
Data-Mining Approaches Reveal Hidden Families of Proteases in the Genome of Malaria Parasite
- Yimin Wu1,4,
- Xiangyun Wang2,
- Xia Liu1, and
- Yufeng Wang3,5
Abstract
The search for novel antimalarial drug targets is urgent due to the growing resistance of Plasmodium falciparum parasites to available drugs. Proteases are attractive antimalarial targets because of their indispensable roles in parasite infection and development, especially in the processes of host erythrocyte rupture/invasion and hemoglobin degradation. However, to date, only a small number of proteases have been identified and characterized in Plasmodiumspecies. Using an extensive sequence similarity search, we have identified 92 putative proteases in the P. falciparum genome. A set of putative proteases including calpain, metacaspase, and signal peptidase I have been implicated to be central mediators for essential parasitic activity and distantly related to the vertebrate host. Moreover, of the 92, at least 88 have been demonstrated to code for gene products at the transcriptional levels, based upon the microarray and RT-PCR results, and the publicly available microarray and proteomics data. The present study represents an initial effort to identify a set of expressed, active, and essential proteases as targets for inhibitor-based drug design.
[Supplemental material is available online at www.genome.org.]
Malaria remains one of the most dangerous infectious diseases in the world. It kills 1–2 million people each year, and is responsible for enormous economic burdens in endemic regions. The development of new antimalarial drugs is urgently needed due to the continuing high mortality and morbidity caused by malaria and the increasing prevalence of drug-resistance in the pathogenic parasite Plasmodium falciparum.
Malarial proteases have long been considered potential targets for chemotherapy due to their crucial roles in the parasite life cycle, and the feasibility of designing specific inhibitors (for reviews, seeMcKerrow et al. 1993; Rosenthal 1998; Blackman 2000; Rosenthal 2002). Efforts to identify functional proteases targeted by inhibition assays are ongoing. Subtilase-1 and Subtilase-2, two homologous serine proteases, are demonstrated to be involved in schizont rupture and merozoite invasion (Blackman et al. 1998; Barale et al. 1999; Hackett et al. 1999). Cysteine proteases have also been implicated in the rupture/invasion process (Salmon et al. 2001). A cluster of Serine Repeat Antigens (SERAs) exhibit limited sequence similarity to cysteine proteases, though their proteolytic activity remains undocumented (Delplace et al. 1988; Miller et al. 2002). A zinc-metallo-aminopeptidase has also been demonstrated to possess enzymatic activity (Florent et al. 1998). Meanwhile, three classes of proteases have been identified to be involved in hemoglobin degradation: (1) Four aspartic proteases (plasmepsin I, II, IV, and HAP) (see Banerjee et al. 2002 for a review); (2) three cysteine proteases (falcipain-1, -2, and -3) (see Rosenthal 2002 for a review); and (3) one metalloprotease (falcilysin; Eggleson et al. 1999). The successful crystallization of plasmepsin II and the expression of recombinant plasmepsin I/II and falcipain-2 represented a significant advance towards a functional understanding and a rational design of inhibitors of these enzymes (Silva et al. 1996; Bernstein et al. 1999;Tyas et al. 1999; Shenai et al. 2000; Dua et al. 2001).
The recent completion of the P. falciparum genome provides a basis on which to identify new proteases. The first pass annotation has predicted 25 proteases that belong to ten families of five catalytic classes (Table1). Despite this initial progress, direct evidence from protease inhibition assays and independent comparisons with other genomes suggest that in addition to the limited number of characterized and predicted proteases, many important proteolytic enzymes remain uncharacterized (Olaya and Wasserman 1991; Southan 2001). The following six sets of experimental data suggest that unidentified proteases are responsible for additional critical hydrolytic activities: (1) A calpain-type protease, which appears to be involved in merozoite invasion of red blood cells (Olaya and Wasserman, 1991); (2) an entire group of threonine proteases in the proteasome complex (Gantt et al. 1998); (3) proteases that catalyze the primary processing of Merozoite Surface Protein (MSP-1; David et al. 1984), Apical Merozite Antigen-1 (AMA-1; Narum and Thomas 1994), and the precursor of SERA (Li et al. 2002); (4) the gp76 and gp68 GPI-anchored serine proteases that cleave host erythrocyte surface proteins in P. falciparum and P. chabaudi, respectively (Braun-Breton et al. 1988); (5) a 75-kD merozoite serine protease (Rosenthal et al. 1987); and (6) a neutral aminopeptidase essential in hemoglobin digestion (Curley et al 1994). Additional supportive evidence that the majority of malarial proteases are unexplored comes from a comparison with the number of proteases found in other organisms. According to the statistics in the protease database Merops (http://www.merops.ac.uk) as released on March 18, 2002, all the model organisms possess a large number of predicted and characterized proteases (human, 493; mouse, 431; Drosophila melanogaster, 529; Caenorhabditis elegans, 360;Arabidopsis thaliana, 568; Baker's yeast, 112;Escherichia coli, 127; Bacillus subtilis, 119). An average of 2.21% of the gene products belong to the protease superfamily in 77 completed genomes. Hence, given the observation that the number of predicted proteases appears to be positively correlated with organismal complexity, one might envisage that a considerable number of malaria proteases have yet to be identified in the ∼23 Mbp Plasmodium falciparum genome that encodes for approximately 5300 gene products.
Ninety-two (92) P. falciparum Protease Homologs Predicted From Comparative Genomic Analysis
Here, we report a complete survey of protease homologs in the predicted and annotated P. falciparum genome (Gardner et al. 2002). Our initial comparative sequence search identified 92 putative malaria proteases, including potentially an interesting calpain, a metacaspase, and a signal peptidase I. Their expressions have been evaluated by microarray and RT-PCR assays. This study helps to develop an integrated view of a number of novel malarial proteases within an organismal, evolutionary, and functional context, and offers an intriguing opportunity to further target expressed and active proteases for chemotherapy.
RESULTS AND DISCUSSION
Ninety-Two Putative Proteases Are Predicted by Comparative Genomic Analysis
To gain further insight into the proteolytic machinery of the malaria parasite, the protein sequences in the annotated P. falciparum genome were subjected to an exhaustive search against the Merops protease database, which has a catalog and a structure-based classification of proteases. We adopted a relatively stringent threshold of E ≤ 1e-04 for BLASTP to ensure the high coverage with low false-positives. Redundant hits and partial sequences were excluded, resulting in a total of 92 protease homologs (Table 1). As highlighted in the Protease nomenclature column in Table 1, all twelve previously characterized proteases with proteolytic activity are included. In addition, as highlighted in the Gene ID column, 23 out of 25 proteases predicted by first-pass annotation published in PlasmoDB are included, among which subtilases 1 and 2 have been demonstrated to possess proteolytic activity; PFI0660c is not included because the E-score (0.39) of its closest homolog (Bacillus anthracis CAAX amino terminal protease, accession number NP_655263) is far below the cut-off 1e-04; PF11_0314 is not included because it is more likely to possess ATP hydrolytic and regulatory function than protoelytic function based on sequence homology.
The domain/motif organization of predicted proteases was revealed by the InterPro Search. For each putative malaria protease, the known protease sequence or protease domain of the highest similarity was used as a reference for annotation; the catalytic type and protease family were predicted in accordance with the classification in the protease database Merops (http://www.merops.co.uk/merops/merops.htm), and the enzyme was named in accordance with the SWISS-PROT enzyme nomenclature (http://www.expasy.ch/cgi-bin/lists?peptidas.txt) and literatures.
New Catalytic Types and Families
Proteases are classified into five major clans (Aspartic, Cysteine, Metallo, Serine, and Threonine) based on their catalytic mechanisms. They can be further grouped into distinct families and subfamilies by intrinsic evolutionary relationships (Rawlings and Barrett 1993). Using the comparative database search, we detected a total of 59 new protease homologs, in addition to 12 characterized proteases with proteolytic activity and 21 predicted by official annotation (Table 1). Moreover, a spectrum of conserved core characteristic domains/motifs for specific catalytic classes has been detected in most of the predicted proteases, indicating their potential activity.
The 92 putative proteases belong to 26 families of five clans, compared to the previously reported 12 proteases that belong to six families of four clans (Rosenthal 2002). The distribution (11% aspartic, 36% cysteine, 22% metallo, 17% serine, and 14% threonine) resembles those in other model organisms, supporting the fundamental premise that a prototype protease system is conserved throughout evolution (Rawlings and Barrett 1993; Southan 2001). Our speculation that a large number of potential proteases remain unexplored in the P. falciparumgenome appears justified. Undoubtedly, some of the uncharacterized proteases will perform crucial functions in the parasite life cycle as discussed below.
Examples of Potentially Important Proteases
Calpain
Calpain is a group of intracellular cysteine proteases that mediate a wide variety of physiological and pathophysiological processes, including signal transduction, cell motility, apoptosis, and cell cycle regulation (Sorimachi et al. 1997; Glading et al. 2002). In P. falciparum, a calpain, yet unidentified, was believed to be essential in merozoite invasion, based on the observation that Calpain inhibitors I and II strongly blocked invasion (Olaya and Wasserman 1991).
We have identified a putative calpain (MAL13P1.310) in the P. falciparum genome, which exhibits high sequence similarity toC. elegans calpain-7 (E=2e-35). Moreover, its ortholog (accession no. EAA19663) has been identified in the newly released genome of the model rodent malaria parasite Plasmodium yoelii yoelii. It possesses a catalytic domain (985–1453) detected by the Hidden Markov Model in the pfam search, with E = 8.0e-13 (Fig.1). The most intriguing aspect of this domain is the presence of three active sites (Cys1035, His1371, and Asn1391) that constitute a cleft crucial for catalytic activity (Arthur et al. 1995). A multiple alignment of the catalytic regions was produced for the putative plasmodial calpain and the representative human calpains. In addition to the invariable Cys-His-Asn triad, a high degree of identity is also observed in its vicinity, reflecting stringent functional and mechanistic conservation (Fig. 1). Indeed, the experimental demonstration that a single catalytic subunit in rat and chicken calpains possesses a full bona fide proteolytic activity (Yoshizawa et al. 1995) reinforces the potential processing capacity of the putative plasmodial calpain.

Multiple alignment of the catalytic domains of the putative P. falciparum calpain (MAL13P1.310) and the representative human calpains using T-coffee program followed by manual correction. The catalytic domain region is predicted to be from amino acid residue 985 to 1453 by pfam HMM algorithm. The three conserved amino acids, C(1035), H(1371), N(1391), that are part of the active sites are highlighted with arrowheads. Graphic presentation of the alignment and the consensus sequence were obtained by the program BOXSHADE 3.21. Conserved residues are shaded with black and gray. The accession numbers of calpain protein sequences used for alignment refer to Figure2.
Our further phylogenetic analysis of the putative P. falciparum calpain revealed its striking origin, which might have attributed to an alternative Ca2+-independent regulatory mechanism. Figure 2 shows the evolutionary tree inferred by the neighbor-joining (NJ) method using Poisson corrected distance (Saitou and Nei 1987). Evolutionary trees based on Parsimony (PAUP4.0) and Maximum Likelihood (PHYLIP) also yielded topologies and clade structures congruent with NJ (data not shown). Apparently, two putative plasmodial calpains belong to a novel monophyletic group of animal calpain-7 proteases, with 61% bootstrap support. They share the common domain architecture in the calpain-7 clade: lacking any significant similarity to the C-terminal EF-hand Ca2+-binding domain present in most of the essential Ca2+-dependent mammalian calpain subtypes (calpains -1, -2, -3, -9, -11, and Mu/M-type) (Franz et al. 1999). Provided that fungi cysteine protease PalB, the nearest neighbor of calpain-7, contains a PBH domain resembling the Ca2+-binding domain (Denison et al. 1995), one could speculate that the loss of Ca2+ dependency in calpain-7 subtype had been derived from evolutionary events such as domain shuffling, which might be associated with the divergence of mRNA splicing sites (Craik et al. 1983). Such events appear to have occurred close to or prior to the origin of the animal kingdom (Fig. 2).
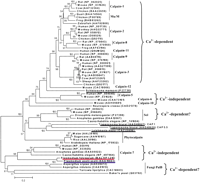
The phylogenetic tree of the calpains, inferred by the neighbor-joining method based on the amino acid sequences with Poisson corrected distance. The option of complete deletion of gaps was used for tree construction. 1000 bootstrap replicates were used to infer the reliability of branching points. Bootstrap values of >50% are presented. The scale bar indicates the number of amino acid substitutions per site. The parasite sequences are underlined. The putative P. falciparum calpain and P. yoelii yoeliiortholog are highlighted in red and blue, respectively. The accession number for each sequence is included in the parentheses after the species name.
The identification of plasmodial calpain has also implicated the existence of calpain-mediated pathways. Its potential cognate targets include host cytoskeletal proteins such as spectrin, integrin, and ezrin. Moreover, the recent discovery of a typical endogenous substrate of calpain, Protein Kinase C (PFL1110c; PFI1685w) in P. falciparum, has provided the support of a parasite-controlled signaling cascade (Doerig et al. 2002).
It is conceivable that the putative protease-active and Ca2+-independent plasmodial calpain may serve as a good antimalarial target for two reasons. First, it may be the central component of crucial signal transduction pathways that affect parasite biology and host-parasite interactions. Second, because it is evolutionarily divergent from the essential subtypes of host calpains, its specific inhibitor may have minimal effects on the host.
Metacaspase
Metacaspase (PF13_0289) is another interesting hypothetical protease. In vertebrates, a cascade of caspases (cysteineaspartyl proteases) is the major modulator of apoptosis (programmed cell death) (Thornberry and Lazbnik 1998; Aravind et al. 1999). Two families of ancient caspase-like proteins (paracaspases and metacaspases) have been found in metazoans, fungi, and protozoa. As shown in the phylogenetic tree (Fig.3), the putative plasmodial metacaspase occupies a distinct clade constituting paracaspases and metacaspases, which are likely to be the primordial form of 14 subfamilies of vertebrate caspases (bootstrap value = 100%). Interestingly, human paracaspase is capable of interacting with the oncogene Bcl10 and triggering NF-kB activation, indicative of the prone-to-apoptosis property of the ancestral caspase (Uren et al. 2000). Moreover, yeast metacaspase has been demonstrated as an effective executor for apoptosis, suggesting the root of apoptosis dates back to unicellular organisms (Madeo et al. 2002). The multiple alignment clearly reveals that the putative plasmodial metacaspase retains the typical caspase fold, which is centered with the His (404)-Cys (460) catalytic dyad conserved in all representative proteolytically active caspases (Fig.4). Conversely, considerable sequence diversity is observed in the vicinity of this active site cleft. In particular, yeast metacaspase and the plasmodial homolog exhibit distinct sequence profile to other vertebrate caspases and human paracaspase. Previously, Uren et al. (2000) have postulated that ancient (paracaspases and metacaspases) and vertebrate subtypes differ in substrate-specificity. We have demonstrated that the experimentally confirmed differential substrate-specificity in major vertebrate subtypes is largely determined by the chemical property and configuration of residues situated in the caspase fold (Wang and Gu 2001). Thus, the observed distinct configuration of residues in the active site proximity could account for parasite-specific substrate-preference.
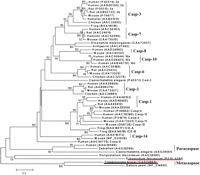
The phylogenetic tree of the caspases, inferred by the neighbor-joining method based on the amino acid sequences with Poisson corrected distance. The option of complete deletion of gaps was used for tree construction. 1000 bootstrap replicates were used to infer the reliability of branching points. Bootstrap values of >50% are presented. The scale bar indicates the number of amino acid substitutions per site. The protozoan sequences are underlined.

Multiple alignment of the catalytic regions of the putative P. falciparum metacaspase (PF13_0289) and the representative proteolytically active caspases using Clustal X1.8 followed by manual correction. The catalytic dyad H (404) and C (460), which are part of the active sites, is highlighted with arrowheads. Graphic presentation of the alignment and the consensus sequence were obtained by the program BOXSHADE 3.21. Conserved residues are shaded with black and gray. The accession numbers of caspase protein sequences used for alignment refer to Figure 3.
In Plasmodium, the physiological process of apoptosis has never been reported, nor the critical components identified. Nevertheless, the detection of the metacaspase homolog will allow us to investigate the role, if any, of apoptosis and/or analogous signal transduction pathway in the parasite. In addition, since metacaspases have only been found in protozoans, yeasts, and possibly in plants, and are phylogenetically distinct from other caspase subtypes (Fig. 3), the putative plasmodial metacaspase may serve as a potential chemotherapeutic target.
Signal Peptidase 1 (SP1)
Signal peptidases (SP) play indispensable roles in protein trafficking and sorting by removing signal peptides from precursors of secretary proteins. This serine protease family consists of two subtypes, SP1 and signalase, based on their distinct structural, functional, and evolutionary features. To date, SPs have been found in bacteria, archaea, fungi, plants, and animals; however, SP has never been reported previously in protists, despite the fact that the dynamic parasite life cycle reflects a need of specific peptidase(s) to process proteins that are translocated across host and parasite membranes. Using the comparative genomic search, we first identified two homologs of signal peptidase, PF13_0118 (SP1) and MAL13P1.167 (signalase) in P. falciparum.
Between two subtypes, SP1 has generated extensive research interest because it represents a novel antibiotic target for its distinct prokaryotic origin and essential functions (Paetzel et al. 2000). We have also identified an ortholog of P. falciparum SP1 in the rodent parasite P. yoelii yoelii genome. Our evolutionary analysis revealed that the two putative plasmodial SP1 have three clusters of homologs: (1) Bacteria SP1; (2) an Arabidopsischloroplast thylakoidal processing peptidase; and (3) mitochondrial inner membrane peptidases (Imp) found in eukaryotes, which appear to be the nearest neighbor to plasmodial SP1 (Fig.5). Given the proposed prokaryotic origin of the chloroplast and mitochondrion, malarial SP1 is likely to have evolved via the prokaryotic-specific lineage. Moreover, the potential of its catalytic activity can be inferred from the comparative sequence analysis. The putative SP1 contains the catalytic dyad (Ser175, Lys274) that is invariable across representative SP1 proteins with confirmed signal peptidase activity (Fig. 6). Most notably, this Ser/Lys catalytic dyad mechanism is unique in SP1, compared with the typical Ser/His/Asp triad system in other serine proteases. It seems plausible that the putative plasmodial SP1 has a fundamental role yet to be determined, and represents a promising target given its distant relatedness to the host.

The phylogenetic tree of the Signal peptidases I (SP1), inferred by the neighbor-joining method based on the amino acid sequences with Poisson corrected distance. The option of complete deletion of gaps was used for tree construction. 1000 bootstrap replicates were used to infer the reliability of branching points. Bootstrap values of >50% are presented. The scale bar indicates the number of amino acid substitutions per site. The putative P. falciparum calpain andP. yoelii yoelii ortholog are highlighted in red and blue, respectively. The SP1 homolog in Arabidopsis is termed Chloroplast thylakoidal processing peptidase. Imp is the abbreviation for mitochondrial inner membrane peptidase.

Multiple alignment of the catalytic regions of the putative P. falciparum SP1 (PF13_0118) and the representative proteolytically active signal peptidase I using T-coffee program followed by manual correction. The catalytic dyad Ser (S175) and Lys (K274), which are part of the active sites, is highlighted with arrowheads. Graphic presentation of the alignment and the consensus sequence were obtained by the program BOXSHADE 3.21. Conserved residues are shaded with black and gray. The accession numbers of SP1 protein sequences used for alignment refer to Figure 5.
Important Protease-Mediated Pathways Implicated inP. falciparum
Our findings suggested at least five new protease-mediated activities: (1) an ATP-dependent ubiquitin-proteasome-mediated cell-cycle control and stress-response system (Verma et al. 2002). Although the mechanism by which proteasomes function in P. falciparum is poorly understood, their importance was suggested by the observed irreversible inhibition on the growth and development of the hepatic and erythrocytic stages of three differentPlasmodium species by Lactacystin, a specific threonine protease inhibitor (Gantt et al. 1998). The identification of the clade of threonine proteases α and β, and a series of ubiquitinyl hydrolases (UCH1 and UCH2) brings new insight into this universally conserved proteasome machinery (Table 1). (2) A lysosomal proteolysis. This selective pathway to degrade cytosolic proteins may involve a number of cathepsins with versatile functions, which are assisted by cytosolic and lysosomal molecular chaperones and receptor proteins in the lysosomal membrane. (3) A calpain-activated signal transduction cascade, which may work in conjunction with upstream modulator and downstream effectors of host or parasite origin. (4) A caspase-mediated apoptosis or apoptosis-like signal transduction pathway. Although yeast metacaspase has been confirmed to induce apoptosis, the classical apoptosis regulators appear to be missing in the yeast genome. Thus, it is desirable yet challenging to identify the key components in this pathway, which may be conserved across organisms, or be parasite-specific. (5) A signal peptidase-initiated precursor protein processing pathway.
Evolutionary Implications
Studying the origin and the evolutionary mechanisms behind plasmodial proteases will contribute to the selection of target proteases to be studied in detail, for which specific inhibitors with no or minimal effect on the host can be designed. A complex evolutionary scenario including gene duplication, domain shuffling, and lateral gene transfer has been implicated in the preliminary analysis of the predicted proteolytic machinery in P. falciparum. Gene duplication is believed to play important roles in the evolution of multigene families by providing raw material for the novel functionality under differential evolutionary constraints (Ohno 1970;Li 1983; Friedman and Hughes 2001; Gu et al. 2002; McLysaght et al. 2002). In P. falciparum, well-characterized falcipains (-1, -2, -3), plesmepsins (I-IV), and subtilases (-1, -2) exemplify the multigene families that arise from gene duplications (Coombs 2001;Rosenthal 2002). We have identified a series of putative proteases that may comprise multigene families (Table 1). Some reflect tandem gene duplications in adjacent chromosome loci. For example, eight SERA homologs aggregate as a cluster in chromosome 2 contig 11953 (Miller et al. 2002). In contrast, some potential paralogs are located in remote chromosome regions. For instance, the UCH2 family with the consensus domain is sparsely distributed over seven chromosomes. This suggests that ancient gene duplications and subsequent functional divergence may result in an extensive repertoire of the present multigene families. In addition to gene duplication, domain shuffling coupled with the splice-site variation, intron loss, and horizontal gene transfer are proposed to be important modes in the evolution of aspartic proteases in the parasite genus Apicomplexa, including P. falciparum (Jean et al. 2001). The proteases encoded by or destined to parasite organelles are of particular interest because organelles represent microenvironments in which proteases may evolve at different rates and thus achieve novel functions (Fast et al. 2001). The first target organelle is the apicoplast, the apicomplexan-specific plastid derived by secondary reduction of a red alga endosymbiont. Since the plastid-encoded gene is of prokaryotic origin, its inhibitor may have only a minor, if any, effect on the vertebrate host and therefore may represent a promising antimalarial target. Our preliminary analysis shows that the putative clpC gene “PF11_0175” matches one apicoplast-encoded gene (Wilson et al. 1996). Moreover, 14 predicted proteases may contain an apicoplast transit peptide, among 511 genes identified in the entire parasite genome by pattern-recognition program PATS (Predict Apicoplast-Targeted Sequences) (Zuegge et al. 2001). From the population genetics perspective, we would anticipate detecting a certain level of polymorphism among putative proteases, due to the ancient origin of P. falciparum as revealed by chromosome-wide SNP analysis (Verra and Hughes 1999; Mu et al. 2002; Wootton et al. 2002). However, the alternative Malaria's eve hypothesis of a severe recent population bottleneck may still be valid (Rich et al. 1998;Volkman et al. 2001). More detailed analysis of the genomics and proteomics of plasmodial proteases will help resolve these fundamental questions about P. falciparum evolution.
Eighty-Three Putative Proteases Are Actively Transcribed in the Intraerythrocytic Stage, and Sixty-Seven Are Actively Translated in the Life Cycle
We are bearing in mind that genome analysis based solely on sequence similarity clearly predicts many unknown putative malaria proteases, however, these are only predictions. Which of the 59 newly predicted proteases, in addition to the 12 characterized proteases and 21 proteases annotated previously, are true protein-encoding genes expressed in the parasite life cycle? This important question was first addressed by analyzing an en masse gene expression profile using microarray chips, and then followed by RT-PCR confirmation.
Microarray
We focused on the parasite expression profiles of the asexual erythrocyte stage not only because this stage is responsible for malaria clinical manifestations, but also because of the accessibility of the research materials. In order to obtain all genes transcribed throughout the erythrocyte stage of the parasite, we extracted and pooled mRNAs from P. falicarpum 3D7 culture samples collected at four 12-h intervals. Figure 7 shows the temporal development of parasites that includes rings, trophozoites, schizonts, and merozoites, indicating that an asynchrony was successfully achieved. Probes were labeled with fluorescent dyes using mRNAs purified from the asynchronous culture as a template, and then hybridized to the microchip arrayed with 6239 Malaria Genome Array Oligomers (Operon Technologies).

Four P. falciparum 3D7 culture samples were collected at 12-h intervals, and pooled to achieve a total asynchrony.
Results, summarized in Table2, clearly demonstrated that 75 predicted proteases have signal intensities higher than those of negative controls. Being aware that the cut-off value for signal intensity is controversial, and that using the average intensity of the negative controls may be somewhat arbitrary, we selected the gametocyte-specific proteins (CS protein TRAP-related protein, Pfs25, Pfs48/45, Pfg377, and a gametocyte-specific var) and three large gene families in which the majorities are silent due to clonal expression switching (var, rifin, and stevor) as internal references (Hayward et al. 2000; Ben Mamoun et al. 2001). As anticipated, all gametocyte-specific genes, 39 of 45 var genes, 99 of 118 rifin genes, and 12 of 14 stevor genes displayed signal intensities below the level of the negative controls (data not shown). These data further support our conclusion that 75 predicted proteases are actively transcribed during the erythrocytic stage. Interestingly, the putative multigene families such as SERA and UCH2 exhibit variable expression levels across paralogous members, reflecting a certain level of functional divergence after gene duplication events.
Expression Profiles of Putative Plasmodial Proteases
We also analyzed two microarray datasets published in the PlasmoDB. The first dataset includes the expression profile of two erythrocyte stages (Trophozoites and Schizonts) using the Oligo Microarray (Hayward et al. 2000). The result of 66 predicted proteases transcribed in at least one stage supported our finding that the majority of the predicted proteases were actively transcribed during the erythrocyte stage (Table2). The second dataset represents the first proof-of-concept experiment of using cDNA microarray to explore the expression profile of five erythrocytic forms and stages (Ben Mamoun et al. 2001). Among 944 elements or gene fragments (317 genes of identifiable homology) included in the probe design, eight corresponded to predicted proteases. The positive signals of seven genes are consistent with our result from asynchronous culture. The stage-specific profile also confirmed the ubiquitous expression of the putative proteosome β6 (PFI1545c), which does not have corresponding 70-mer in the Oligo Microarray.
Reverse Transcription Polymerase Chain Reaction
Among the 17 remaining predicted proteases that are not detected using microarray hybridization, seven showed signal intensity below the negative controls. One possibility is that some of them are expressed in stages other than the asexual erythrocytic ones. This could be further investigated by using RNAs extracted from the intraerythrocytic and extraerythrocytic stages. The remaining ten predicted proteases were not included in the oligomer set printed on the array slides because only ∼90% of the P. falciparum genome data was available when the oligomers were designed. To examine whether these predicted proteases were also expressed in the erythrocyte stage, we designed specific primers and performed RT-PCR using the RNA extracted from the asynchronous culture (Fig. 7) as templates. Data shown in Figure 8A clearly suggests that all ten predicted genes were actively transcribed.
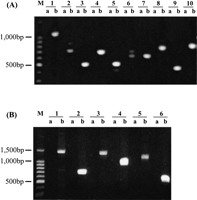
(A) Expression of ten putative proteases without corresponding oligomers in the microarray set. RT-PCR was conducted to examine the transcriptional expression of the ten putative proteases using specific primers based on the prediction. Lane a in each sample represents the negative control in which RT-PCR was conducted without reverse transcriptase. Lane 1b: PF14_0281; Lane2b: PFL1635w; Lane 3b: MAL6P1.153; Lane 4b: PFL1925w; Lane 5b: PFC0950C; Lane 6b: MAL8P1.16; Lane7b: MAL6P1.88; Lane 8b: MAL8P1.128; Lane 9b: PFA0400c; Lane 10b: PFI1545c.M indicates 1 kb DNA ladder. (B) Expression of putative calpain, caspase, and SP1 genes using two pairs of primers. RT-PCR was conducted to confirm the transcriptional expression of putative calpain, metacaspase, and SP-1 genes, using 2 pairs of specific primers for each gene. Lanes “a” represent negative controls in which RT-PCR was conducted without reverse transcriptase.M indicates 1 kb DNA ladder. Lanes1b and 2b: MAL13P1.310; Lanes 3b and4b: PF13_0289; Lanes 5b and 6b: PF13_0118. See Suppl. Table 1 for the predicted size of RT-PCR products.
As mentioned above, P. falciparum calpain, metacaspase, and signal peptidase1 are of particular interest due to the potential biological roles they may play. The microarray analysis suggested that the predicted genes for these proteases were actively transcribed (Table 2). We also performed RT-PCR to further confirm their expression (Fig. 8B).
The microarray and RT-PCR data only indicated the active transcription of 85 predicted proteases. In order to examine expression at the level of translation, we analyzed the proteomics data published in PlasmoDB (Florens et al. 2002). It appeared that 67 out of 92 predicted proteases are translated at some point during the life cycle (Table 2). Some proteases are ubiquitous, whereas others show stage-specific expression. It was notable that the three predicted proteases that did not have detectable transcription from the microarray assay did show positive translation in specific stages including intraerythrocytic stages.
Combining the complementary results from microarray, RT-PCR, and proteomics analysis, we found that of the 92 putative proteases identified by scanning the genome, 88 were transcribed and 67 were translated at some stage in the life cycle. The remaining four may be expressed at extraerythrocytic stages or may be pseudogenes, a result due to the frameshift in the open reading frame (Triglia et al. 2001).
Conclusions
The exhaustive homology search and comparative sequence analysis have resulted in the delineation of 92 putative proteases, including 59 that had not been previously recognized in the P. falciparumgenome. This set includes potentially important proteases such as calpain, metacaspase, and signal peptidase, and indicates protease-mediated activity that may be vital for parasite life cycle. Furthermore, 88 are demonstrated to be actively transcribed proteins by the microarray, RT-PCR data, and proteomics. This study is an initial attempt at the systematic identification of novel malaria proteases that have essential functions and assessment of their evolutionary relationship to the vertebrate host. By combining in silico genomics-based predictions with experimental confirmation, there is an increased likelihood of identifying new therapeutic targets.
METHODS
Genome Sequences, Homology Search, and Comparative Sequence Analysis
A total of 5865 nonredundant query sequences of characterized and predicted proteases from 1066 organisms were obtained from the Merops database (http://www.merops.ac.uk, release 5.8 of March 19, 2002), which has a catalog and a structure-based classification of proteases. The BLASTP searches with default setting were targeted to the predicted and annotated Plasmodium falciparum genome that was published in the PlasmoDB (http://plasmodb.org/; Kissinger et al. 2002). A cut-off criteria of E-value <1e-04 was adopted to define protease homologs. Partial sequences (<80% of full-length) and redundant sequences were excluded. Conserved domains/motifs in P. falciparum sequences were identified by searching InterPro release 5.1, which integrates Pfam 7.3, PRINTS 33.0, PROSITE 17.5, ProDom 2001.3, SMART 3.1, TIGRFAMs 1.2, and the current SWISS-PROT + TrEMBL data.
Multiple alignments were obtained by the program T-coffee (Notredame et al. 2000), followed by manual editing according to the structure information. Graphic presentation of the alignment and consensus sequences were deduced by the program BOXSHADE 3.21 (http://www.ch.embnet.org/software/BOX_form.html). Phylogenetic trees were inferred by the neighbor-joining method (Saitou and Nei 1987) using MEGA2.0 (http://www.megasoftware.net/). Unweighted Maximum Parsimony (as implemented in PAUP 4.0) and Maximum Likelihood (as implemented in PHYLIP) were used to examine whether the inferred phylogeny is sensitive to any tree-making method. The bootstrap resampling with 1000 pseudoreplicates was carried out to assess support for each individual branch. Bootstrap values of <50% were collapsed and treated as unresolved polytomies.
Microarray Expression Analysis Using Asynchronous ErythrocyticP. falciparum Culture
An en masse gene expression profile was obtained using microarray chips arrayed with 6239 Malaria Genome Array Oligomers (Operon Technologies), designed by Dr. Joe DeRisi of the University of California at San Francisco (http://derisilab.ucsf.edu/). These 6239 70-mers mapped to 4407 predicted open reading frames which covered >90% of the available P. falciparum genome sequences. In order to obtain all genes transcribed throughout the erythrocyte stage of the parasite, we extracted and pooled mRNAs from P. falciparum 3D7 culture samples (Trager and Jensen 1976) collected at four 12-h intervals to achieve an asynchrony (shown in Fig. 3). Probes were labeled with fluorescent dyes using mRNAs purified from the asynchronous culture as a template. Messager RNAs were purified using oligo T cellulose, and reverse transcription was conducted to incorporate aminoallyl dUTP into the cDNAs. The Cy3 and Cy5 NHS esters were then coupled to amine groups of the cDNA, and dye-labeled probes were hybridized with the microarray slides under standard conditions (3×SSC, 50% formamide, 0.1% SDS, 10 mg/mL salmon sperm DNA, 68°C). The slide was scanned with a GenePix 4000B (Axon Instrument) at default PMT settings, 100% power. The array data were analyzed initially with GenePixPro software (Axon Instrument), then with global normalization. The expression level is indicated by the mean signal intensity of all corresponding oligomers in triplicates on the microarray slides (MRA-452) obtained from Malaria Research and Reference Resource Center (http://www.malaria.mr4.org/). Two sets of negative controls were included in the DeRisi design: (1) 20 oligomers from yeast intergenic region with the mean intensity 529, (2) 33P. falciparum genes cloned into a plasmid, including 16 ribosomal proteins, 17 tRNA genes, LSU, Clp, andtufA. Their mean intensity was 598.
Reverse Transcription Polymerase Chain Reaction
RT-PCR was performed using the same mRNA described above as template. Reverse transcription was conducted using SuperScript II (Invitrogen). The PCR cycle: 95°C 1 min; (95°C 1 min, 54°C 30 sec, 52°C 30 sec, 65°C 1 min) x 35, 65°C 10 min, hold at 4°C. The primer sequences used to amplify 10 target genes without corresponding oligomers in the array set, and putative calpain, metacaspase, and signal peptidase I are included in the Supplemental Table 1.
WEB SITE REFERENCES
http://www.merops.ac.uk; a catalogue and structure-based classification of proteases.
http://www.expasy.ch/cgi-bin/lists?peptidas.txt; classification of peptidase (protease) families in SWISS-PROT.
http://plasmodb.org; official database of the malaria parasite genome project.
http://www.ch.embnet.org/software/BOX_form.html; software for printing and shading of multiple alignment files.
http://www.megasoftware.net/; software package for molecular evolutionary genetics analysis.
http://derisilab.ucsf.edu/; microarray resources provided by Dr. Joseph DeRisi at University of California, San Francisco.
http://www.malaria.mr4.org/; Malaria Research and Reference Reagent Resource Center.
Acknowledgments
We thank Lois Blaine, David Emerson, and Thomas Nerad for their critical comments during the manuscript preparation, Truc Nguyen for computational support. This study is supported by an ATCC start-up fund to Y.W., and an NIH-grant (1R21AI49300) to Y.W. We thank the scientists and funding agencies comprising the International Malaria Genome Project for making sequence data from the genome of P. falciparum (3D7) public prior to publication of the completed sequence. The Sanger Centre (UK) provided sequence data for chromosomes 1, 3–9, and 13, with financial support from the Wellcome Trust. A consortium composed of The Institute for Genome Research, along with the Naval Medical Research Center (USA), sequenced chromosomes 2, 10, 11 & 14, with support from NIAID/NIH, the Burroughs Wellcome Fund, and the Department of Defense. The Stanford Genome Technology Center (USA) sequenced chromosome 12, with support from the Burroughs Wellcome Fund. The Plasmodium Genome Database is a collaborative effort of investigators at the University of Pennsylvania (USA) and Monash University (Melbourne, Australia), supported by the Burroughs Wellcome Fund.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
-
↵4 Present address: Malaria Vaccine Development Unit, National Institute of Allergy and Infectious Diseases, National Institutes of Health, Bethesda, MD 20892, USA.
-
↵5 Corresponding author.
-
E-MAIL ywang{at}atcc.org; FAX (703) 365-2740.
-
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.913403.
-
- Received October 16, 2002.
- Accepted January 28, 2003.
- Cold Spring Harbor Laboratory Press








