered, particularly low-frequency SNPs. The increased polymorphism in segmental duplications can be explained either by gene conversion with other LCRs found in the genome (Giordano et al. 1997; Hurles 2002; Hurles et al. 2004) or by selective pressure on sequence diversification and suppression of deleterious recombination between LCR22-2 and LCR22-4.
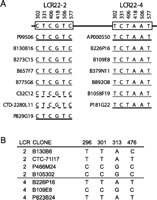
Experimental confirmation of selected PSVs and SPSs examples. (A) Paralogous sequence variants in LCR22-2 and LCR22-4 in the GGT pseudogene loci. Interval 17132218–17132600 on LCR22-2 was chosen because it contained PSV sequences identified in Figure 2. The PCR primers (F, TAGTCAGCATCAAGGTGGAG; R, CAGCACAGTAGTAGC GGATTT) amplified a 383-bp segment from the GGT locus, 4 kb downstream from the 3′-UTR. Clones were selected from different genomic libraries (genome assembly, black; RPCI PAC library, black; RPCI 11, gray; CTD library, dashed line; LL22NC03 cosmid library, C32C12, dashed line) mapping to LCR22-2 and LCR22-4 identified from the genome assembly, BAC end pairs, GenBank, and a previous report (Edelmann et al. 1999a). This report described a 4.4-kb resolution physical map of genomic clones spanning LCR22-2 and LCR22-4. This map was constructed experimentally with pure genomic clone DNA. Clones P99506 (AC008132), B130B16, B273C15, C32C12, and P829G19, for LCR22-2 and clones B226P16, B109E8, B379N11 (AC008018), and P181G22 for LCR22-4 were integrated into this map. All these clones were experimentally anchored to their respective LCR22s. Clone B657F7 is anchored to LCR22-2 because its 3′-end is in unique sequences outside the LCR. CTD-2280L11 is anchored to LCR22-2 because its 5′-end, oriented as it is, lies in the junction region of dupA and dupB (Fig. 1). The 3′-end is in dupC. This pattern is unique. It is a similar situation as described for the sequenced clone, AC007981. The 5′-end of B1058F19 is in unique, non-LCR sequences; thus, it is correctly anchored to LCR22-4. Thus, only B775G6 and B892O8 cannot be unequivocally placed into LCR22-2 or LCR22-4, but were placed in their respective LCR22s computationally (UCSC browser; http://genome.ucsc.edu/). The presumed ancestral locus in LCR8 contains TCCTAC (Supplemental Fig. 2AS). (B) Shared polymorphic sites in LCR22-2 and LCR22-4 in the GGT pseudogene loci (Fig. 2). Interval 17149097–17149647 was chosen for analysis of shared polymorphic sites. This region is duplicated in LCR22-4 and LCR22–8. The PCR primers (F, TGCCTGTTGAAAAGGCAGGA; R, CAGGCTGGCCTTTGC CAG) amplified a 551-bp segment from the GGT locus. This interval contains SPSs in genomic clones obtained from different libraries: (B) RPCI 11; (P) RPCI PAC library; CTC library. The putative ancestral locus in the GGT genomic interval contains CTAC at the same sites (Supplemental Fig. 2BS).

Shared polymorphism of a 153-bp-long indel between LCR22-2 and LCR22-4. (A) Alignment of the indel region from LCR22-2/LCR22-4 clones. These breakpoints correspond to positions 77473–77635 in the clone alignment (Fig. 2). LCR22-2 clone AC008132 and two LCR22-4 clones, AC008018 and AC000928, share the same gap. The rearrangement was caused by homologous recombination between two AluSx monomers and led to duplication of one Alu monomer in the middle. Alternatively, the indel could be a result of deletion in a pre-existing Alu element with a duplicated monomer. (B) Alignment of the duplication breakpoints. We compared the duplicated monomer with the two parental Alu monomers. The first 37 bp in the product is nearly identical to the right monomer. The similarity to the left monomer starts from position 27 and, with the exception of a 12-bp minideletion, continues until the end of the monomer. In a 15-bp region (boxed) the duplicated monomer is identical to both the parental monomers. The original break was probably located within this segment.
The overwhelming majority of positions polymorphic in both LCR22s represent shared polymorphism, indicating interdependence of polymorphism between the LCR22-2/LCR22-4 segmental duplications. Taking into account the presence of several LCR22-specific insertions/deletions and ∼1% divergence along the homologous segments, the potential contribution of ancestral polymorphism seems limited because of the relatively ancient origin of the duplications (Shaikh et al. 2000). Furthermore, the negative correlation between the concentration of PSVs and SPSs is consistent with exchanges of genetic information between LCRs (concerted evolution) rather than with random fixation/preservation of ancestral polymorphism. Several large regions of potential recombination between the two LCR22s were discovered, and they cover more than half of the LCR22-2/LCR22-4 homologous region.
Our BAC clones-based method cannot formally distinguish between crossovers and gene conversion. Several recent approaches addressed this difficulty by different approaches including sperm typing (Jeffreys and May 2004), analysis of loci in the non-pseudoautosomal region of Chromosome Y escaping meiotic crossovers (Bosch et al. 2004; Hurles et al. 2004), and by analysis of individuals with fully homozygous genomes (Fredman et al. 2004). Experimental evidence suggests that gene conversion is a prominent homology-repair mechanism of double-stranded breaks in mammalian cells (Johnson and Jasin 2000). In the same vein, interallelic gene conversion seems to be four to 15 times more frequent than crossovers (Jeffreys and May 2004). We can, therefore, extrapolate that gene conversion is the main mechanism behind extensive exchanges of genetic information between LCRs 22-2/LCR22-4 during meiosis.
More complicated is the situation with shared indels between LCR22-2/LCR22-4, since one is 1470 bp long. Typical interallelic gene conversion tracts detected in the human genome are relatively short, with a range estimated to be somewhere between dozens and several hundred base pairs (Bosch et al. 2004; Jeffreys and May 2004). In this context, however, it is worth noting that experiments in mammalian cells indicate repair of double-stranded breaks by long-track gene conversions, often transferring several kilobases of sequence (Richardson et al. 1998; Johnson and Jasin 2000; Richardson and Jasin 2000). Many of these events in vitro are associated with additional transfer of DNA from one strand (unbroken) to the other (broken) and may represent an elegant mechanism of genomic duplications (Babcock et al. 2003). It is also possible that such long gene conversions can convert large segments between LCR22s, but also indels, including the two shared indels we detected. Another possibility is that occasional double crossovers transfer indels between the duplicated segments. While the precise mechanism remains unclear, our results indicate that indel transfer between segmental duplications is possible. In turn, indels cannot be considered as specific markers for detection of individual LCR22s.
One of our major goals was to predict potential hotspots of deleterious 22q11.2 rearrangements. Both gene conversion and crossover hotspots tend to colocalize in the human genome (Jeffreys and May 2004). Along the same lines, direct links between gene conversion and rearrangements' hotpots were recently reported for the AZFa locus (Hurles et al. 2004). Therefore, even if the clusters of SPSs detected during our analysis were mostly created by gene conversion, we can use them to predict hotspots for crossovers. Regions of high concentration of SPSs and low concentration of PSVs detected in this work are the best candidates for meiotic deletion/duplication hotspots in 22q11.2 rearrangements. Regions depleted in SPSs and rich in PSVs, on the other hand, can be used for construction of LCR-specific markers. Their presence/absence in patients can help to narrow the search for chromosome rearrangement breakpoints. An analogous approach can be applied to other genomic segmental duplications.
Current evidence indicates that recent segmental duplications may exchange genetic information, preferably via gene conversion (Rozen et al. 2003; Fredman et al. 2004; Jeffreys and May 2004; Stankiewicz et al. 2004). In this paper, we performed the first study of DNA polymorphism in full-length LCRs. We uncovered extensive traffic of genetic information between large regions in LCR22-2 and LCR22-4. We have defined several hotpots of recombination, which will help us to map the most probable unstable regions stimulating rearrangements leading to 22q11.2 rearrangement disorders. Importantly, we have developed a new approach for detection of recombination/gene conversion hotspots in LCRs. This method uses sequenced, well-mapped BAC clones to obtain information about LCR polymorphism even for human-specific LCRs, which are inaccessible for comparisons with other primates. Interestingly, our approach can be used to distinguish SNPs from paralogous sequence variants and other complex polymorphic positions, and in turn to curate records in SNP databases. In genomic regions with good clone coverage (i.e., with two or more clones representing at least two different alleles), our methodology can be directly applied without any requirements for further experiments. Currently, ∼62% of the human genome is covered by two or more BAC clones representing two or more alleles (Krzywinski et al. 2004). BAC sequencing may thus permit a global in silico analysis of recombination between LCRs on the genomic scale.
Methods
Sequence analysis
DNA and protein sequences were aligned by BLAT (Kent 2002), MAVID (Bray and Pachter 2004; http://baboon.math.berkeley.edu/mavid/), Dialign2.2 (Schmollinger et al. 2004; http://bibiserv.techfak.uni-bielefeld.de/dialign/), and MAFFT (Katoh et al. 2002; http://bioinformatics.uams.edu/mafft/), and edited in Seaview (Galtier et al. 1996; http://pbil.univ-lyon1.fr/software/seaview.html). Repetitive elements were detected by Tandem Repeat Finder (Benson 1999; http://tandem.bu.edu/trf/trf.html), Censor (Jurka et al. 1996; http://www.girinst.org/Censor_Server.html), and RepeatMasker (A.F. Smit, R. Hubley, and P. Green, “RepeatMasker Open-3.0.1996–2004”; http://www.repeatmasker.org) with Repbase Update libraries (Jurka 2000; http://www.girinst.org/Repbase_Update.html).
Detection of SPSs clusters
First we excluded all positions located within simple repeat regions from the BAC alignment. For each possible pair of shared
polymorphism sites (SPSs) in the alignment, we first counted the total number of SPSs within the interval between the positions
including the terminal SPSs. Then we calculated the probability that a region of this length will contain this number of SPSs
or higher, assuming a random distribution of SPSs. For a window of length n bp, the probability of observing exactly r events is given by the Binomial distribution 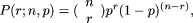 where p is the average probability of an event at each point. For example, if there are a total of R events observed in a region of N bp, we can define p = R/N. The probability that at least r events occur in an interval is given by the incomplete β-function Ip(r, n – r + 1) such that P{Sn ≥ r} = Ip(r, n – r + 1). Hence for any window, we can count the number of events and evaluate the probability that at least this many would
have occurred in that interval by random chance. The lower the probability, the higher is the significance of the window.
Probabilities were calculated in this way for all possible windows spanning SPSs. The windows were ordered from most to least
significant (lowest to highest probability). Each position in the BAC alignment was then assigned the value of the lowest
SPS probability window within which it falls.
where p is the average probability of an event at each point. For example, if there are a total of R events observed in a region of N bp, we can define p = R/N. The probability that at least r events occur in an interval is given by the incomplete β-function Ip(r, n – r + 1) such that P{Sn ≥ r} = Ip(r, n – r + 1). Hence for any window, we can count the number of events and evaluate the probability that at least this many would
have occurred in that interval by random chance. The lower the probability, the higher is the significance of the window.
Probabilities were calculated in this way for all possible windows spanning SPSs. The windows were ordered from most to least
significant (lowest to highest probability). Each position in the BAC alignment was then assigned the value of the lowest
SPS probability window within which it falls.
PCR analysis of BAC clones
The DNA from BAC clones anchored to 22q11.2, spanning parts of LCR22-2 and LCR22-4, was isolated and used as template for PCR amplification using primers flanking putative paralogous sequence variants and gene conversion sites as shown in Figures 5A and 5B. Each of the PCR products was subject to DNA sequence analysis using an ABI 3730 automated sequencing instrument.
Acknowledgments
We thank Melanie Babcock for helpful discussions. This work was supported by the NIH, P01 HD039420-04S2 (B.E.M.).
Footnotes
-
[Supplemental material is available online at www.genome.org.]
-
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.4281205. Freely available online through the Genome Research Immediate Open Access option.
-
↵4 Corresponding authors. E-mail morrow{at}aecom.yu.edu; fax (718) 430-8778. E-mail jurka{at}girinst.org; fax (650) 961-4473.
-
- Accepted August 10, 2005.
- Received June 14, 2005.
- Cold Spring Harbor Laboratory Press








