
Evolution and Comparative Genomics of Odorant- and Pheromone-Associated Genes in Rodents
Abstract
Chemical cues influence a range of behavioral responses in rodents. The involvement of protein odorants and odorant receptors in mediating reproductive behavior, foraging, and predator avoidance suggests that their genes may have been subject to adaptive evolution. We have estimated the consequences of selection on rodent pheromones, their receptors, and olfactory receptors. These families were chosen on the basis of multiple gene duplications since the common ancestor of rat and mouse. For each family, codons were identified that are likely to have been subject to adaptive evolution. The majority of such sites are situated on the solvent-accessible surfaces of putative pheromones and the lumenal portions of their likely receptors. We predict that these contribute to physicochemical and functional diversity within pheromone-receptor interaction sites.
Genome sequences are providing an opportunity to investigate evolutionary phenomena that are absent from the paleontological record. Whereas adaptation is consistently invoked in accounting for morphological changes that are apparent in extant and fossilized organisms (Hughes 1999), its contribution to the distinctive behavioral responses of animals is less understood. The diversifying effects of adaptive evolution on genes can be revealed from comparisons of their sequences. Analysis of virtually complete sets of genes from the two rodents Rattus norvegicus and Mus musculus (Waterston et al. 2002; Rat Genome Sequencing Project Consortium 2004), which diverged ∼12–24 million years ago (Mya; Adkins et al. 2001), now allows observations of the impact of diversifying selection on each of their genes during this relatively short time period.
Prominent among genes that have been associated with rodent behavior are the pheromone receptors expressed in the vomeronasal organ (VNO; Dulac and Torello 2003). This is an olfactory organ for the detection of chemical cues, situated in the septum of the nose. Deletion of a cluster of mouse vomeronasal type-1 receptor-like (V1R-like) genes (Dulac and Axel 1995; Ryba and Tirindelli 1997) results in significant modification of male and female behavioral responses, including reduced male libido and inappropriate maternal aggressive behavior (Del Punta et al. 2002). V1Rs are seven-transmembrane (7TM) proteins and are members of the G-protein coupled receptor (GPCR) superfamily.
V1Rs are closely related to a second family of GPCRs, the olfactory receptors (ORs), that are expressed in the olfactory epithelium. This is one of the largest gene families identified. The mouse genome has ∼1500 OR genes, and the human ∼900 (Young and Trask 2002). The dynamic nature of the evolution of this family is characterized by rapid expansion, gene duplication, extensive gene loss by pseudogene creation, and diversifying selection (Lane et al. 2001; Waterston et al. 2002; Young et al. 2002). In mouse, 20% of ORs are pseudogenes, whereas this fraction is much higher (∼60%–70%) in humans (Rouquier et al. 1998; Glusman et al. 2001; Zhang and Firestein 2002). The reduction in the human OR gene repertoire is postulated to be reflected in a poorer sense of smell (Young et al. 2002). A family of OR-like molecules have been found to be expressed in the testes of humans and dogs (Vanderhaeghen et al. 1997), and members have been localized to the spermatozoa where they have been linked to chemotaxis (Spehr et al. 2003).
V2Rs are members of family C GPCRs and are also expressed in the VNO (Matsunami and Buck 1997; Ryba and Tirindelli 1997). These receptors are similar to metabotropic glutamate and calcium-sensing receptors in possessing an N-terminal extracellular region and a C-terminal 7TM GPCR region (Matsunami and Buck 1997; Ryba and Tirindelli 1997). Based on studies of a goldfish V2R-like molecule which binds arginine and lysine (Speca et al. 1999), it appears possible that V2Rs bind protein rather than volatile organic compounds. This is consistent with observations that the V2R extracellular regions are homologous to bacterial proteins with amino acid-binding properties (Kunishima et al. 2000; Hermans and Challiss 2001).
In common with other GPCR superfamily members, ORs, V1Rs, and V2Rs present considerable challenges in the elucidation of their structures, functions, and evolution. GPCRs are characterized by an extracellular N-terminus, a cytosolic C-terminus, three cytosolic loops, and three extracellular loops (Filipek et al. 2003). The GPCR fold is represented by crystal structures of a single eukaryotic protein, bovine rhodopsin (Okada et al. 2002; Filipek et al. 2003), to which many GPCR family members such as V1Rs and V2Rs cannot be modeled with accuracy. In general terms, however, structure-function relationships for GPCRs are well established: extracellular ligands bind within the 7TM region and induce conformational changes that are coupled to the binding of intracellular G-protein heterotrimers (Wess 1997; Filipek et al. 2003). Nevertheless, the understanding of structure-function relationships for specific GPCRs is far from complete. This has lead to many sequence- and structure modeling-based studies dependent on the assumption that amino acids within functional sites are particularly well conserved (Floriano et al. 2000; Singer 2000; Oliveira et al. 2002; Vaidehi et al. 2002; Cavasotto et al. 2003).
Several pheromonal cues are known to directly affect rodent behavior. Aphrodisin, a female hamster lipocalin homolog present in the vaginal discharge, induces copulatory behavior in the male (Singer et al. 1986; Briand et al. 2000). Urinary and other scent compounds influence rodent mate selection, estrus synchronization (Whitten effect; Whitten 1958; Novotny et al. 1999), estrus suppression (Lee-Boot effect; Lee and van der Boot 1955; Ma et al. 1998), male-induced termination of pregnancy (Bruce effect; Bruce 1968), and puberty acceleration (Vandenbergh effect; Vandenbergh 1976, 1989).
Mouse major urinary proteins (MUPs) and their counterparts in rat, termed α2u-globulins, have also been shown to influence puberty acceleration. These genes show a high degree of sequence polymorphism, including differences between laboratory mouse strains (Robertson et al. 1996). MUPs and α2u-globulins are lipocalins whose tertiary structure consists of an eight-stranded antiparallel β-barrel enclosing an internal ligand binding site (Flower 1996; Cavaggioni and Mucignat-Caretta 2000). MUPs and α2u-globulin genes are largely expressed in the liver, under stimulation by androgens, and become concentrated in the urine by filtration from the blood serum (Knopf et al. 1983; Mucignat-Caretta et al. 1995; Flower 1996; Novotny et al. 1999; Cavaggioni and Mucignat-Caretta 2000). They are thought to stimulate behavioral responses either directly at close proximity (Mucignat-Caretta et al. 1995; Hurst et al. 2001) or by air-borne delivery of volatile compounds (Novotny et al. 1999).
Odorant binding proteins (OBPs) are additional members of the lipocalin family. They are expressed in glands producing mucus for the olfactory and vomeronasal organs. Although their functions remain unclear, they appear to bind small molecules either for presentation to specific receptors, or to remove odorants from the proximity of the receptors to allow detoxification (Miyawaki et al. 1994; Lobel et al. 1998; Steinbrecht 1998). Mouse chromosome X contains a cluster of eight OBP-like lipocalin genes. The syntenic region on human chromosome X reveals no evidence of a functional OBP-like gene. Rather it contains an OBP-like pseudogene (positions 4463681–4464381 on the April 2003 assembly) almost identical to another pseudogene on the Y chromosome (nucleotides 17822583–17823282). Whereas OBP genes have proliferated and diversified in the rodent lineage, it would appear that they have been lost in the human lineage. This is likely to be associated with the loss of other genes involved in olfaction such as ORs (Gilad et al. 2003; Menashe et al. 2003), V1Rs (Mundy and Cook 2003), and TRPC2 (Wes et al. 1995).
We sought to estimate the effects of evolutionary selection on the potential ligands, receptors, and accessory molecules involved in odorant detection by exploiting the newly sequenced rat and mouse genomes. We infer selection by estimating the ratio of KA, the number of nonsynonymous substitutions per nonsynonymous site, to KS, the number of synonymous substitutions per synonymous site (Miyata and Yasunaga 1980; Yang and Nielsen 2000; Hurst 2002). Typically KA/KS is small (<0.15) for genes subject to purifying selection, whereas for pseudogenes, which are not subject to selection, this ratio relaxes towards 1. Those genes that contain several sites that have been subject to diversifying selection and rapid amino acid substitution tend to possess higher KA/KS values. In exceptional circumstances, KA/KS values may far exceed 1, providing unequivocal support for positive diversifying selection.
However, positive selection is often found to be acting on only a few sites, with the majority of sites remaining under purifying selection. In these cases, selection will not necessarily be identified by analyzing gene-averaged KA/KS values. In order to identify such instances, sequence data can be fitted to pairs of codon-based substitution models of evolution that either allow or disallow sites with KA/KS > 1. Results from such nested models can be compared to provide a statistically significant estimate of their relative likelihood (Yang et al. 1998). The predictive accuracy and power of these methods have been demonstrated by computer simulations (Anisimova et al. 2001, 2002). Their likelihood-ratio tests (LRTs) have been shown to act conservatively, suggesting that predictions are most likely to represent real evolutionary processes, rather than being artifacts of the method. The accessibility of these methods in PAML (Yang 1997) has afforded much opportunity to apply these techniques to biological data. Results have been very encouraging, and the LRT methods implemented in PAML have become well established in predicting sites that have been subject to positive selection (Zanotto et al. 1999; Bishop et al. 2000; Yang et al. 2000b; Peek et al. 2001; Schaner et al. 2001; Yang and Nielsen 2002; Yang and Swanson 2002; Jansa et al. 2003).
Here we used maximum likelihood models of sequence evolution to analyze OR, V1R, and V2R families. We performed a similar analysis on potential extracellular ligands of these receptors (MUPs, LUPs, and OBPs) and MHC class Ib molecules thought to interact with V2Rs (Ishii et al. 2003; Loconto et al. 2003). These analyses predict single codons that are likely to have been subject to positive selection. We interpret the rapid evolution of these sites as indicative of their participation in binding interactions. This method represents the complement of the more usual approach where purifying selection and sequence conservation are harnessed to predict functional sites.
RESULTS
We performed evolutionary analyses of 16 families of rat and mouse genes that are known to be involved in odorant-mediated G-protein signaling pathways (Table 1). With the exception of the G-protein subunits, each of these families yielded median KA/KS values that exceed the median KA/KS value (0.11) for the set of all mouse–rat orthologs (Rat Genome Sequencing Project Consortium 2004). In particular, V1Rs, odorant-binding proteins (OBPs and MUPs/α2u-globulins) and MHC M10 molecules possess unusually high median KA/KS values, indicating that they might have been subject to adaptive evolution.
Summary Information of Families Analyzed
Multiexon genes were predicted from the rat and mouse genome assemblies using hidden Markov models derived from multiple protein sequence alignments and Genewise (Birney and Durbin 2000). This ensures accurate prediction of full-length genes containing the appropriate complement of exons where the genome sequence allows. For each family (with the exceptions of OBPs and G-protein subunits), representative genes were chosen by virtue of their involvement in multiple recent gene duplication events. This is because gene duplication is often associated with sequence diversity (Emes et al. 2003), and because it provided sufficient gene numbers for per-codon evolutionary analysis. Detailed analyses of all members of all of these families are beyond the scope of this study. However, our findings that adaptive evolution has occurred to varying extents among the gene sets investigated suggests that adaptive evolution is likely to have acted differentially within and between these families.
With the exception of G-protein subunits, each family was investigated for sites predicted by the codeml algorithm to have been subject to adaptive evolution (see Methods). To ensure consistency of predictions among the different families, only nonconserved alignment positions that were predicted to have been subject to positive selection by one codeml model with a posterior probability P > 0.90, and by another codeml model with P > 0.50, were considered (Table 1). We will use the term “ω+ sites” to describe these alignment positions. For interpretative purposes, ω+ sites have been mapped to homologous positions within representative protein crystal structures using plausible multiple sequence alignments (Supplemental Table 2 available online at www.genome.org). From the fractional solvent accessibility estimates of these sites (Table 2) it is evident that ω+ sites are underrepresented among buried amino acids, and overrepresented among exposed amino acids. Thus, the majority of these ω+ sites are available to participate in binding interactions. We provide multiple alignments, ω+ sites, genome coordinates, and gene prediction methodology information in Supplemental Table 2.
Relative Accessibility of ω+ Residues
Olfactory Receptors
Approximately 1500 OR genes are present in the mouse genome (Young and Trask 2002), and a similar number is expected in the rat genome. Performing an evolutionary analysis of all sites for all of these genes is prohibitive in terms of computational resources. Consequently, we decided to identify OR genes that have been particularly prone to gene duplication since the divergence of rat and mouse lineages. Using a set of 672 well described mouse ORs and 810 homologous rat genes, we constructed a phylogenetic tree from a matrix of gene pairs' KS values (data not shown). Using a KS upper bound of 0.2, which represents the approximate median KS value for 1:1 rat–mouse orthologs (Rat Genome Sequencing Project Consortium 2004), we dissected clades from the tree that contained 10 or more ORs (Table 1). Five such clades were found. Codeml analysis of these five OR families revealed a wide variation in ω+ site number, ranging from just two sites for two families, and 9, 14, and 22 sites for the remaining three clades (Fig. 1C,D; Table 1; Supplemental information).
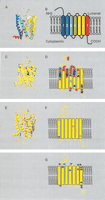
Site-specific KA/KS analysis of olfactory receptors, candidate testis odorant receptors, and V1Rs. ω+ codons that were predicted to be under positive selection are mapped to a ribbon representation of the structure of bovine rhodopsin (PDB 1L9H; Okada et al. 2002; for full details see Suppl. Table 2). (A) Ribbon diagram of bovine rhodopsin chain A colored by secondary structure succession. (B) Schematic representation of secondary structures. Ribbon diagram (C) and secondary structure schematic (D) of ω+ sites for olfactory receptors. ω+ sites predicted for OR family A are highlighted in green, B in black, C in purple, D in blue, and E in red. The analysis of these ORs indicates that positive selection appears to be confined largely to the lumenal half of the molecule. Seventy-five percent of the ω+ sites are located in the amino terminal region, extracellular loops, and the extracellular half of the transmembrane helices. Ribbon diagram (E) and secondary structure schematic (F) of ω+ sites for the candidate testis-specific odorant receptors (posterior probability > 0.99, in a single model). Three ω+ sites identified when analyzing the rat genes alone are highlighted in green. A single ω+ site identified in analysis of both the mouse and rat lineages is highlighted in blue. (G) Schematic of ω+ sites for the V1R family (posterior probability > 0.90 in one model and > 0.5 in at least one other model) highlighted in blue. The V1R proteins could not be reliably aligned to the rhodopsin sequence, and so positions of ω+ sites are relative to published predictions of secondary structures (Dulac and Axel 1995; Ryba and Tirindelli 1997). Swiss-PDBviewer (www.expasy.org/spdbv/; Guex et al. 1999) was used for all structural manipulations, and POVRAY (www.povray.org) was used to generate images.
We were interested in analyzing homologs of ORs that are expressed in mammalian testes, as these have been suggested to mediate sperm chemotaxis (Parmentier et al. 1992) and might, therefore, exhibit relatively high evolutionary rates (Branscomb et al. 2000). We collated a set of rat and mouse ORs that are predicted, by synteny and phylogenetic analyses (data not shown), to be orthologous to hOR17–4. This is a human testicular OR that has been shown to mediate chemotaxis activity (Spehr et al. 2003). Four ω+ sites were identified among rat genes of this family, of which one was also predicted for mouse genes (Table 1; Fig. 1 E,F).
Vomeronasal Receptors
We identified 14 ω+ sites for a set of mouse V1R genes, and their likely rat orthologs, that, when deleted, results in modified behavioral responses (Del Punta et al. 2002). The majority of these sites map to extracellular loops and four of the seven TM regions (Fig. 1G). We then performed a similar analysis of those V2R genes that are the closest homologs to V2R1, the founder member of this family (Ryba and Tirindelli 1997), and identified 12 ω+ sites. Because V2Rs contain two distinct structural regions, these sites were mapped to crystal structures either of rat metabotropic glutamate receptor subtype 1 (mGluR1; PDB 1EWK, (Kunishima et al. 2000) or to the secondary structure of bovine rhodopsin. Eight of the 12 ω+ sites map to the extracellular mGluR1-homologous domain (Fig. 2).

Site-specific KA/KS analysis for V2Rs. (A) Ribbon diagram showing ω+ sites. (B) Representation of the extracellular domain of metabotropic glutamate receptors with mapped ω+ sites. Predicted ω+ sites are mapped as described in Figure 1 and colored blue. Residues are mapped to the structure of rat metabotropic glutamate receptor subtype 1 complexed with glutamate here colored purple (PDB 1EWK, Kunishima et al. 2000). ω+ sites in the carboxy-terminal domain were mapped to the secondary structures of bovine rhodopsin 1EWK (for full details see Suppl. Table 2).
Pheromones and OBPs
Three lines of evidence strongly suggest that mouse MUPs and rat α2u-globulins arose via separate gene expansions that have been driven by adaptive evolution. First, the topology of the MUP/α2u-globulin phylogenetic tree consists of two well separated clades, one specific to mouse MUPs and the other specific to rat α2u-globulins (data not shown). Second, rat α2u-globulin, but not mouse MUP, gene duplications have often been accompanied by a `hitchhiking' zinc finger-pseudogene (Rat Genome Sequencing Project Consortium 2004). Finally, α2u-globulin exonic sequence is less conserved than their intronic sequence (Fig. 3A), again implying rapid coding sequence diversification.
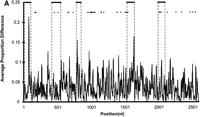
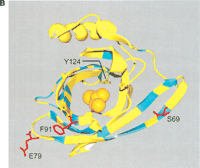
(A) Sequence divergence of rat α2u-globulin genes and pseudogenes. Genomic DNA from 22 rat α2u-globulin genes, including 12 suspected of being pseudogenes, identified as described in Methods. Nucleotide sequences were highly conserved (mean 93%±2% identity calculated from ungapped columns), and were aligned using HMMer (Eddy 1998) with manual adjustments. The proportion difference was calculated as the fraction of nucleotides in each column that differ from the consensus nucleotide. The average proportion difference from a sliding window of 10 nt is plotted against initial nucleotide position. Alignment columns with >50% gaps are shown as diamonds and were ignored for these calculations. Exons are shown as solid horizontal lines, with intron-exon boundaries defined by dashed vertical lines. (B). Site-specific KA/KS analysis of the MUP/α2u-globulin family, lipocalin homologs. ω+ codons with a posterior probability >0.90 by one codeml model, and >0.5 by at least one other model, are shown mapped to the crystal structure of murine MUP complexed with 2-(Sec-butyl) thiazoline (PDB 1MUP; Bocskei et al. 1992). Residues corresponding to ω+ codons are highlighted in blue; 2-(Sec-butyl) thiazoline is shown in orange. A single interior ω+ site corresponds to 1MUP:Y124 (shown in green). Although there are no data available to support a direct role in binding, this tyrosine residue neighbors, and has a similar orientation to, 1MUP:R126, which is required for thiazoline-binding (Bocskei et al. 1992). Y124 is as close to 2-(Sec-butyl) thiazoline as 1MUP:F60, a proposed ligand-binding residue (Darwish Marie et al. 2001). All mouse, and most rat, sequences contain a tyrosine in the position equivalent to 1MUP:Y124. However, two rat genes (mup_rn11 and mup_rn20) possess substitutions (to leucine and tryptophan, respectively) that might modify the geometry of the internal ligand-binding cavity. Consequently, these two genes might have evolved either to bind a ligand different from those of their paralogs, or else the same ligand with a different affinity. We also identified three codons (1MUP:S69, E79, and F91) that have been subject to rat lineage-specific positive selection. These are shown here in ball-and-stick format and colored red. These sites are the only alignment positions where amino acid substitutions are not conserved in rat α2u-globulins but are conserved in mouse MUPs. One rat α2u-globulin (mup_rn5) possesses unusual substitutions at all three positions (for S69, two substitutions at second and third codon positions [R→T]; for E79, a first position transversion [P→A]; and for F91, a second position transition G→E). mup_rn18 shares the R→T substitution at S69 with mup_rn18. This suggests that at least three sites in α2u-globulins may be under positive selection in the rat lineage alone, possibly reflecting the functional divergence of two genes (mup_rn5 and mup_rn18) from the other rat paralogs.
Site-specific analysis of the MUP/α2u-globulin family showed strong evidence for positive selection at an exceptionally high proportion of possible sites (32 of 157 sites; Table 1). All but one of these ω+ sites map to the surface of the mouse MUP tertiary structure (Bocskei et al. 1992), rather than to its 2-(Sec-butyl) thiazoline-binding pocket (Fig. 3B). This observation is consistent with previous amino acid conservation studies (Darwish Marie et al. 2001). We have also obtained evidence for rat α2u-globulins, but not their MUP counterparts in mouse, that three codons (S69, E79, and F91) have been subject to rat lineage-specific positive selection (Fig. 3B).
Site-specific analysis of OBP genes also predicts a large number of ω+ sites (Table 1). Mapping these onto a known crystal structure (1OBP; Bianchet et al. 1996) reveals that the majority of sites are located within the β-sheets, and project outwards towards the solvent (Fig. 4A). When the 1MUP-Family 5 alignment and 1OBP-Family 6 alignment are superimposed, it is apparent that nine ω+ sites are shared by both the MUP and OBP families. Seven of these sites cluster in a region of the 1MUP protein surface, formed by two β-strands and a loop (Fig. 4B), that is distinct from both its ligand-binding pocket and the OBP dimerization surface (data not shown; Bianchet et al. 1996).

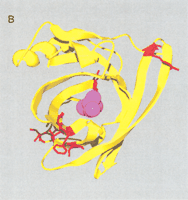
(A) Site-specific KA/KS analysis of OBPs. ω+ codons with a posterior probability >0.90 by one codeml model, and >0.5 by at least one other model, were mapped to the crystal structure of cow OBP (PDB: 1OBP; Bianchet et al. 1996) and colored blue. Three ω+ site side-chains, shown in ball-and-stick format and colored red, are predicted to project into the interior of the OBP β-barrel. Two of these side-chains (1OBP:T38 and V69) are within 5 Å of the ligand that was cocrystalized in the 1OBP X-ray structure, whereas the third side-chain (1OBP:E84) is more distant from the ligand (∼8 Å), and closer to the cavity edge. (B) Positively selected sites shared by both MUP/α2u-globulins and OBPs. A multiple alignment of MUP/α2u-globulins and OBPs was used to identify nine ω+ codons shared by both families. These sites are spatially clustered and therefore represent a hypervariable region likely to be important for functional diversity. The nine ω+ codons were mapped to the crystal structure of mouse MUPs (PDB 1MUP) and are colored red. 2-(Sec-butyl) thiazoline is shown in purple.
Following the successful identification of high numbers of ω+ sites in OBPs, mouse MUPs, and rat α2u-globulins, our attention was drawn to another family of rodent urinary proteins. Two Ly-6 homologous proteins were originally identified in rat urine (Southan et al. 2002). Mapping their genes to the same region of the rat genome resulted in the identification of a large family of Ly-6 homologous proteins, which we shall refer to as Ly-6-homologous urinary proteins (LUPs). Investigation of the syntenic regions of mouse and human genomes revealed evolutionary characteristics similar to those seen from MUPs (Fig. 5A). These include independent expansions of LUPs in both rodent genomes and elevated KA/KS values (Table 1). The phylogenetic tree of LUP sequences indicates that only five rat–mouse–human 1:1:1 orthologous relationships can be resolved (Fig. 5A; data not shown). Of the remaining 22 sequences, eight rat LUP genes appear to have undergone a recent rapid expansion, with only one close mouse relative, and no orthologous human sequence apparent.
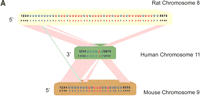
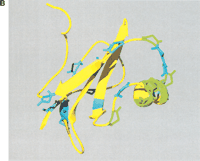
(A) Gene order of Ly-6 homologous urinary protein (LUP) genes. Syntenic regions of mouse chromosome 9 (February 2003 assembly), human chromosome 11 (November 2002 assembly), and rat chromosome 8 (June 2003 assembly) are shown, approximately to scale. Genes are indicated in blue letters, and pseudogenes in red. Strand orientation is indicated by arrowheads. Homologous rat–mouse–human relationships are represented by pink lines. A single mouse–rat homologous relationship that does not have a detectable human counterpart is represented by a green line. Numbers represent genes not homologous to LUPs: 1, BOC an immunoglobulin superfamily member; 2, DEAD box polypeptide 25; 3, FLJ32915 hypothetical protein; 4, pseudouridine synthase 3; 5, checkpoint kinase 1; 6, oligosaccharyl transferase STT3 subunit (B5); 7, P53-induced protein 8; and 8, zygin I. LUP genes and pseudogenes scored greater than 0 bits using GeneWise, default parameters [excepting a substitution error rate (subs) of 0.1], and a hidden Markov model of LUP homologs' amino acid sequences. Pseudogenes, as opposed to genes, were assigned on the basis of incomplete sequences, frameshifts and in-frame stop codons. (B) Sitespecific KA/KS analysis of LUP homologs. ω+ codons with a posterior probability >0.80 by at least two codeml models are mapped to the NMR structure of human CD59 (PDB 1CDQ; Kieffer et al. 1994). Residues corresponding to ω+ codons are highlighted in blue. Several residues that have been experimentally shown to be important for species selectivity of human CD59 (i.e., H44, N48, D49, T51, T52, R55, and E58; Zhao et al. 1998) are shown in green.
Site-specific KA/KS analysis of this LUP family provided support for positive selection across mouse, human and rat lineages for 25 sites (Table 1; Suppl. Table 1). For more stringent and better-fitting (M2 selection and M3 discrete) models, 15 ω+ sites were predicted with P > 0.9 by one codeml model and P > 0.8 by an additional codeml model. Of these, nine form a cluster on the structure of human complement regulatory protein CD59 (Kieffer et al. 1994), an LUP and Ly-6 homolog (Fig. 5B). A functional role for this region has not yet been proposed. An additional four ω+ sites map to the opposite face of CD59, which has been shown to be critical for species specificity (Fig. 5B; Zhao et al. 1998).
MHC M10 and G-Protein Subunit Genes
Analysis of the MHC M10 genes again predicted an exceptionally large number (50) of ω+ sites (Table 1). Mapping these sites onto the structure of mouse MHC class I H-2DD, chain A (PDB 1BII; Fig. 6; Achour et al. 1998) shows that 26 of these sites are present in the two α-helices of the antigen-binding site. Strikingly, no ω+ sites were detected in the immunoglobulin (IG) domain (Bjorkman et al. 1987b; Achour et al. 1998; see Discussion).

Site-specific KA/KS analysis of MHC-M10 genes. Codons that were predicted to be under positive selection with a posterior probability >0.90 by one codeml model, and >0.5 by at least one other model, are mapped to the crystal structure of mouse MHC class I H-2DD, chain: A in complex with the HIV-1 derived peptide p18–110, shown in green (PDB identifier 1BII (Achour et al. 1998). ω+ sites, shown in blue, are predominantly located along the α-helices of the peptide recognition region. No ω+ sites were mapped to the immunoglobulin domain of the protein, shown here in orange.
Due to the small number of orthologs known for each of the Gαβγ families relevant to olfaction, site-specific KA/KS analysis was not conducted. However, using yn00 (Yang and Nielsen 2000), the gene pair median KA/KS values for Gα, Gβ, and Gγ families were found to be 0.028, 0.025, and 0.081, respectively, which are lower than the median KA/KS value (0.11) for all 1:1 mouse:rat orthologs (Rat Genome Sequencing Project Consortium 2004). This strongly suggests that G-proteins, including those relevant to odorant signal transduction, have been subject to strong purifying and not diversifying selection.
DISCUSSION
ω+ Sites as Functionally Important Sites
The inference of function from sequence information frequently assumes that conserved residues are of the greatest functional importance, because these have been subject to strong purifying selection over long periods of evolution (Sander and Schneider 1993; Lichtarge et al. 1996; Mirny and Shakhnovich 1999; Wood and Pearson 1999; Friedberg and Margalit 2002; Madabushi et al. 2002). However, there are two reasons why this approach is not appropriate for genes that evolve rapidly over shorter time periods. First, for orthologs and paralogs that have diverged relatively recently, insufficient time has elapsed for significant sequence divergence due to genetic drift (even for human–mouse alignments, nucleotide identity at selectively neutral sites is high, ∼67% [Waterston et al. 2002]). Thus residue identity is more likely to be due to chance rather than purifying selection.
Secondly, there are reasons to believe that some functionally important sites on rapidly evolving proteins are characterized by amino acid substitution, rather than conservation. Sites that have experienced more unusual and greater numbers of nucleotide substitutions present evidence for adaptive, or Darwinian, selection: Substitutions are fixed in the population more rapidly when advantages accrue to the individual in which they occur (Bock 1980; Golding and Dean 1998; Hughes 1999). Evidence for positive selection implicates these single sites as mediators of function.
Receptors and Positive Selection
We have obtained evidence for positive selection acting on sites within OR, V1R, and V2R subfamilies, and MUP/α2u-globulin and LUP putative pheromone ligands. It is striking that the majority of these ω+ sites are solvent-accessible (Table 2) and, for V1Rs and V2Rs, lumenal (10 of 14 ω+ sites in V1Ra, and 10 of 12 ω+ sites in V2Rs, are lumenal). Thus, predicted ω+ sites on receptors and their putative extracellular ligands are available to participate in potential receptor-ligand interactions.
The V2R ω+ sites form two distinct clusters (Fig. 2). The first of these is within the amino-terminal ligand binding domain (LB1) and forms a cluster with residues involved in dimer formation (Kunishima et al. 2000). Thus, variation of codons at this site may influence the affinities of V2R hetero- or homodimer formation (Fig. 2B). The second cluster lies distant from known ligand binding or dimerization sites in LB2 (Kunishima et al. 2000; Tsuchiya et al. 2002).
Relatively few ω+ sites map to the 7TM regions of V1Rs and V2Rs. This contrasts with 72.7% of ω+ sites in the five OR families that lie within TM helices. It is notable that 75.0% of ω+ sites occur within the lumenal half of the OR molecule (the amino-terminal and extracellular loops, and the lumenal halves of TM helices; Fig. 1C,D). This portion of the OR structure is thought to contain the odorant binding pocket within TMs 3–7 (Floriano et al. 2000; Singer 2000; Vaidehi et al. 2002). Two residues K164 (TM4) and F209 (TM5) predicted to bind the ligand in rat OR-17 (Singer 2000) are predicted ω+ sites for at least one of the OR families analyzed here. Additional ω+ sites lie close to TM3 residues which influence ligand binding (Vaidehi et al. 2002). Thus, the receptors' ω+ sites predicted in this study represent excellent candidates for mediating ligand interactions.
For ORs (Fig. 1 C–F), V1Rs (Fig. 1G), and V2Rs (Fig. 2), ω+ sites were predicted at cytoplasmic regions of TMs 1 and 7, which spatially cluster in three dimensions (Fig. 1A). G-proteins are thought to bind GPCRs following a conformational change involving TM7 (for review, see Filipek et al. 2003). The clustering of ω+ sites at the TM1 and TM7 cytoplasmic surface of these GPCRs thus suggests that these sites may bind G-protein subunits. However, it is notable that we find no evidence for positive selection and gene duplication of G-protein subunit genes. Thus, the rapidity of sequence change within these receptors' sites might have arisen from adaptive requirements for higher association or dissociation constants with their cognate G-proteins.
Ligands and Positive Selection
External residues are also overrepresented among ω+ sites in putative pheromones (Table 2), and thus these may represent possible binding sites. Only one ω+ site occurs within the interior of MUPs that may participate in binding the volatile odorant (Fig. 3B; Timm et al. 2001). The identification of these ω+ sites should assist in teasing apart the pheromonal functions of MUPs. Both the MUP protein without a bound ligand, and a recombinant MUP have pheromone activities (Mucignat-Caretta et al. 1995; Hurst et al. 2001). Moreover, a rat α2u-globulin lacking a ligand induces the activation of GO, a G-protein that is coexpressed with V2Rs in the basal half of the VNO neuroepithelium (Krieger et al. 1999). This strongly suggests that mouse MUPs and rat α2uglobulins bind V2Rs, and that ω+ sites in MUPs/α2u-globulins and V2Rs might participate in these MUP-V2R and α2u-globulin-V2R heterodimeric interactions. A V1R is known to bind 2-heptanone (Boschat et al. 2002), whose analog is a volatile ligand for MUPs (Timm et al. 2001). Thus, each component of the odorant-MUP/α2u-globulin complex may be recognized by one of the two VNO receptor types: the odorant by V1Rs and the MUP/α2u-globulin by V2Rs.
We also investigated LUPs whose members, like MUPs, are expressed in the male rat liver (evidence from EST BQ094064), and are not known to be expressed in females. Like MUPs they possess the unusual property of being present at high levels in male rat urine (Southan et al. 2002). Moreover, their rapid sequence diversification, pseudogenization and gene duplication in rodents mirror those seen for MUPs and are strong evidence for adaptive evolution. Together, the evolutionary and expression similarities between MUPs and LUPs, and their unusually high concentrations in urine, suggest to us that these two families of proteins possess comparable functions. Consequently, we suggest that rodent LUPs, like MUPs, may be pheromones which contain hypervariable solvent-accessible binding sites.
M10s and Positive Selection
We identified 50 ω+ sites in MHC M10 genes. M10s are nonclassical class I MHC molecules which possess a comparable structure to class 1 heavy chains. The class I heterodimer consists of the heavy chain, containing a peptide-binding domain and an IG domain, and a β2-microglobulin light chain. M10s are not known to possess immunity functions comparable to those of classical class I MHC molecules, but the finding that β2-microglobulin is required for the cell surface expression of M10s argues that the molecular functions of nonclassical and classical class I MHC molecules are highly similar.
Twenty-six of the 50 ω+ sites are located within the two α-helices that, in class 1 molecules, form the peptide-binding groove. For class 1 molecules those residues that bind antigen, and others that bind a T-cell receptor (TCR), have been determined (Bjorkman et al. 1987a). Four ω+ sites in α -helix 1, and six in α-helix 2, map to predicted TCR-binding residues, whereas seven ω+ sites in α-helix 1, and four in α-helix 2, are located in the equivalent antigen-binding site (Fig. 6). M10s appear, from their high propensities of gene duplication and sequence diversification, to have been subject to strong adaptive constraints during their recent evolution. Selection appears to have driven sequence diversity within a peptide-binding site of M10s, in a similar manner to that of classical class 1 molecules (Hughes and Nei 1988; Swanson et al. 2001).
Several functional roles have been proposed for M10s (Dulac and Torello 2003; Ishii et al. 2003; Loconto et al. 2003). M10s as accessory molecules in V2R-mediated pheromone recognition does not account for the exceptionally strong adaptive evolution that we have observed. As pheromone recognition itself is well described for V2Rs only, this leaves a role for M10s in binding protein pheromones during their cellular internalization, which we assume is necessary for VNO detoxification. These observations contribute to the ongoing debate regarding whether the MHC mediates pheromone-recognition events and odorant-induced behavior (Singh et al. 1987; Potts et al. 1991; Schaefer et al. 2001).
Selective Pressures
Underlying the positive selective pressures exerted on these pheromone and odorant receptors is competition among individuals. Interspecific competition in terms of foraging and predator avoidance may have driven OR sequence diversification and gene duplication. Conspecific competition, however, such as among adults for mate selection (Alley 1982; Bolnick et al. 2003), or among siblings for sustenance (Stockley and Parker 2002), may have acted on all other genes considered in the present study. This is because olfactory cues are costly in terms of fitness (Gosling et al. 2000), and have been implicated in individual recognition (mediated by MUPs; Hurst et al. 2001) and sexual behavior (Lee and van der Boot 1955; Whitten 1958; Bruce 1968; Vandenbergh 1976, 1989; Ma et al. 1998; Novotny et al. 1999), and thus provide a potent mechanism for sexual selection. We hypothesize that conspecific competition has resulted in the propagation, within rodent populations, of pheromones that are well adapted to their receptors, and receptors that are well adapted to their pheromones. Adaptations consequently involve amino acids situated at the interface between pheromone and receptor, and these are represented among the ω+ sites identified in this study. This hypothesis is well suited to experimental investigation by molecular, site-directed mutagenesis, and ethological studies.
METHODS
Sequence Data Collection
Mouse and Rat Olfactory Receptors
GenBank identifiers of predicted full-length mouse olfactory receptors were obtained from ORDB, a database of olfactory receptors (Crasto et al. 2002). Subsequently, the corresponding DNA sequences were mapped to the mouse genome (Feb 2002 mmbuild2) and known Ensembl (Hubbard et al. 2002) mouse genes using BLAT, default parameters and a minimum acceptance threshold of 95% sequence identity over 40 nucleotides (Kent 2002) at the UCSC genome browser (http://genome.ucsc.edu/). The corresponding protein sequences of these genes were multiply aligned using CLUSTAL W (Thompson et al. 1994) and edited manually to minimize gap positions. Sequences that appeared to be incomplete were removed from the alignment. Orthologous rat genes were then obtained using relationships available from Ensembl's Compara database (Clamp et al. 2003).
Vomeronasal Receptors, MUPs, LUPs, OBPs, MHC-M10, and G-Proteins
Families of genes were collected for further analysis using a protocol of gene identification, mapping of the gene to the genome, and gene building (Suppl. Table 2). Briefly, a starting gene set was identified containing either known reference sequences or else homologs identified by database searching. The gene set was then mapped to the relevant genome via EnsMart (http://www.ensembl.org/EnsMart/) or BLAT and the UCSC genome browser. To obtain fulllength gene predictions, genomic DNA in the region of the mapped genes was then used as a target for gene prediction using GeneWise (Birney and Durbin 2000). The initial step of gene mapping using BLAT dramatically reduces the search space for the gene building programs, adding greater reliability in gene prediction. For OBPs, identified protein sequences were used in a PSI-BLAST search (Altschul et al. 1997) to obtain likely orthologs from species other than mouse and rat. G-protein-α, β, and γ sequences were obtained by text searching EnsMart at Ensembl (http://www.ensembl.org/EnsMart/). FASTA-formatted sequence files, alignments, and trees for each family are available as Supplemental information.
The solvent accessibility of ω+ residues was calculated using structural information from PDB (http://www.rcsb.org/pdb/). For each structure, any ligand was removed prior to accessibility score prediction using the DSSP algorithm (Kabsch and Sander 1983). Relative accessibility scores were produced by dividing by amino acid-specific maximal accessibility values. These scores were fractionated into three states: buried (<9% relative accessibility), intermediate (9%–35% relative accessibility), and exposed (≥36% relative accessibility), as described (Rost and Sander 1994).
Estimation of Evolutionary Rates and Orthology Relationships
Evolutionary rates were estimated for all gene pairs in each of the 16 families. When multiple transcripts were predicted for a single gene, the longest transcript was used. The corresponding DNA sequences were aligned according to the amino acid multiple alignment. All pairs of these sequences were extracted from the multiple alignments and used to calculate values of KA and KS using yn00 (Yang and Nielsen 2000) and default parameters.
Site-Specific KA/KS Analysis
Site-specific KA/KS analysis was carried out using codeml, an application from the PAML package (v1.31) of phylogenetic software for applying different models of evolution to sequences (Yang 1997). For each analysis, cDNA sequences were collected as described previously. Phylogenetic trees were constructed from the KS matrices by the neighbor-joining algorithm implemented in the PHYLIP Neighbor application (Felsenstein 1989). In order to ensure that the site-specific KA/KS analysis was not influenced by sites with a large number of gaps, all columns with gaps in greater than 25% of the sequences were removed from the cDNA alignment. The KS-derived tree file and alignment were used as input files for codeml. Codeml uses maximum likelihood to predict sites in a group of cDNA sequences that have been subject to positive selection. Simple models where sites are predicted to have a KA/KS or ω ratio between 0 and 1 can be compared with more complex models that also allow for ratios that are greater than 1. Log likelihood values (l) are calculated for each model by maximum likelihood. This enables a likelihood ratio test (LRT) to be used to provide a statistically significant comparison between a simple model, and a more complex nested model. If the complex model indicates an estimated ω ratio that is greater than 1, and the test statistic (2Δl) is greater than critical values of the Chi square (χ2) distribution with the appropriate degree of freedom (Yang et al. 1998), then positive selection can be inferred. Bayesian probabilities are used to predict which sites (codons) from the original data are most likely to have been under positive selection. We used three pairs of simple and complex models: M0 (one-ratio; Goldman and Yang 1994) versus M3 (discrete; Yang et al. 2000a); M1 (neutral) versus M2 (selection; Nielsen and Yang 1998); and M7 (β) versus M8 (β + ω; Yang et al. 2000a). M0 allows a single per-site ω value for all sequences, which is allowed to vary between 0 and 1, whereas M3 attempts to fit the data to three ω values estimated from the data. M1 allows an ω value of 0 or 1, whereas M2 allows an additional site class which is estimated from the data. M7 attempts to fit the data to a beta distribution of ω values between 0 or 1, with 10 site classes, whereas M8 allows an additional site class which is estimated from the data. According to previous studies, these tests are of increasing stringency, with M8 having the most statistical power, and M3 the least (Anisimova et al. 2002). The M0/M3 LRT is compared to χ2 with four degrees of freedom (df), whereas both the M1/M2 and M7/M8 LRTs have two df; the latter two pairs thus possess a greater statistical power. Consequently, predictions by M0/M3 alone should be viewed with caution in the absence of corroborating evidence from other model pairs. It was previously noted that the iterative estimation of ω values by both M2 and M8 are susceptible to local minima. This has the result that the most likely numerical solutions may not always be found (http://abacus.gene.ucl.ac.uk/software/paml.html). To circumvent this problem, we initiated M2 and M8 codeml runs with different initial ω values (ω = 0.03, 0.3, and 1.3), and present here only those results with the greatest log likelihood values.
Protein Structure Analysis
Representative protein tertiary structures were chosen for each gene family based on the most similar sequence for which a crystal structure is available (Table 2). The amino acid sequence corresponding to the chosen structure was aligned to the family alignment using HMMer or BLAST, depending on the degree of similarity, and manually adjusted to ensure conservation of secondary structure elements. Consistency between different evolutionary models was taken into account, so that, where appropriate, only nonconserved alignment positions predicted to be under positive selection with a posterior probability >0.90 by one codeml model (i.e., M2, M3, or M8), and >0.50 by at least one other model, were highlighted. We have termed these alignment positions “ω+ sites”. Swiss-PDBviewer (www.expasy.org/spdbv/; Guex et al. 1999) was used for all structural manipulations, and POVRAY (www.povray.org) was used to generate images.
Acknowledgments
We thank Eitan Winter, and other CPP group members, for helpful discussions, and Prof. Ziheng Yang for advice. We also thank Ensembl, George Weinstock, and the Rat Genome SequencingProject Consortium for valuable assistance. This work was funded by the UK Medical Research Council. S.A.B. is supported by a Royal Commission for the Exhibition of 1851 Fellowship.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
-
[Supplemental material is available online at www.genome.org.]
-
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.1940604.
-
↵1 These authors contributed equally to this work.
-
↵2 Present address: Division of Immunity & Infection, University of Birmingham. Birmingham, B15 2TT, UK.
-
↵3 Corresponding author. E-MAIL Chris.Ponting{at}anat.ox.ac.uk; FAX 44 (0)1865 272420.
-
- Accepted November 17, 2003.
- Received September 5, 2003.
- Cold Spring Harbor Laboratory Press








