
The Role of Lineage-Specific Gene Family Expansion in the Evolution of Eukaryotes
Abstract
A computational procedure was developed for systematic detection of lineage-specific expansions (LSEs) of protein families in sequenced genomes and applied to obtain a census of LSEs in five eukaryotic species, the yeasts Saccharomyces cerevisiae andSchizosaccharomyces pombe, the nematode Caenorhabditis elegans, the fruit fly Drosophila melanogaster, and the green plant Arabidopsis thaliana. A significant fraction of the proteins encoded in each of these genomes, up to 80% in A. thaliana, belong to LSEs. Many paralogous gene families in each of the analyzed species are almost entirely comprised of LSEs, indicating that their diversification occurred after the divergence of the major lineages of the eukaryotic crown group. The LSEs show readily discernible patterns of protein functions. The functional categories most prone to LSE are structural proteins, enzymes involved in an organism's response to pathogens and environmental stress, and various components of signaling pathways responsible for specificity, including ubiquitin ligase E3 subunits and transcription factors. The functions of several previously uncharacterized, vastly expanded protein families were predicted through in-depth protein sequence analysis, for example, small-molecule kinases and methylases that are expanded independently in the fly and in the nematode. The functions of several other major LSEs remain mysterious; these protein families are attractive targets for experimental discovery of novel, lineage-specific functions in eukaryotes. LSEs seem to be one of the principal means of adaptation and one of the most important sources of organizational and regulatory diversity in crown-group eukaryotes.
[Supplemental material is available online at ftp://ncbi.nlm.nih.gov/pub/aravind/expansions, and http://www.genome.org.]
The eukaryotic crown group (the unresolved assemblage of lineages in the eukaryotic tree, which includes plants, animals, fungi, and some protists, as opposed to early branching eukaryotes, which are all unicellular protists), although only representing the proverbial tip of the eukaryotic phylogenetic iceberg, encompasses a remarkable variety of organisms (Patterson 1999; Dacks and Doolittle 2001). This diversity is apparent in both morphological and biochemical features of the crown group that spans the entire range from unicellular yeasts and chlorophytes, through facultatively multicellular slime molds, to genuine multicellular organisms, plants, animals, and fungi (Sogin et al. 1996; Patterson 1999). The complete, or nearly complete, genome sequences from three major branches of the crown group, plants, animals, and fungi are starting to provide the first molecular explanations for both their unity and diversity. From one viewpoint, the crown-group eukaryotes are remarkably uniform in that they share a large set of conserved orthologs in the core components of their essential functional systems, such as those involved in DNA replication and repair, most aspects of RNA metabolism, cytoskeletal organization, protein degradation, and secretion (Chervitz et al. 1998; Rubin et al. 2000; Lander et al. 2001). Furthermore, components of the signal transduction pathways, structural and regulatory components of the nucleus, and pre-mRNA processing complexes, although showing clear differences between the major crown-group lineages, are largely constructed from the same set of protein domains, and are based on the same architectural principles (Chervitz et al. 1998; Aravind and Subramanian 1999; Rubin et al. 2000;Lander et al. 2001).
This unity notwithstanding, preliminary comparative studies on the sequenced eukaryotic genomes also provided clues as to what evolutionary phenomena might underlie their diversity. At the level of the protein sets encoded in the crown-group genomes, the main contributing forces appear to be the emergence of new domain architectures through domain accretion and domain shuffling, lineage-specific gene loss, and lineage-specific expansion of protein families (Aravind and Subramanian 1999; Aravind et al. 2000; Rubin et al. 2000; Lander et al. 2001). Lineage-specific expansion (LSE) is defined in relative terms, as the proliferation of a protein family in a particular lineage, relative to the sister lineage, with which it is compared (Jordan et al. 2001). Thus, if two sister lineages, for example, Drosophila and Caenorhabditis representing insects and nematodes, respectively, are compared, all protein-family proliferation events (duplications to n-plications) that occurred in either of these lineages after their separation are considered LSEs.
Preliminary analysis of proteins from the crown-group eukaryotic genomes revealed some tangible correlations between LSE and emergence of new biological functions, response to diverse environmental pressures, and organizational complexity. Some of the most striking cases of LSE are related to pathogen and stress response and include, among other families, expansions of the immunoglobulin superfamily associated with the vertebrate immune system, AP-ATPases involved in plant disease resistance (Hulbert et al. 2001), and the cytochrome P450 family, which participates in detoxification systems in both plants and animals (Nelson 1999; Tijet et al. 2001). Transcription factors represent another functional category of proteins that tend to show widespread LSE: the independent expansions of the POZ–C2H2 and C4DM–C2H2 fusions in insects, the nuclear hormone receptors in nematodes, and the KRAB-domain-fused Zn-fingers in vertebrates, apparently made substantial contributions to the evolution of developmental and differentiation features specific to each of these lineages (Sluder et al. 1999; Aravind et al. 2000; Riechmann et al. 2000; Coulson et al. 2001; Lander et al. 2001).
Despite a wealth of anecdotal information, we are unaware of a systematic comparative analysis of LSEs in eukaryotic genomes. With this objective, we devised a procedure to systematically detect LSEs. Having identified LSEs in five eukaryotic proteomes, those ofSaccharomyces cerevisiae, Schizosaccharomyces pombe,Caenorhabditis elegans, Drosophila melanogaster, andArabidopsis thaliana, we predicted, wherever feasible, the biochemical or biological functions of the lineage-specific clusters (LSC) and explored their potential roles in the diversification of the crown group. Here, we present a systematic analysis of the demography of LSEs and provide evidence for a major involvement of LSEs in the generation of the diversity of biological functions in multicellular eukaryotes.
RESULTS AND DISCUSSION
Identification and Validation of Candidate Lineage-Specific Clusters
Using the clustering procedure described in the Methods section, we delineated candidate LSCs for five eukaryotic genomes. The automatically generated LSCs were further surveyed for false positives, that is, proteins that were unrelated to the rest of the proteins in the cluster, by using BLAST searches and multiple alignments. A subset of false-positives arose from compositionally biased segments that escaped filtering during the automatic process. The presence of some false-positives was mainly due to one or more of the proteins in a cluster containing multiple domains or being artificially fused to another protein. The majority of such false-positives were detected among A. thaliana proteins, in which gene prediction errors resulted in artificial fusions of distinct genes. On several occasions, these artificial gene fusions resulted in an erroneous merger of one or more distinct clusters; these were manually separated. Additionally, a few smaller clusters that belonged to a larger LSE-specific expansion were merged. On average, ∼9% of the LSCs of size greater than two were subjected to manual corrections.
The automatic procedure used for delineating candidate LSCs included single-linkage clustering of proteins by sequence similarity and an ultrametric tree construction using UPGMA (see Methods). These methods accurately reproduce phylogenetic relationships only under the strict molecular clock hypothesis. Therefore, to verify the phylogenetic coherence of the candidate clusters, 10 of the candidate LSCs from each analyzed species that consisted of 4 or more members and had homologs in other species were chosen for phylogenetic analysis. In each tree, the proteins from the candidate LSC grouped together and, in 48 of the 50 cases, this grouping was strongly supported by bootstrap analysis (>70%) to the exclusion of homologs from other species and paralogs from the same species that do not belong to the given LSC (Fig. 1; Supplementary Material available online at ftp://ncbi.nlm.nih.gov/pub/aravind/expansions, andhttp://www.genome.org). Thus, the clusters generated by the automatic procedure used here appeared to represent predominantly, if not exclusively, authentic LSEs and, therefore, could be utilized reliably for quantitative and qualitative analyses of this phenomenon. Certain limitations related to the current state of sequencing and annotation of the eukaryotic genomes need to be kept in mind when interpreting these clusters. Only one genome, that ofS. cerevisiae, should be considered truly complete, whereas in others, some genes are obviously still missing, for example, those that reside in heterochromatinic regions. Furthermore, given the known problems with gene prediction in plant and animal genomes, we removed nearly identical sequences prior to the LSC analysis (see Methods). This eliminated potential redundancy, but some true (nearly identical) paralogs resulting from recent duplications could have been lost in the process. Given this procedure, the results presented here should be considered conservative estimates of the number of genes in LSCs. On the other end of the spectrum, extremely diverged members of LSCs (or even entire LSCs), which retain minimal sequence conservation, could have been missed by this analysis.
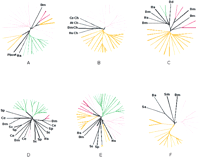
Phylogenetic analysis of selected eukaryotic lineage-specific expansions. Groups supported by a bootstrap value >70% are colored pink for Drosophila melanogaster, red for Homo sapiens, orange for Caenorhabditis elegans, green forArabidopsis thaliana, and yellow for Schizosaccharomyces pombe. (A) Prolyl hydroxylases. (B) Small molecule kinases (Ch stands for choline kinase). (C) Patched-like protein. (D) MAP-Kinases. (E) P450 family hydroxylases. (F) MBOAT membrane acyltransferases. (At)Arabidopsis thaliana; (Bs) Bacillus subtilis; (Ce)Caenorhabditis elegans; (Dd) Dictyostelium discoideum; (Dm) (Drosophila melanogaster; (Hs) Homo sapiens; (Pbcv1) Paramecium bursaria Chlorella virus 1; (Rs) Ralstonia solanacearum; (Sa) Staphylococcus aureus; (Sc) Saccharomyces cerevisiae; (Sm)Sinorhizobium meliloti; (Sp) Schizosaccharomyces pombe. Complete tree descriptions (full lists of GI numbers or gene names, and bootstrap values) are available in the Supplementary Material online atftp://ncbi.nlm.nih.gov/pub/aravind/expansions, andhttp://www.genome.org..
The two ascomycete yeasts, S. pombe and S. cerevisiae, were the closest pair of sister lineages compared. The two animals, D. melanogaster and C. elegans, represented a slightly greater phylogenetic divergence relative to each other, whereas the plant A. thaliana represented an even deeper branch with respect to animals and fungi. Thus, the LSCs from each of these species enabled us to examine the role of LSEs in diversification of eukaryotes at different levels of evolutionary divergence.
Proteome-Wide Demography of Lineage-Specific Family Expansion
The detected LSEs encompassed between ∼20% of the proteome (the yeasts) and ∼80% (A. thaliana) (Fig.2A). One of the causes for this diverse range of LSEs appears to be the phylogenetic distance factor; the two yeast species have accrued far fewer LSEs after diverging from their common ancestor compared with A. thaliana, which has no close sister lineages in the analyzed set of genomes and has, accordingly, gained the greatest number of expansions after its divergence from the common ancestor with fungi and animals. Positive linear correlations, with moderate-to-strong significance, were observed between the proteome size and each of the following: (1) fraction of proteins contained in LSCs (Fig. 2A), (2) number of LSCs (Fig 2B), and (3) average number of proteins per LSC (Fig. 2C). The majority of the clusters in each species consisted of two members. In each case, the number of two-member clusters showed a negative correlation with the proteome size, whereas the number of clusters with three or more members showed a positive correlation with the proteome size (Fig. 2D). Thus, larger proteomes had more proteins in larger LSCs at the expense of two-member LSCs. For each species, the distribution of the LSCs by the number of members followed the negative power law:P(k) = ck −γ in which P(k)is the frequency of families with exactly k members and c and γ are constants (Fig. 3). The differences between the slopes of these power law distributions (in double-logarithmic coordinates) were compatible with the aforementioned correlations between the degree of clustering and proteome size, that is, the yeast LSCs showed the steepest decay, whereas those fromA. thaliana had the flattest distribution (Fig. 3). This is also consistent with earlier observations that, in general, the size distribution of paralogous protein families in proteomes followed the power law decay (Huynen and van Nimwegen 1998; Qian et al. 2001). These findings suggest that LSCs evolved largely through a stochastic process of gene duplication whereby the probability of duplication within a cluster at any given time is proportional to the size of the cluster, rather than through genome-scale duplications.
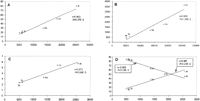
Linear correlation between the proteome size and parameters of eukaryotic lineage-specific expansion (LSE) in five eukaryotic species. Correlation coefficients (r) and significance levels (P) were determined using ordinary least square linear regression. (At)Arabidopsis thaliana; (Ce) Caenorhabditis elegans; (Dm) Drosophila melanogaster; (Sc) Saccharomyces cerevisiae; (Sp) Schizosaccharomyces pombe. (A) The proteome size (X-axis) is plotted against the percentage of the proteome made up of LSEs. (B) The proteome size (X-axis) is plotted against the number of lineage-specific clusters. (C) The proteome size (X-axis) is plotted against the mean number of proteins in lineage-specific clusters. (D) The proteome size (X-axis) is plotted against the percentage of duplication (♦) and the percentage of n-plication (n> = 3) (□) among the LSCs.
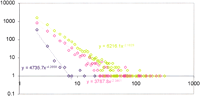
Size distribution of the lineage-specific clusters in three eukaryotic species. (Blue) Schizosaccharomyces pombe; (pink)Caenorhabditis elegans; (green) Arabidopsis thaliana. Cluster size (X-axis) is plotted against the number of LSCs in double logarithmic coordinates. The equations of the power law distribution fitting the linear part of the data are shown on the graph.
To characterize the role of LSEs in the evolution of the respective classes of paralogous proteins in each lineage, we devised the expansion coefficient (EC), which is the ratio of the number of proteins in LSCs to the total membership of the given class of paralogs in a given proteome. The EC is a measure of the fraction of a given paralogous class that has evolved through LSE after the divergence of the given lineage from the closest sister lineage included in the analysis. LSCs with EC = 1 are those families that have been invented de novo and proliferated thereafter in a particular lineage. The relative abundance of LSCs in the EC range between 0 and 0.9 is roughly constant for all taxa considered here, with slightly >5% of the LSCs in each of the bins of size 0.1 in this range (Fig.4). Notably, ∼40% (on average) of the LSCs present in a given proteome were in the EC range of 0.9 to 1 (Fig.4). Thus, nearly one-half of the paralogous protein clusters encoded in eukaryotic genomes have been generated almost entirely through LSE. This applied to the full range of evolutionary distances explored here and there was no obvious dependence on the evolutionary depth at which LSEs were identified; the fraction of paralogous classes contained in these exclusive LSCs was even greater in the yeast S. cerevisiae than it was in A. thaliana (Fig. 4). This observation, together with the correlations between proteome size and different parameters of LSEs (Fig. 2), suggests that the ancestral core set of proteins inherited by the crown-group lineages from their last common ancestor contained few paralogs compared with the extant proteomes. Subsequent to the divergence of the individual lineages, many genes inherited from the common ancestor as well as gene families invented de novo have undergone one or more rounds of duplication. This process seems to have been particularly active in the generation of the large proteomes of multicellular eukaryotes and probably provided them with the raw material for their cellular differentiation. In principle, it could be argued that the ancestor had as many paralogous families as the most complex of the extant genomes or even more, and the appearance of LSE had been created by lineage-specific gene loss, which is common in the evolution of at least some eukaryotic lineages (Aravind et al. 2000; Braun et al. 2000). However, apart from the general implausibility of a highly complex common ancestor for the crown group, this mechanism for the evolution of apparent LSEs would necessarily entail independent gene losses in the same paralogous family in multiple lineages, as opposed to a single expansion. Therefore, the lineage-specific duplication scenario is more parsimonious than the scenario based on the lineage-specific losses.
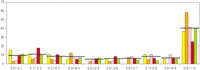
Distribution of lineage-specific clusters by Expansion Coefficient (EC).The X-axis shows ranges of EC values (see text) and the Y-axis shows the percentage of LSCs within each EC range. (Yellow)Schizosaccharomyces pombe; (orange) Saccharomyces cerevisiae; (pink) Drosophila melanogaster; (red)Caenorhabditis elegans; (green) Arabidopsis thaliana.In each class, the average value of the five species is indicated by a horizontal line.
Analysis of the top 25 LSEs with EC = 1 from all proteomes pooled together, indicated that the majority of them are α-helical proteins or have conserved patterns of histidines and cysteines. Typical examples include the α-helical nonspecific lipid-transfer protein in plants, the C4Dm domain in D. melanogaster that chelates cations through conserved cysteines, and the T20D4.15 family of disulfide-bonded secreted proteins from C. elegans. Thus, de novo emergence of protein domains that substantially contributed to LSEs appears to have involved primarily invention of structurally simple folds. These folds could have evolved through compaction of long α-helical coiled coils or through disulfide-bond- or metal-supported stabilization mediated by a few strategically placed, conserved cysteines and/or histidines. Invention of such simple domains could have been more expedient than emergence of complex α/β structures that require several specific stabilizing interactions to be fixed (Aravind and Koonin 2000).
Biological Significance of Lineage-Specific Expansions
The above observations show that, quantitatively, LSEs are a major component of the differences between the proteomes of various eukaryotic taxa. New paralogous families could provide the material for specific adaptations and for evolution of new functional systems. In qualitative terms, we sought to investigate the biological significance of LSEs by identifying conserved domains, subcellular localization signatures, such as signal peptides and transmembrane regions, and other features of proteins in LSCs that might allow prediction of their functions (when less than obvious). These identifications for the top five LSCs in each organism are shown in Table1. We categorized the LSCs into broad functional classes to discern global functional trends and also investigated individual LSCs in an attempt to gain a more detailed understanding of their actual biological roles (Table 2; Supplementary Material available online.).
The Top Five Lineage-Specific Gene Family Expansions in Five Eukaryotes
Functions of Selected Lineage-Specific Protein Clusters in Five Eukaryotes
Although LSEs occurred in most biological functional classes, LSCs with predicted organism-specific functions, such as pathogen and stress response, transcription regulation, controlled protein degradation mediated by the ubiquitin system, protein modification, signal transduction, chemoreception, and small molecule metabolism were most abundant (Tables 1 and 2). A typical example of an expansion related to an organism-specific function is that of the C. eleganscollagens, which are required for cuticle formation, a characteristic adaptation of the nematodes (Johnstone 2000). Similarly, in D. melanogaster and Arabidopsis, prominent LSEs are, respectively, the insect cuticular proteins (Andersen et al. 1995) and pectin/cellulose biosynthesis enzymes (Willats et al. 2001), both of which are critical for the formation of morphological features unique to these lineages. Typically, these proteins are required in large amounts as structural components of the respective organisms; hence, these lineage-specific expansions could principally help in increased production of these proteins. Extending this analogy, it is possible that several of the LSCs with no detectable homologs elsewhere could represent as yet uncharacterized, but abundant, lineage-specific structural proteins (Table 2).
Many of the identified LSCs had predicted biochemical characteristics that pointed to roles in stress and pathogen response. Particularly striking in this category was the expansion of proteases of the pepsin-like and subtilisin-like families in A. thaliana, trypsin-like proteases in D. melanogaster, and Zn-metalloproteases in C. elegans (Table 2). All of these proteases are predicted to be secreted molecules, and their repeated, independent expansion suggests that they are widely utilized either for direct degradation of pathogen proteins or as components of stress-triggered proteolytic cascades broadly analogous to the vertebrate complement and clotting systems (Bouchard and Tracy 2001;Southan 2001). Alternatively, in the case of plants, they could aid in protein digestion in the process of germination. Better-understood cases of similar lineage-specific expansions related to stress/pathogen-response components include the massive proliferation of apoptotic (AP-) ATPases and the accompanying moderate expansion of metacaspases in plants, and the parallel expansion of caspases in vertebrates (Aravind et al. 2001; Holub 2001). These proteins are either known or predicted to participate in multiple pathways associated with apoptosis or hypersensitive response. In this context, also of interest are the expansions of molecules containing modules functioning in extracellular adhesion. Prominent examples of these include the C-type lectins (D. melanogaster, C. elegans), PR1 proteins (C. elegans, A. thaliana), CUB domain proteins (C. elegans), and the bulb-lectin domain (A. thaliana). As with the immunoglobulin domain protein, that are highly expanded in vertebrates, these molecules probably participate in the recognition and binding of specific pathogens as a part of defense mechanisms of the corresponding organisms (Table 2).
Earlier analysis of the LSEs involving transcription factors had suggested that they included proteins regulating critical aspects of the development of the organism (Aravind and Koonin 1999; Riechmann et al. 2000; Lander et al. 2001). For example, the proteins belonging to the POZ and SAZ-type Myb domain expansions in D. melanogaster(Table 2) regulate as diverse functions as maintenance of the antero-posterior Hox gene expression pattern, neurite outgrowth and pathfinding, and organogenesis (Aravind and Koonin, 1999; Lander et al. 2001). Thus, it appears that proliferation of new transcription factor families, followed by their recruitment as upstream or downstream regulators with respect to core conserved developmental pathways, have contributed substantially to the evolution of morphological diversity in animals. The generality of this observation was reinforced by the evidence of massive, lineage-specific expansion and diversification of various transcription-factor families in the plant A. thaliana(Table 2). Many of these include well-characterized DNA-binding proteins, such as the MADS box and MYB domain proteins, that have been shown previously to participate in plant-specific functions, including development of flowers and other structures, meristemal differentiation, and organ-specific gene expression (Riechmann et al. 2000). In this study, we detected certain unexpected expansions of DNA-binding proteins in plants that might point to previously unrecognized transcription regulators. Examples include the proteins homologous to the mitochondrial transcription termination factor, which, in other eukaryotes, is present in a single copy that functions in the mitochondrion (Fernandez-Silva et al. 1997). The additional paralogs in plants have probably acquired different transcription-related functions because they form a tight cluster, distinct from the ancestral mitochondrial version. Plants also show an expansion of the DNA-binding replication factor A (RF-A), with >40 copies in A. thaliana, in contrast to the one-three copies observed in other eukaryotes. The expansion and divergence of RF-A in plants suggest that the plant-specific paralogs are probably utilized as transcription factors rather than in their usual capacity in replication (Wold 1997). These and other such examples (Table 2) illustrate that transcription factors are recruited from a wide variety of pre-existing sources and diversify to occupy new functional niches via LSE.
We observed a major role of LSE in the elaboration of the ubiquitin pathway, which is involved in the degradation and regulatory modifications of proteins (Hershko and Ciechanover 1998). Evidence of LSE was obtained for several components of the ubiquitin system, in particular, E3 subunits of ubiquitin ligases containing the F-box domain (Kipreos and Pagano 2000) (A. thaliana and C. elegans) and the RING-finger (A. thaliana). Because the E3 proteins are specificity determinants that are involved in targeting the conserved ubiquitin-ligation machinery system to specific substrates (Jackson et al. 2000), their diversification through LSE probably provides a means of harnessing an otherwise conserved system to regulate the degradation of diverse sets of targets. In a similar vein, both nematodes and plants also show independent LSEs of the MATH domain. This domain, which tends to form fusions to ubiquitin carboxy-terminal hydrolases or RING-finger E3s (Aravind et al. 1999;Polekhina et al. 2002), might serve as an additional adaptor that mediates de/ubiquitination of specific targets. A. thalianahas a prominent proliferation of the adenovirus-like thiol protease superfamily whose members (e.g., Smt4/Ulp1) in yeast and in vertebrates, remove ubiquitin-like proteins from their targets (Li and Hochstrasser 2000; Nishida et al. 2000). Thus, in plants, this LSC probably contributes to further diversification of the regulation of ubiquitin-dependent protein degradation. Targeting of proteins for degradation has been shown to occur through the recognition of hydroxyproline by ubiquitin ligase complexes (Ivan et al. 2001). Thus, the LSE of 2-oxoglutarate-dependent prolyl hydroxylases (Aravind and Koonin 2001) detected in D. melanogaster and A. thaliana could represent another case in which the range of the core ubiquitination pathway is expanded via diversification of the terminal effectors.
The role of LSE in the diversification of proximal components of signal transduction systems, receptors, had been noticed previously in the cases of independent expansions of odorant receptors/7-transmembrane chemoreceptors seen in different animal lineages (Dryer 2000; Glusman et al. 2001) and plant receptor kinases containing extracellular leucine-rich repeats, bulb lectin, or EGF-like extracellular domains (Shiu and Bleecker 2001). Here, we detected other analogous expansions of upstream signaling proteins, such as potassium channels, innexin family channels (both in C. elegans), and tetraspanins and degenerin-type channels in D. melanogaster (similar LSEs of K-channels and tetraspanins are also seen in humans). The proteins involved in these expansions are linked to the organism's responses to external as well as internal homeostatic stimuli. Thus, such expansions could serve as the raw material for the behavioral and physiological adaptation of organisms to their specific environments. Lineage-specific expansions are also seen in a range of protein-modifying enzymes of different signal transduction cascade, such as protein kinase families in most lineages, SET-domain protein-methylases in D. melanogaster, and PP2C phosphatases in plants. As with the ubiquitin system, these appear to be a means of linking well-conserved stems of signaling pathways to distinct sets of terminal targets.
Another aspect of the involvement of LSEs in the evolution of signal-transduction networks is the extensive proliferation of families of proteins containing adaptor domains. Along with their expansion, many adaptor domains have also recombined with a variety of other domains, probably allowing the emergence of new networks of interactions. A striking example is the major expansion of proteins containing the small Ca-binding octicosapeptide (OOP) module (Ponting 1996) in A. thaliana. Some OOP modules are fused to VIV1-like plant-specific DNA-binding proteins and a specialized class of GAF domains, suggesting that they link transcription regulation and small molecule interactions to Ca-dependent signaling. Another notable case is a novel adaptor domain, typified by the amino-terminal domain of the Caspase-1A isoform, which so far was detected only in C. elegans. Altogether, the C. elegans genome encodes >40 members of this domain family, which, in addition to the caspase fusion, also form multidomain proteins with SET-domain methylases, PHD fingers, and kinases. Given the α-helical structure predicted for this domain, and enrichment in charged residues, it probably functions as a protein–protein interaction module.
Another, somewhat unexpected generalization that emerged from the present analysis is the prevalence of small molecule-modifying enzymes among the LSEs. In plants, the proliferation of such enzymes, namely methylases of the caffeic acid O-methylase family, dioxygenases of the gibberellin-hydroxylase family, and a variety of lipases and acyltransferases, correlates with the plethora of secondary metabolites, such as pigments, volatile aromatic compounds alkaloids, and waxes that are produced by plants (Seigler 1998). However, their large numbers suggest that the entire diversity of metabolites produced even by plants such as A. thaliana with relatively simple genomes is under-appreciated to a large extent. Interestingly, animals also have several LSEs associated with small molecule metabolism. Some of these, such as glycosyltransferases and acyltransferases, suggest there might be an as yet unexplored, lineage-specific diversity of carbohydrates and lipid moieties that are associated with glycoproteins, lipoproteins, and other cellular metabolites. The two independent expansions of predicted small-molecule kinases related to ethanolamine and aminoglycoside kinases (Hon et al. 1997) (in D. melanogaster and, to a lesser extent, in C. elegans) and the expansion of secreted methylases in C. elegans are particularly enigmatic. Given the role of the related bacterial kinases and methylases in xenobiotic resistance (Haggblom 1990), these enzymes might be used to modify a range of xenobiotics encountered by the animals in their specific environments. Alternatively, they could modify various environmental substances to convert them to forms more easily sensed by the chemoreceptors of these organisms.
Conclusions
A computational procedure for systematic detection of lineage-specific expansions of protein families was developed and applied to obtain a comprehensive census of LSEs in five eukaryotic genomes. LSEs appear to have played an important role in the growth and differentiation of the proteomes of multicellular eukaryotes. Many paralogous gene families in crown-group eukaryotes appear to have evolved almost entirely through LSE after the divergence of the examined sister lineages from their ancestors. This fundamental process of gene family expansion was active at a wide range of phylogenetic distances, from the relatively close species of yeasts to the much earlier separation of plants from the rest of the crown-group taxa. Generally, the fraction of proteins found in LSCs and the fraction of large families among LSCs positively correlate with the size of eukaryotic proteomes.
Examination of the known and predicted functions of the detected LSEs reveals certain general principles. Genes encoding proteins typically required in large quantities as components of an organism's morphological structures are often subject to LSE and appear to be fixed versions of the common phenomenon of gene amplification, with fine-tuning added through sequence diversification (Kondrashov et al. 2002). Another major set of LSCs consists of proteins involved in recognition and binding of pathogens and xenobiotics and withstanding environmental stress. These LSCs probably provide the raw material for generating the diversity required to counter rapidly changing pathogens and to respond to other variable environmental factors. Expansion followed by diversification of the proteins in the LSCs appears to be a common means of generating new specificities in signaling pathways. In particular, in the ubiquitin system, a large number of the E3 components of the ubiquitin ligase, which target it to specific proteins, are drawn from LSEs. Expansions of adaptor modules followed by their fusion to diverse domains probably result in the emergence of novel interactions that contribute to signaling and transcription regulation. Several expanded enzyme families also point to the existence of an, as yet, undiscovered diversity of small molecule metabolites in various lineages. Thus, LSE seems to be one of the most important sources of structural and regulatory diversity in crown-group eukaryotes, which was critical for the tremendous exploration of the morphospace seen in these organisms.
METHODS
The protein set for the nematode C. elegans was from the WormPep20 data set (http://www.sanger.ac.uk/Projects/C_elegans/wormpep); the protein sets for other analyzed eukaryotic species were extracted from the NCBI (NIH) nonredundant (nr) protein sequence database. The human protein set was not systematically analyzed because of extensive problems with gene predictions, resulting in fragmentary proteins, artificial fusions, and inclusion of pseudogene translations and translation of noncoding DNA.
Identical or nearly identical (98% or greater) sequences were removed from the data sets using the BLASTCLUST program. For documentation on its use, seeftp://ftp.ncbi.nlm.nih.gov/blast/documents/README.bcl. LSCs were identified using the following procedure: BLASTcomparisons for all proteins in the analyzed set of complete eukaryotic genomes were run against the database consisting of the same set of proteins. Symmetrical relative similarity scores (R AB = R BA = max(S AB/S AA,S BA/S BB), in which S AB is the BLAST bit score for query A and subject B were recorded. Such scores range from 0 (no significant hit found) to 1 (identical proteins). For each protein A in a given genome X (e.g., C. elegans), a set of candidate family comembers {B} was defined as a set of proteins from the same genome X satisfying the condition (R AB>R AC; for ⦡C⊄X) (i.e., similarity between the given protein A and another C. elegansprotein B is greater than that between A and any protein C from any other genome). Then, all such sets from X were merged if they shared at least one member (single-linkage clustering), resulting in grouping all proteins from X into clusters {A} (many of which might contain only a single protein). This procedure leads to heavy overclustering because, even if only one pair of proteins in two distinct LSCs passes the comembership condition (e.g., due to fluctuations in the observable similarity), the two LSCs are merged by the single-linkage algorithm. This over-inclusive set of clusters was refined through identification of the most closely related proteins from other genomes. For each A⊄{A}, the best alien hit C was identified as [C ‖ max(R AC); C⊄X]. Sets {A}∪{C} (i.e., candidate LSC members and their closest alien relatives) were subject to UPGMA clustering on the basis of relative similarity scores. Under this procedure, proteins from other genomes that show high similarity to some candidate LSC members may intrude into the cluster and split it apart. Subclusters {A′} satisfying [A′⊂X] (i.e., UPGMA subtrees consisting of proteins exclusively from the currently analyzed genome X) and including more than one protein were considered to represent LSCs.
Protein sequence similarity searches were performed using the gappedBLASTP program against the nonredundant protein sequence database (NCBI, NIH). Iterative profile searches to detect more distant relationships were performed using thePSI-BLAST program (Altschul et al. 1997), with the inclusion threshold typically set at E = 0.01; only predicted globular regions from proteins were used as seeds for PSI-BLAST searches. Proteins were partitioned into probable globular and nonglobular regions using theSEG program (Wootton 1994). Conserved domains were detected using domain-specific PSSMs constructed using thePSI-BLAST program (Chervitz et al. 1998). Multiple alignments were constructed using the T_Coffee(Notredame et al. 2000) and ClustalX (Thompson et al. 1997) programs and corrected manually on the basis ofPSI-BLAST search results, which, on some occasions, correctly detect conserved sequence motifs missed by multiple alignment methods. These alignments were used to construct Neighbor Joining phylogenetic trees (Saitou and Nei 1987) using thePAUP* (Swofford 1998) and PHYLIP(Felsenstein 1996) package (the evolutionary distances were calculated using the PROTDIST program of PHYLIP), and the support for nodes of interest was evaluated by use of 1000 bootstrap replicates. Secondary structure of proteins was predicted using the PHD program, with multiple alignments used as input for prediction (Rost and Sander 1994). Signal peptides were predicted using theSignalP program (Nielsen et al. 1997).
The supplementary material available online atftp://ncbi.nlm.nih.gov/pub/aravind/expansions, andhttp://www.genome.org includes: (1) Complete lists of proteins in the identified lineage-specific clusters from five eukaryotic species (Format: text files). 2). The phylogenetic trees that were constructed to verify the ability of the above reported procedure to correctly detect lineage specific expansions (Format: text file containing trees that can be visualized with the Treeview program; Roderic Page; URL:http://taxonomy.zoology.gla.ac.uk/rod/treeview.html (3). A detailed version of table 2 with references for the entries wherever possible (Format: PDF).
WEB SITE REFERENCES
ftp://ftp.ncbi.nlm.nih.gov/blast/documents/README.bcl; Documentation for the BLASTCLUST program.
http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=Genome; Source of the analyzed protein sequence set except for those of C. elegans.
http://www.sanger.ac.uk/Projects/C_elegans/wormpep; Wormpep database, the source of the C. elegans proteins.
ftp://ncbi.nlm.nih.gov/pub/aravind/expansions; Supplementary material.
http://taxonomy.zoology.gla.ac.uk/rod/treeview.html; TreeView program for phylogenetic tree visualization.
Acknowledgments
We thank I. King Jordan and Kira Makarova for their help in developing the procedures for identifying the LSCs.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
-
↵1 Corresponding author.
-
E-MAIL koonin{at}ncbi.nlm.nih.gov; FAX (301) 435-7794.
-
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.174302.
-
- Received February 8, 2002.
- Accepted May 8, 2002.
- Cold Spring Harbor Laboratory Press








